

Summary
Aims
Thioredoxin‐interacting protein (TXNIP) is associated with activation of oxidative stress through inhibition of thioredoxin (Trx). However, some evidences point out that TXNIP acts as a scaffolding protein in signaling complex independent of cellular redox regulation. The autophagy‐lysosomal pathway plays important roles in the clearance of misfolded proteins and dysfunctional organelles. Lysosomal dysfunction has been involved in several neurodegenerative disorders including Parkinson's disease (PD). Although researchers have reported that TXNIP inhibited autophagic flux, the specific mechanism is rarely studied.
Methods
In this study, we investigated the effects of TXNIP on autophagic flux and α‐synuclein accumulation by Western blot in HEK293 cells transfected with TXNIP plasmid. Further, we explored the influence of TXNIP on DA neuron survival in substantia nigra by IHC.
Results
We found that TXNIP induced LC3‐II expression, but failed to degrade p62, a substrate of autophagy. Also, TXNIP aggravated α‐synuclein accumulation. We also found that TXNIP inhibited the expression of ATP13A2, a lysosomal membrane protein. Moreover, we found that overexpression of ATP13A2 attenuated the impairment of autophagic flux and α‐synuclein accumulation induced by TXNIP. Furthermore, overexpression of TXNIP in substantia nigra resulted in loss of DA neuron.
Conclusion
Our data suggested that TXNIP blocked autophagic flux and induced α‐synuclein accumulation through inhibition of ATP13A2, indicating TXNIP was a disease‐causing protein in PD.
Keywords: α‐synuclein, ATP13A2, autophagy, Parkinson's disease, TXNIP
1. INTRODUCTION
Degeneration of dopaminergic neuron in the substantia nigra pars compacta is a characteristic feature of Parkinson's disease (PD), one of the most common neurodegeneration diseases.1 The appearance of α‐synuclein‐containing Lewy bodies in DA neurons is the pathological hallmark of PD.2 Accumulation of α‐synuclein is due to disrupted protein degradation pathways.3 A main route for intracellular α‐syn degradation is autophagy, a lysosomal protein degradation pathway.4, 5 Autophagy pathway includes four steps: initiation, elongation, maturation, and fusion with the lysosome.6 Unobstructed autophagic flux is an indicator that the autophagy process is functional. However, increased number of autophagosomes is sometimes a result of inhibition of autophagosome clearance instead of indication of an activation of autophagy.7 Emerging evidences point out that dysfunction of autophagy was demonstrated to cause intracellular accumulation of proteins and neurodegeneration in in vitro and in vivo experiments.8 ATP13A2 gene encodes a transmembrane lysosomal ATPase and is essential to maintain lysosomal function. Reduced ATP13A2 expression resulted in lysosomal dysfunction and decreased lysosomal proteolysis.9 Overexpression of ATP13A2 in primary DA neurons suppressed α‐synuclein toxicity.10
Thioredoxin‐interacting protein (TXNIP) is the endogenous inhibitor of reactive oxygen species (ROS) elimination, by binding to the active cysteine residue of thioredoxin, resulting in oxidative stress.11 TXNIP overexpression renders cells more susceptible to oxidative stress and promotes apoptosis.12 It was reported that downregulation of TXNIP prevented retinal neurodegeneration.13 However, research reporting the role of TXNIP in PD is rare. Although it was reported that TXNIP inhibited autophagic flux, the mechanistic link between TXNIP and autophagy remains to be clarified.
This study was performed to investigate the mechanism of autophagic flux dysregulation induced by TXNIP. We showed that overexpression of TXNIP resulted in the impairment of autophagic flux via inhibition of ATP13A2. Meanwhile, we found that TXNIP induced α‐synuclein accumulation in α‐synuclein‐transfected cells. Furthermore, we found that TXNIP contributed to the loss of DA neurons in mouse midbrain. In summary, our study demonstrated that TXNIP was a key regulator for autophagy and played important roles in DA neuron degeneration.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
DMEM cell culture medium was purchased from Hyclone. Fetal bovine serum (FBS) was purchased from BI. p‐AMPK, AMPK, LC3, and p62 antibodies were purchased from Cell Signaling Technology. TXNIP was purchased from Abcam. β‐Actin, goat anti‐rabbit, and rabbit anti‐goat IgG horseradish peroxidase (HRP)‐conjugated secondary antibodies were purchased from Santa Cruz. Lip3000 was purchased from Thermo Fisher Scientific (Temecula, CA, USA).
2.2. Cell culture
HEK293 and SH‐SY5Y cells were cultured in DMEM supplemented with 10% FBS. Cells were cultured as a monolayer in 5% CO2 in a humidified incubator at 37°C.
2.3. Plasmids and transfection
The plasmids expressing TXNIP, RFP‐LC3, and ATP13A2 were purchased from GenePharma (Suzhou, China). HEK293 cells were cultured in 96‐well plates (1×104 cells/well) or in 6‐well plates (5×105 cells/well). 70%‐80% confluent HEK293 cells were transfected with TXNIP, RFP‐LC3, and ATP13A2 using Lip3000 in DMEM containing 10% FBS for 48 hours. The concentration of plasmid was 1 μg/mL for TXNIP, RFP‐LC3, and ATP13A2.
2.4. Assay of MTT conversion
Cells were seeded into 96‐well plates at a density of 104 cells/well in 200 μL culture medium. After treatment, the medium was replaced with 200 μL DMEM containing 0.5 mg/mL MTT and incubated at 37°C for 4 hours. Afterward, the supernatant was sucked out, and cells were lysed in 200 μL DMSO for 10 minutes at 37°C. The optical density (OD) values were measured at 490 nm using a plate reader. The obtained values were presented as fold of the control group.
2.5. Western blot analysis
Cells were washed three times with ice‐cold PBS and lysed with RIPA lysate containing PMSF on ice for 30 minutes. Samples were centrifuged for 25 minutes at 15 000 g at 4°C, and the supernatants were collected. Protein (60 μg for each extract) was resolved by 12% SDS‐PAGE, electroblotted to PVDF membrane, and blocked in 5% nonfat milk at room temperature. Membranes were incubated with primary antibodies overnight at 4°C. Membranes were washed by TBST and probed with HRP‐conjugated anti‐rabbit or anti‐goat IgG, respectively.
2.6. Mice and stereotaxic injections
C57BL/6 mice (23‐25 g), purchased from SLAC Laboratory Animal Ltd (Shanghai, China), were used in the study. Mice were kept under standard conditions according to governmental rules and regulations. We generated lentiviruses (LVs) encoding human TXNIP (GenePharma). Plasmid quality was tested by Western blot and sequencing. Mice were placed in a stereotaxic frame and anesthetized with 4% chloral hydrate. Viral solutions (1×109 viral particles/mL) were injected bilaterally into the substantia nigra using the following coordinates: (from bregma) anterior=−3.0 mm, lateral=1.0 mm, and (from skull surface) height=−4.4 mm. A 2 μL volume was injected stereotaxically over 10 minutes (injection speed: 0.2 μL/min) using Hamilton syringe. To prevent reflux around the injection track, the syringe was maintained in situ for 10 minutes, slowly pulled out halfway, and kept in position for an additional 2 minutes.
2.7. Immunohistochemistry
Mice were sacrificed 28 days after LVs injection. Mice were perfused with 4% paraformaldehyde (PFA). Brain samples were collected and postfixed in 4% PFA at 4°C overnight and then transferred to 15% sucrose and 30% sucrose in PBS for 2 days, respectively. The brain tissues were sectioned at 30 μm. Briefly, the sections were permeabilized in PBST (containing 0.3% Triton X‐100) for 1 hours and blocked with 5% BSA in PBS for 1 hours at room temperature. Then, the sections were incubated with primary antibody (anti‐TH, 1:800; Millipore, Waltham, MA, USA) at 4°C overnight and appropriate anti‐rabbit secondary antibody for 1 hours at room temperature. Immunostaining was visualized by DAB solution for 10 minutes.
2.8. Statistical analysis
Data were presented as mean±SEM. Statistical comparisons were analyzed by one‐way ANOVA and LSD test using the SPSS 16.0 (SPSS Inc, Chicago, IL, USA) software. P<.05 was considered as statistically significant.
3. RESULTS
3.1. TXNIP was upregulated in A53T mice and PD cellular model
We detected TXNIP in WT or A53T mouse midbrain. As shown in Figure 1A, TXNIP significantly increased in the midbrain of 5‐month‐old A53T mice compared with WT mice (P=.044; Figure 1A). Moreover, we evaluated TXNIP in α‐synuclein‐transfected HEK293 cells. TXNIP increased about 1.5‐fold in α‐synuclein‐transfected cells (P=.017; Figure 1B). Furthermore, we used the vector/α‐synuclein‐transfected cellular supernatant to stimulate SH‐SY5Y cells for 48 hours. The supernatant of α‐synuclein‐transfected HEK293 cells significantly induced TXNIP expression in SH‐SY5Y cells (P=.042; Figure 1C). These results indicated that TXNIP may play important roles in PD.
Figure 1.
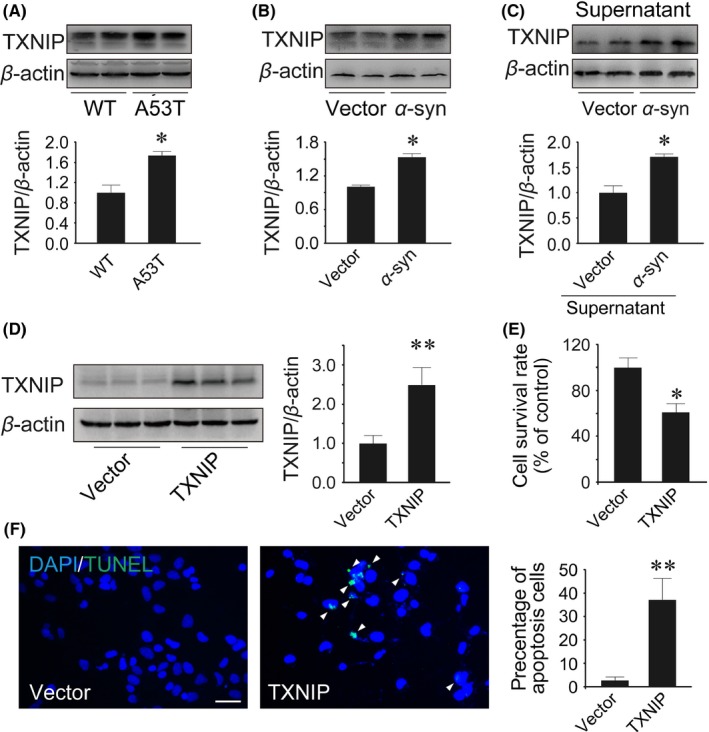
Thioredoxin‐interacting protein (TXNIP) increased in the midbrain of A53T mice and in Parkinson's disease (PD) cellular model. (A) Western blot analysis of TXNIP in 5‐month‐old WT and A53T mice. (B) Western blot analysis of TXNIP in HEK293 cells transfected with α‐synuclein plasmid. (C) The supernatant of HEK293 cells transfected with vector or α‐synuclein plasmid was collected to stimulate SH‐SY5Y cells for 48 hours. Then, TXNIP was detected by Western blot. HEK293 cells were cultured in 6‐well or 96‐well plates and transfected with TXNIP or α‐synuclein plasmid for 48 hours. (D) Western blot analysis of TXNIP. (E) Cytotoxicity was measured by MTT conversion. (F) Cell apoptosis was detected by TUNEL staining. *P<.05, **P<.01 vs WT mice or vector‐transfected cells. n=4 in (A); n=3 in (B‐F). Scale bar, 50 μm
3.2. Overexpression of TXNIP induced cell death
Moreover, we transfected TXNIP plasmid into HEK293 cells to upregulate TXNIP expression. TXNIP was remarkably increased by 2.5‐fold in HEK293 cells following the transfection with 2 μg TXNIP plasmid after 48 hours (6‐well plate) (P=.007; Figure 1D). To investigate the cytostatic effect of TXNIP, MTT assay was used to determine cell growth rate. Compared with control cells, TXNIP inhibited about 40% of cell growth (P=.049; Figure 1E). Moreover, we used TUNEL dyeing to evaluate the impact of TXNIP on apoptosis. The results showed that the percent of apoptosis cells was significantly increased by 7‐fold after TXNIP overexpression (P=.003; Figure 1F). These results indicated that TXNIP contributed to cellular apoptosis.
3.3. TXNIP inhibited autophagy
To determine the effect of TXNIP on autophagy, HEK293 cells were cotransfected with LC3‐RFP and TXNIP plasmids. We observed that upregulation of TXNIP significantly induced LC3 punctation in HEK293 cells (P=.011; Figure 2A). Meanwhile, we monitored changes in the processing of LC3 after TXNIP transfection. Endogenous LC3 transformation into PE‐conjugated LC3‐II was dramatically increased by overexpression of TXNIP (P=.002; Figure 2B). These results indicated that autophagosome was induced by TXNIP. On the contrary, p62, a marker of autophagic degradation, was significantly elevated after TXNIP transfection (P=.011; Figure 2B). What we observed pointed out that TXNIP blocked autophagic flux.
Figure 2.
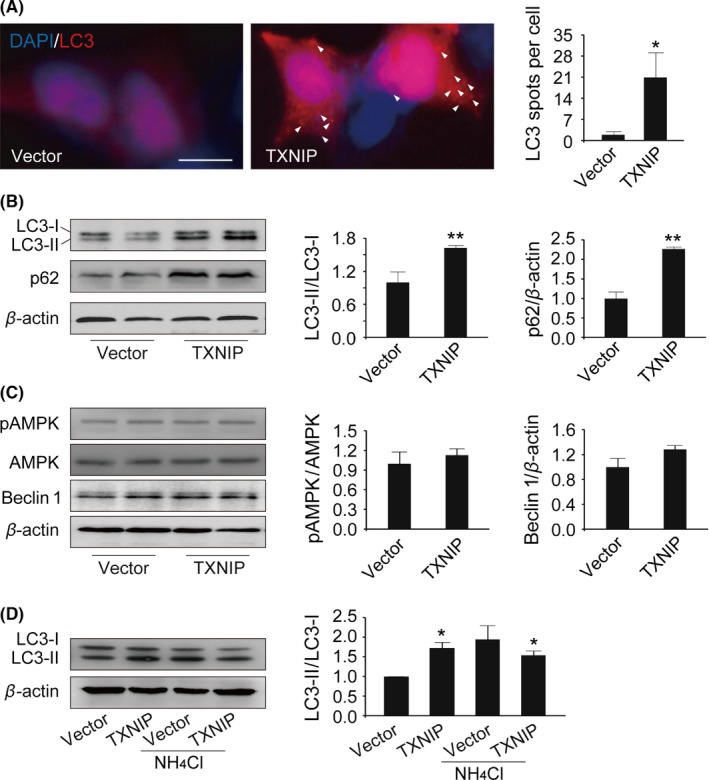
Thioredoxin‐interacting protein (TXNIP) blocked autophagic flux. (A) HEK293 cells were cotransfected with RFP‐LC3/TXNIP plasmids. LC3 spots were detected by fluorescence microscopy. To inhibit lysosome, HEK293 cells were treated with 20 μmol L−1 NH 4Cl from 7 to 48 hours after TXNIP transfection. (B‐D) LC3, p62, p‐AMPK, AMPK, and Beclin 1 were determined by Western blot analysis. *P<0.05, **P<0.01 vs vector‐transfected cells. n=3. Scale bar, 10 μm
3.4. TXNIP had no influence on AMPK‐mTOR pathway
Having identified that TXNIP inhibited autophagic flux, we next tried to explore the possible mechanisms. Given AMPK‐mTOR pathway is a key regulatory pathway in the upstream of autophagy, we detected the phosphorylation of AMPK by Western blot. Unfortunately, overexpression of TXNIP had no impact on the phosphorylation of AMPK (P=.588; Figure 2C). Meanwhile, Beclin 1 is another key regulator to initiate autophagy (P=.138; Figure 2C). However, we found that Beclin 1 had no change in TXNIP‐transfected cells. These findings indicated that TXNIP blocked autophagic flux not via inhibition of the upstream pathway of autophagy.
3.5. Overexpression of TXNIP resulted in lysosomal damage
Further, we focused on lysosome, which plays important roles in autophagy. First, HEK293 cells were incubated in NH4Cl, a lysosome inhibitor, 7 hours after TXNIP transfection. There was no further increase in LC3‐II in NH4Cl‐incubated cells transfected with TXNIP (Figure 2D). These results suggested that TXNIP blocked autophagic flux maybe via lysosome damage. Emerging evidences point out that ATP13A2, a lysosomal membrane protein, is critical to maintain lysosome function. Interestingly, ATP13A2 was significantly decreased in cells transfected with TXNIP (P=.034; Figure 3A). Then, we transfected ATP13A2 plasmid into HEK293 cells to increase ATP13A2 expression. As shown in Figure 3C, we found that LC3‐II was increased significantly in cells cotransfected with TXNIP and ATP13A2 plasmids compared with the control group (P=.013; Figure 3C). Meanwhile, p62, a substrate of autophagy, was decreased after ATP13A2 overexpression in cells transfected with TXNIP plasmid (P=.002; Figure 3C). These results indicated that TXNIP impaired lysosome via downregulation of ATP13A2.
Figure 3.
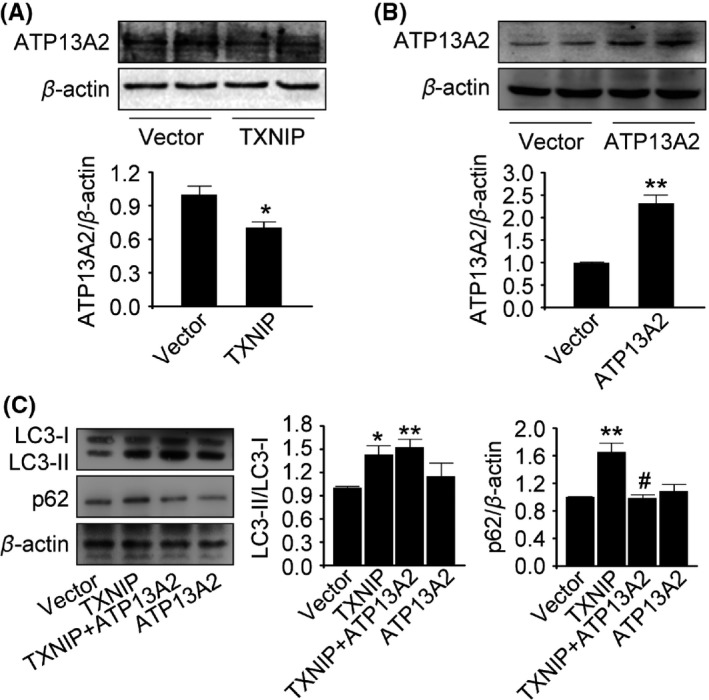
ATP13A2 improved dysfunction of autophagy induced by Thioredoxin‐interacting protein (TXNIP). (A‐C) ATP13A2, LC3, and p62 were detected by Western blot analysis. *P<0.05, **P<0.01 vs vector‐transfected cells; # P<0.05 vs TXNIP‐transfected cells. n=3
3.6. Overexpression of TXNIP resulted in α‐synuclein accumulation
Several studies reported that ATP13A2 deficiency leads to α‐synuclein accumulation. We further explored the impact of TXNIP on α‐synuclein. We transfected α‐synuclein plasmid into HEK293 cells using Lip3000. As shown in Figure 4A, α‐synuclein expression was significantly increased in α‐syn‐transfected cells (P=.001; Figure 4A). Upregulation of TXNIP in α‐syn‐transfected cells remarkably increased α‐synuclein more than 2‐fold compared with vector‐transfected cells (P=.002; Figure 4B). In contrast, increased ATP13A2 significantly attenuated α‐synuclein accumulation induced by TXNIP (P=.005; Figure 4B). These results showed that the impairment of autophagic flux induced by TXNIP resulted in α‐synuclein accumulation, suggesting that TXNIP was a harmful molecule in PD.
Figure 4.
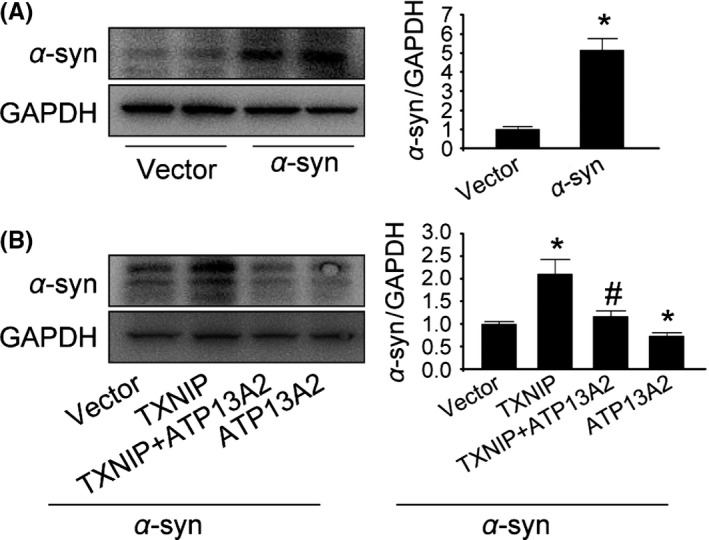
ATP13A2 attenuated α‐synuclein accumulation induced by Thioredoxin‐interacting protein (TXNIP). (A) HEK293 cells were transfected with α‐synuclein plasmid. α‐Synuclein was detected by Western blot analysis. (B) TXNIP and ATP13A2 were cotransfected into α‐syn‐transfected HEK293 cells to explore their effects on α‐synuclein accumulation. α‐Synuclein was detected by Western blot analysis. *P<0.05 vs vector‐transfected cells; # P<0.05 vs TXNIP‐transfected cells. n=3
3.7. Overexpression of TXNIP in SNc caused DA neuron loss
To further study the relation between TXNIP and PD, we stereotactically injected LVs containing TXNIP into mouse substantia nigra. Next, we examined the effect of TXNIP on TH‐positive neuron numbers. Increased TXNIP in SNc induced a 30% loss of DA neurons compared with control mice (P=.038; Figure 5A). Similar to the cellular results, our data showed that TXNIP significantly elevated LC3‐II level in the midbrain (P=.018; Figure 5C), but LC3‐II upregulation failed to decrease p62 (t=6.554, P=.001; Figure 5C). Moreover, lysosomal membrane protein ATP13A2 was decreased by 70% in TXNIP‐injected mice (P<.001; Figure 5D). α‐Synuclein, the primary component of the Lewy body, has been considered as a critical hallmark of PD. Meanwhile, TXNIP significantly increased α‐synuclein expression (P=.003; Figure 5D). These data suggested that TXNIP was a disease‐causing protein for PD and accelerated the progression of PD.
Figure 5.
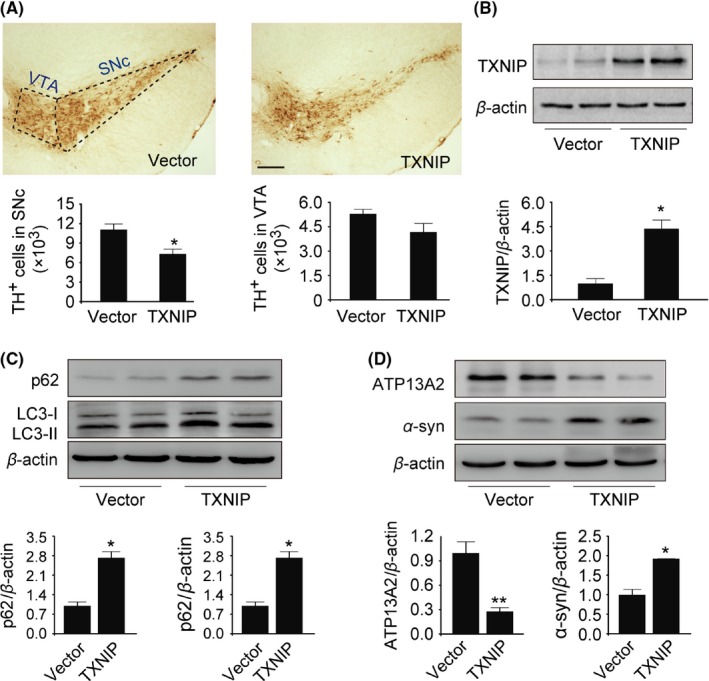
Thioredoxin‐interacting protein (TXNIP) resulted in DA neuron loss in mouse midbrain and induced autophagic dysfunction. (A) TH immunohistochemistry and stereological counts of TH‐positive cells in the midbrain. (B‐D) TXNIP, LC3, p62, ATP13A2, and α‐synuclein were detected by Western blot analysis. *P<.05 vs control mice. n=4. Scale bar, 200 μm
4. DISCUSSION
The present study was conducted to investigate the influence of TXNIP on autophagy and α‐synuclein accumulation. The results demonstrated increased formation of autophagosome and reduced autophagic clearance in TXNIP‐transfected cells. Meanwhile, we found that upregulation of TXNIP inhibited ATP13A2, a lysosomal membrane protein, and induced α‐synuclein accumulation. Furthermore, upregulation of ATP13A2 attenuated the impairment of autophagic flux and α‐synuclein accumulation induced by TXNIP. Moreover, stereotaxic injection of TXNIP into substantia nigra resulted in DA neuron loss in mice. Our findings indicated that TXNIP resulted in α‐synuclein accumulation by blocking autophagic flux via lysosome damage.
TXNIP, also known as thioredoxin‐binding protein‐2, is an extensively expressed protein which is known to promote oxidative stress by binding to and inhibiting Trx.14 TXNIP overexpression suppresses thioredoxin activity and induces apoptosis.15 It was found that TXNIP has a specific arrestin‐like domain, contributing to protein‐protein binding, to interact with several proteins, such as Jab1, E3 ubiquitin ligase itch,16 and NOD‐like receptor protein 3 (NLRP3).17, 18, 19 These findings point out that TXNIP may act as a scaffolding protein in signaling complex independent of the redox regulation.20 A few studies reported the impact of TXNIP on autophagy.13, 21 Xin‐Ming Chen reported that TXNIP mediates dysfunction of tubular autophagy in diabetic kidneys through inhibition of autophagic flux.13 Similarly, we also found that overexpression of TXNIP induced accumulation of LC3‐II, a marker of autophagosome, while it failed to degrade p62, a substrate of autophagy. These results confirmed that TXNIP blocked autophagic flux.
Autophagy mediates the degradation of misfolded/aggregated protein and dysfunctional organelles in cells, maintaining organism balance. Either excessive autophagy beyond a certain threshold or reduced autophagy flux will compromise cell survival.22 Autophagy pathway includes four key stages: initiation, phagophore elongation, autophagosome formation, and fusion with the lysosomes. It was pointed out that AMPK is an important regulator to initiate autophagy through inhibition of mTOR activity and phosphorylation of Ulk1.23, 24, 25 Ning Wu reported that AMPK activation resulted in TXNIP phosphorylation, leading to its rapid degradation.26 However, in our study, we found that increased TXNIP had no influence on AMPK phosphorylation. Beclin 1 also has a central role in autophagy, by interacting with Vps34 to induce autophagy.27, 28 We found that overexpression of TXNIP had no impact on Beclin 1 expression. These results suggested that TXNIP impaired autophagy not via inhibition of the upstream pathway of autophagy.
Autophagy is a lysosomal protein degradation pathway. Lysosome dysfunction results in failure to degrade the inclusions in autophagolysosome. In the present study, we found that TXNIP induced LC3‐II expression; however, NH4Cl, a lysosome inhibitor, did not increase LC3‐II expression further. This indicated that lysosome was damaged in TXNIP overexpression condition. Growing evidences indicate that ATP13A2, a lysosomal membrane protein, may have a profound role in maintaining the stability of the lysosome membrane structure and promoting the clearance of misfolded proteins.29, 30 Furthermore, studies have shown that loss of ATP13A2 impairs the stability of the lysosome membrane and suppresses the clearance of substrates in neurons. Fortunately, we found that ATP13A2 was inhibited in TXNIP‐transfected cells. Several studies reported that deficiency of ATP13A2 leads to the accumulation of α‐synuclein, a pathological protein in PD.31, 32 We further explored the impact of TXNIP on α‐synuclein. TXNIP significantly increased α‐synuclein expression in α‐synuclein‐transfected cells. Moreover, upregulation of ATP13A2 attenuated α‐synuclein accumulation induced by TXNIP. Similarly, overexpression of TXNIP in substantia nigra resulted in autophagy dysfunction and downregulation of ATP13A2. Fortunately, we also found that overexpression of TXNIP in substantia nigra induced DA neuron loss. Additionally, given the important role that TXNIP plays in inflammasome, we surmise that neuroinflammation triggered by TXNIP also contributes to DA neuron loss.33 Together, these results suggested that TXNIP blocked autophagic flux and was harmful in PD via the promotion of α‐synuclein accumulation.
We demonstrated for the first time that TXNIP inhibited autophagic flux and induced α‐synuclein accumulation through inhibition of ATP13A2, indicating TXNIP accelerated the progression of PD. Our data suggested that TXNIP might be a promising therapeutic target for PD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20150302) and the National Science Foundation of China (No. 81601098, No. 81603181).
Su C‐J, Feng Y, Liu T‐T, et al. Thioredoxin‐interacting protein induced α‐synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson's disease. CNS Neurosci Ther. 2017;23:717–723. 10.1111/cns.12721
Cun‐Jin Su and Yu Feng are co‐first authors of this work.
Contributor Information
Tong Liu, Email: liutong80@suda.edu.cn.
Yun‐Li Yu, Email: haoyyl0902@163.com.
REFERENCES
- 1. Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386(9996):896‐912. [DOI] [PubMed] [Google Scholar]
- 2. Kalia LV, Kalia SK. alpha‐Synuclein and Lewy pathology in Parkinson's disease. Curr Opin Neurol. 2015;28(4):375‐381. [DOI] [PubMed] [Google Scholar]
- 3. Winslow AR, Chen CW, Corrochano S, et al. alpha‐Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190(6):1023‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha‐synuclein: relevance to Parkinson's disease and related synucleopathies. Mov Disord. 2016;31(2):178‐192. [DOI] [PubMed] [Google Scholar]
- 5. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983‐997. [DOI] [PubMed] [Google Scholar]
- 6. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13(7):805‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martini‐Stoica H, Xu Y, Ballabio A, Zheng H. The autophagy‐lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39(4):221‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsui H, Sato F, Sato S, et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint‐like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. 2013;587(9):1316‐1325. [DOI] [PubMed] [Google Scholar]
- 10. Schultheis PJ, Fleming SM, Clippinger AK, et al. Atp13a2‐deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha‐synuclein accumulation and age‐dependent sensorimotor deficits. Hum Mol Genet. 2013;22(10):2067‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alhawiti NM, Al Mahri S, Aziz MA, et al. TXNIP in metabolic regulation: physiological role and therapeutic outlook. Curr Drug Targets. 2017;18(9):1095‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wei M, Jiao D, Han D, et al. Knockdown of RNF2 induces cell cycle arrest and apoptosis in prostate cancer cells through the upregulation of TXNIP. Oncotarget. 2017;8(3):5323‐5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang C, Lin MZ, Cheng D, et al. Thioredoxin‐interacting protein mediates dysfunction of tubular autophagy in diabetic kidneys through inhibiting autophagic flux. Lab Invest. 2014;94(3):309‐320. [DOI] [PubMed] [Google Scholar]
- 14. Nishiyama A, Matsui M, Iwata S, et al. Identification of thioredoxin‐binding protein‐2/vitamin D(3) up‐regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274(31):21645‐21650. [DOI] [PubMed] [Google Scholar]
- 15. Zhou J, Chng WJ. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion. 2013;13(3):163‐169. [DOI] [PubMed] [Google Scholar]
- 16. Zhang P, Wang C, Gao K, et al. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin‐interacting protein for ubiquitin‐dependent degradation. J Biol Chem. 2010;285(12):8869‐8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshihara E, Fujimoto S, Inagaki N, et al. Disruption of TBP‐2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat Commun. 2010;1:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou R, Tardivel A, Thorens B, et al. Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136‐140. [DOI] [PubMed] [Google Scholar]
- 19. Jeon JH, Lee KN, Hwang CY, et al. Tumor suppressor VDUP1 increases p27(kip1) stability by inhibiting JAB1. Cancer Res. 2005;65(11):4485‐4489. [DOI] [PubMed] [Google Scholar]
- 20. Yoshihara E, Masaki S, Matsuo Y, et al. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang C, Zhang Y, Kelly DJ, et al. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci Rep. 2016;6:29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rothermel BA, Hill JA. Myocyte autophagy in heart disease: friend or foe? Autophagy. 2007;3(6):632‐634. [DOI] [PubMed] [Google Scholar]
- 23. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ha J, Guan KL, Kim J. AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med. 2015;46:46‐62. [DOI] [PubMed] [Google Scholar]
- 25. Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381‐400. [DOI] [PubMed] [Google Scholar]
- 26. Wu N, Zheng B, Shaywitz A, et al. AMPK‐dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nazarko VY, Zhong Q. ULK1 targets Beclin‐1 in autophagy. Nat Cell Biol. 2013;15(7):727‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Usenovic M, Krainc D. Lysosomal dysfunction in neurodegeneration: the role of ATP13A2/PARK9. Autophagy. 2012;8(6):987‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dehay B, Martinez‐Vicente M, Ramirez A, et al. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy. 2012;8(9):1389‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha‐synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32(12):4240‐4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kett LR, Stiller B, Bernath MM, et al. alpha‐Synuclein‐independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35(14):5724‐5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye X, Zuo D, Yu L, et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem Biophys Res Commun. 2017;485(2):499‐505. [DOI] [PubMed] [Google Scholar]