

Summary
Introduction
Impaired dopamine D1 receptor (D1R) function in prefrontal cortex (PFC) is believed to contribute to the PFC hypofunction that has been hypothesized to be associated with negative symptoms and cognitive deficits in schizophrenia. It is therefore critical to understand the mechanisms for modulation of D1R function.
Aims
To investigate the physical interaction and functional modulation between D1R and GSK‐3β.
Results
D1R and GSK‐3β physically interact in cultured cells and native brain tissues. This direct interaction was found to occur at the S(417)PALS(421) motif in the C‐terminus of D1R. Inhibition of GSK‐3β impaired D1R activation along with a decrease in D1R‐GSK‐3β interaction. GSK‐3β inhibition reduced agonist‐stimulated D1R desensitization and endocytosis, the latter associated with the reduction of membrane translocation of β‐arrestin‐2. Similarly, inhibition of GSK‐3β in rat PFC also resulted in impaired D1R activation and association with GSK‐3β. Moreover, in a NMDA antagonist animal model of schizophrenia, we detected a decrease in prefrontal GSK‐3β activity and D1R‐GSK‐3β association and decreased D1R activation in the PFC.
Conclusions
The present work identified GSK‐3β as a new interacting protein for D1R functional regulation and revealed a novel mechanism for GSK‐3β‐regulated D1R function which may underlie D1R dysfunction in schizophrenia.
Keywords: Desensitization, Dopamine D1 receptor, GSK‐3β, Internalization, Schizophrenia, β‐arrestin
Introduction
Abnormal dopaminergic activity is associated with a number of neurological and psychiatric diseases such as Parkinson's disease, schizophrenia, and addictive disorders. Dopamine (DA) receptors are members of the G protein‐coupled receptor (GPCR) family and consist of five subtypes, D1‐D5. Like other GPCR receptors, DA receptors, in response to agonist binding, activate respective G proteins, resulting in the activation of second messenger systems. The activated DA receptor subsequently may undergo desensitization, internalization, and resensitization 1, 2, 3. Agonist‐mediated internalization is believed to be an important mechanism for discharging bound receptors and making receptor sites available again on the cell membrane. Thus, studying the regulatory mechanism of DA receptor trafficking and recycling is critical for understanding receptor function and regulation. Phosphorylation of intracellular domains of DA receptors plays an important role in this process.
Recent information indicates that many cellular proteins regulate the functions of GPCR via direct interaction with the receptors 4, 5, 6, 7, 8, 9, 10. Similarly, DA receptor function is regulated via the concerted efforts of a cohort of cytoskeletal, adaptor, and signaling proteins called DA receptor‐interacting proteins (DRIPs) 4, 5, 6, 7, 8, 9, 11. Altered DRIPs are associated with abnormal DA transmission in some pathological processes 5, 10, 11, 12, 13, 14. For instance, our laboratory and others have previously shown that PSD‐95 is physically associated with D1 and NMDA receptors and regulates receptor functions 6, 9. Dysfunction of the D1R is a common feature in a number of neuropsychiatric disorders 14, 15, 16, 17, and altered DRIPs may play an important role in this D1R dysfunction. Altered PSD‐95 expression has been associated with some neuropsychiatric disorders 12, 13. Furthermore, the D1R has been shown to associate with protein phosphatase 1, and selective hypofunction of D1Rs in frontal cortex of prenatal cocaine‐exposed rabbits was associated with the D1 receptor hyperphosphorylation 10. The conclusion from these studies is that alterations in expression or function of DRIPs play critical roles in the regulation of dopaminergic signaling and function.
In an effort to elucidate the underlying mechanisms for prefrontal D1R dysfunctions in schizophrenia, glycogen synthase kinase‐3 (GSK‐3) has drawn our particular attention as it has been proposed that schizophrenia is a GSK‐3β‐related disorder 18. GSK‐3 is an evolutionary conserved, constitutively active, serine/threonine kinase consisting of two isoforms, α and β. GSK‐3β has a central role in regulating neuronal differentiation, survival, and neurotransmission 18, 19, 20. Meanwhile, the Akt/GSK‐3β pathway is a converging target for many antipsychotics and mood stabilizers 21, 22. Accumulating evidences indicate that GSK‐3β pathway is critical in the regulation of DA and serotonin (5‐HT)‐mediated neurochemical and behavioral responses 23, 24, 25. It was shown that some psychostimulants inhibit Akt, consequently activating GSK‐3β 21, 22. Antagonism of dopamine D2 receptor has been found to activate Akt and result in GSK‐3β inhibition 26. Inhibition of GSK‐3β has been reported to attenuate D1 receptor agonist‐induced hyperactivity in mice 27. However, the mechanisms of the Akt/GSK‐3β pathway in D1R functional modulation and in D1R dysfunction in some psychotic conditions such as schizophrenia remain unknown.
In this study, we identified GSK‐3β as a novel interacting protein for D1Rs in cultured cells and in brain tissues. We further identified the S(417)PALS(421) motif of the C‐terminal in D1R as the site at which GSK‐3β interacts. Inhibition of GSK‐3β reduced the association between D1R and GSK‐3β and attenuated D1R internalization, ultimately resulting in D1R dysfunction. In a chronic phencyclidine (PCP)‐treated schizophrenia‐like animal model 28, we detected a decreased activation of GSK‐3β, a reduced association of D1R and GSK‐3β, and impaired D1R activation in prefrontal cortex (PFC). Collectively, our data demonstrated that GSK‐3β is an important interaction protein of D1R in the modulation of D1R function. Alterations in GSK‐3β that lead to D1R dysfunction may provide a new mechanism for understanding receptor dysfunction‐related diseases such as schizophrenia.
Materials and Methods
Preparations of cDNA Constructs
The full‐length human D1R cDNA cloned in pcDNA3.1 with a HA tag in N‐terminus was obtained from UMR cDNA Resource Center at University of Missouri‐Rolla (Rolla, MO, USA). Site‐directed mutagenesis of human D1R was made by substituting single or multiple serine residues with alanine using the wild‐type D1R as template. The mutated D1R cDNAs were subsequently cloned into pcDNA3.1 at the KpnI and XhoI restriction sites. CFP‐tagged D1R was constructed by replacing the stop codon of D1R complementary DNA construct with a BamHI restriction site for in‐frame fusion to the pECFP‐N1 vector 9. The full‐length human GSK‐3β cDNA was generated using reverse‐transcription PCR from total RNA extracted from HEK‐293 cells, which was also used as the template for construction of site‐directed GSK‐3β mutants. YFP‐tagged GSK‐3β was constructed by replacing the stop codon of GSK‐3β complementary DNA construct with a KpnI restriction site for in‐frame fusion to the pEYFP‐N1 vector. All the sequences of wild‐type or mutated cDNA were verified by DNA sequencing before cell transfection. Plasmids of Akt‐Wt, Akt‐myr, and Akt‐K179M were kindly provided by professor Cheng‐Xin Gong, CUNY, NY.
Cell Culture and Transfection
HEK‐293 cells were maintained in Dulbecco's modified Eagle's medium (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone), penicillin (100 unit/mL), and streptomycin (100unit/mL). Transfection was performed when cells reached 80% confluence. For transient transfection, plasmids containing the respective constructs were transfected by calcium phosphate method. If not indicated specifically, cells were used for experiments after 24–48 h posttransfection. For the selection of stable HEK‐293‐D1R cells, G418 (Sigma, St Louis, MO, USA) was added at the concentration of 300 μg/mL and was maintained at 100 μg/mL as described previously 9. Primary‐cultured striatal neurons were prepared from Sprague Dawley (SD) rats at embryonic day 18 and maintained in neurobasal medium supplemented with B27 (Invitrogen, Carlsbad, CA, USA) and 0.5 mM glutamine at medium density. The primary‐cultured neurons were transfected at DIV7 with cDNA constructs using Lipofectamine 2000 (Invitrogen).
Co‐immunoprecipitation and Western Blotting
Approximately 48 h after transfection, HEK‐293 cells were collected and solubilized on ice in a lysis buffer (20 mM HEPES, 150 mM NaCl, 2 mM EDTA, 10% glycerol, and 0.5% NP‐40, pH7.4) containing protease inhibitor cocktail (Sigma). The supernatants were collected by centrifugation for 10 min at 5000 × g. The prefrontal cortex tissues from SD rats were homogenized in the lysis buffer containing protease inhibitor cocktail and incubated on ice for 30 min before centrifuged at 20,000 × g for 10 min. The protein contents of the supernatants were determined by bicinchoninic acid (BCA) method (PIERCE). The same amount of protein in each sample was incubated with 1 μg anti‐HA‐tag antibody (Sigma) for transfected HEK‐293 cells or anti‐D1R antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for brain tissues at 4°C overnight. Immunocomplexes were subsequently captured by Protein A/G PLUS‐Agarose (Santa Cruz Biotechnology) and incubated at 4°C on a rocker platform for 2 h. Immunoprecipitates were finally collected after centrifugation and washed for 5 times, and the pellets were then resuspended in 1× electrophoresis sample buffer. All protein samples were resolved by SDS‐PAGE and probed with a primary antibody against GSK‐3β (Cell Signaling, Danvers, MA, USA) and a species‐specific HRP‐conjugated secondary IgG antibody (Santa Cruz Biotechnology). The immunoreactive signals were visualized by ECL/HRP method (PIERCE), and protein expression level was analyzed using the ImageJ program (NIH, USA).
Measurement of cAMP Accumulation
Stable HEK‐293‐D1R cells were reseeded onto 96‐well plates (1 × 104 cells/well) and incubated over night. For the assay of cAMP accumulation 9, cells were preincubated with 5 mM LiCl (Sigma) for 4 h or 5 μM SB216763 (Sigma) for 20 min prior to stimulation with various concentrations of dopamine (RBI, MA, USA) containing 500 μM isobutyl methylxanthine (IBMX, Sigma) in serum‐free DMEM for additional 10 min. The reaction was then terminated on ice by adding 100 μL of 1M trichloroacetic acid, followed by the addition of 20 μL 2M K2CO3. The samples were centrifuged, and the supernatants were kept (diluted in 1:50) for determining cAMP content using the [125I] cAMP assay kit from the Shanghai University of Traditional Chinese Medicine (Shanghai, China). All experiments were performed in duplicates, and each experiment was repeated at least three times.
Cell Membrane Preparation
Stable HEK‐293‐D1R cells were grown to confluence before LiCl (5 mM for 4 h) and SB216763 (5 μM for 20 min) treatment. For the assay of receptor desensitization, cells were further stimulated with 10 μM dopamine for 1 h in the presence of LiCl or SB216763. After washed twice with ice‐cold phosphate‐buffered saline (PBS), cells were harvested and collected through centrifugation at 100 × g for 10 min. The cells were then lysed in hypotonic buffer (5 mM Tris–HCl, pH7.4, 2 mM EDTA), containing a protease inhibitor mixture (Sigma). The lysate was centrifuged at 100 × g for 10 min, and the supernatant was further centrifuged at 40,000 × g for 30 min at 4°C. The pellet was kept for [35S]GTPγS‐binding assay. The plasma membrane fraction and the light vesicular membrane of crude membranes were separated using a method previously described 9. Briefly, crude membranes were resuspended in hypotonic buffer with protease inhibitor mixture and layered on top of a 35% sucrose cushion and centrifuged at 150,000 × g for 90 min at 4°C. The plasma membrane fractions at the bottom of the sucrose cushion were collected for radioligand saturation binding assays to evaluate the amount of membrane receptors according to our previous report 9.
Brain Membrane Preparation
PFC of rat brain was cut into 300 × 300 × 300 μm prisms using a Mcilwain tissue chopper (Brinkmann Instruments, Westbury, NY, USA) as reported previously 10. The prisms were washed in oxygenated Krebs‐Ringer's buffer (KRB) which had the following composition: 25 mM HEPES, pH7.4, 143 mM NaCl, 4.8 mM KCl, 20 mM NaHCO3, 1.3 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose, 100 μM ascorbic acid, oxygenated for 30 min. Tissues were subsequently incubated at 37°C for 30 min with 10 μM SB216763 and then put on ice to terminate the reactions. Brain tissues were homogenized in 1 mL ice‐cold buffer containing 50 mM Tris–HCl, 0.32M sucrose, 1 mM EDTA with a protease inhibitor mixture. The homogenate was centrifuged at 800 × g for 10 min, and the supernatant was further centrifuged for 30 min at 36,000 × g. The pellets were washed and resuspended in 50 mM Tris–HCl buffer (pH7.4) and centrifuged for 30 min at 36,000 × g. The pellets (brain membrane fraction) were stored at −80°C.
[35S]GTPγS‐Binding Assay
Agonist‐stimulated [35S]GTPγS binding was performed as described previously 9 in reaction buffer contained 50 mM Tris, pH7.4, 5 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 1 mM DTT. The assay mixture (200 μL) containing cell or brain membrane protein, 0.1 nM [35S]GTPγS (PerkinElmer, Boston, MA, USA), 20 μM GDP (Sigma) in the absence or presence of various concentrations of dopamine was incubated for 30 min at 30°C. Then reaction was stopped with 3 mL of ice‐cold reaction buffer, and samples were filtered rapidly through GF/B filter through a 24‐well cell harvester (Brandel, Montreal, QC, Canada) and rinsed three times with 3 mL reaction buffer. Filters were dried, and the binding activity was measured using a Beckman LS 6500 scintillation counter. Nonspecific binding was detected in the presence of 100 μM GPPNHP (Sigma).
Radioligand Receptor Binding Assay
For radioligand saturation binding assays, plasma membrane fractions were resuspended in binding buffer (50 mM Tris–HCl, pH7.4, 1 mM EDTA, 5 mM KCl, 1.5 mM CaCl2, 4 mM MgCl2 and 120 mM NaCl), 5–6 μg protein was added to assay tubes containing 0.1–10 nM [3H]SCH23390 (PerkinElmer) in a total volume of 0.2 mL. (+)‐Butaclamol (Sigma) was added at the final concentration of 1 μM to determine nonspecific binding. After 60‐min incubation at 30°C, the reaction was terminated by rapid filtration onto Whatman GF/B filters and filters were washed with 4 mL of ice‐cold washing buffer (50 mM Tris–HCl, pH7.4, 2 mM EDTA) three times. Radioactivity bound to the filters was quantified by liquid scintillation counting. All experiments were performed in duplicates, and each experiment was repeated at least three times.
Fluorescence Intracellular Accumulation Assay
Fluorescent‐based internalization assay was performed as described 29, 30 with minor modifications. HA‐tagged surface D1R were “live” labeled with rabbit anti‐HA polyclonal antibody by incubating coverslips in conditioned medium containing the antibody (10 μg/mL) for 20 min at 37°C. After brief washing in prewarmed DMEM, striatal neuron transfected with HA‐tagged D1R or stable HEK‐293‐D1R cells were either returned to conditioned medium (for control incubation) or stimulated for 20 min with 1 μM SKF81297. For testing inhibitor affection, LiCl (1 mM for 2 h) or SB216763 (5 μM for 20 min) was preincubated before agonist was added. Cells were immediately fixed in 4% formaldehyde for 10 min at room temperature, and surface‐remaining receptors were visualized with Alexa488‐conjugated secondary antibody. Internalized receptors were detected with Cy3‐conjugated secondary antibody after permeabilizing cells in methanol (−20°C) for 1 min. Confocal images were acquired using an Olympus confocal microscope (Fluoview 500IX71) and quantified with Fluoview 500 software (Olympus, Tokyo, Japan). Red fluorescence intensities indicative of internalization were divided by total (red+green) fluorescence intensities to control for cell density. Units of internalization were measured as red/total fluorescence normalized to untreated controls.
β‐arrestin‐2 Translocation Assay
β‐arrestin‐2 translocation assay was performed as described below. HEK‐293 cells were transiently transfected with β‐arrestin‐2‐GFP and D1R plasmids. One day after transfection, the cells were seeded onto 35‐mm dishes containing 1‐cm hole which was mounted with a glass coverslip. The medium was replaced with serum‐free DMEM for 2 h. LiCl (5 mM) or SB216763 (5 μM) was added for 4 h and 20 min, respectively. The β‐arrestin‐2 translocation was observed under the confocal microscopy (Leica, Wetzlar, Germany). Images were collected sequentially using single‐line excitation (488 nm). Saturating concentrations of agonist were applied directly over the selected cells. The relative level of β‐arrestin‐2‐GFP fluorescence was measured in a fixed area of the cytoplasm over the duration of the experiment. Data were analyzed using a “plateau with exponential decay” nonlinear regression function in GraphPad Prism. For Western blotting, stable HEK‐293‐D1R cells were treated with DA (10 μM) or vehicle for 5 min, the reactions were immediately terminated on ice. Cells were collected and membrane fractions were prepared according to the method described previously 9. Protein concentration was determined using the BCA protein assay kit, and 20 μg of protein was used for detection of β‐arrestin‐2 by immunoblotting.
Data Analysis
The Kd and Bmax for radioligand binding parameters as well as the EC50 and Emax values for dopamine‐stimulated cAMP production were fitted and calculated using Prism program (GraphPad Software, San Diego, CA, USA). The curves presented in this article represented the best fits to the data. Data were expressed as the mean ± SEM. For all data, except specified, one‐way ANOVA or Student's paired t‐test was applied. Significance was considered at the P < 0.05.
Results
Dopamine D1 Receptor is Constitutively Associated with GSK‐3β
We first checked the subcellular localization of D1R and GSK‐3β in transfected HEK‐293 cells with confocal immunofluorescence microscopy. D1R and GSK‐3β displayed different distribution patterns when expressed separately in HEK‐293 cells. Cells expressing GSK‐3β‐YFP alone exhibited diffuse staining throughout the cytoplasm, whereas GSK‐3β was translocated to the cell membrane and colocalized with D1R on the plasma membrane in GSK‐3β‐YFP‐ and D1R‐co‐expressed cells (Figure 1A), the fraction of membrane‐bound GSK‐3β increased from 22.3 ± 3.02% to 57.4 ± 5.86% of total GSK‐3β protein. The colocalization of GSK‐3β with D1R was also evidenced in primary‐cultured rat striatal neurons (Figure 1B). This was confirmed in striatal neurons that were transfected with D1R‐YFP and stained with GSK‐3β (Figure 1C). The quantification measurement using Mander's overlap coefficient analysis indicated that co‐expression of D1R and GSK‐3β in Figure 1B is 66.7 ± 7.38% and 67.2 ± 6.87% in Figure 1C, respectively. The physical association between D1R and GSK‐3β was also detected in stable HEK‐293‐D1R (HA‐tagged) cells and rat frontal cortex tissue (Figure 1D,E), and multiple control experiments indicated that the association between D1R and GSK‐3β is specific. Taken together, the D1R is constitutively associated with GSK‐3β in either constructed HEK‐293 cells or in native brain tissues.
Figure 1.
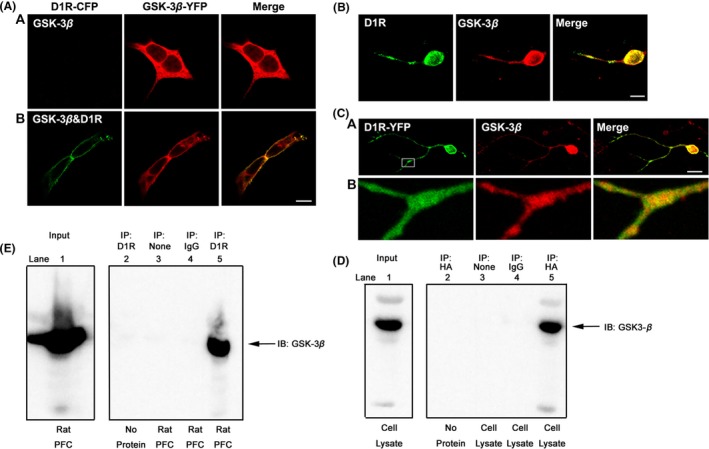
D1R constitutively associated with GSK‐3β. (A) D1R‐dependent translocation of GSK‐3β to the plasma membrane. HEK‐293 cells were transiently transfected with cDNA encoding for both D1R‐CFP and GSK‐3β‐YFP or GSK‐3β‐YFP alone. GSK‐3β (red) was diffusely distributed in the cytoplasm when HEK‐293 cells expressed GSK‐3β‐YFP alone (panel a). However, when HEK‐293 cells were cotransfected with D1R‐CFP and GSK‐3β‐YFP, GSK‐3β translocated to the plasma membrane and colocalized with D1R (green) (panel b). (B) D1R and GSK‐3β colocalized in primary‐cultured striatal neurons. Primary‐cultured striatal neurons were stained with anti‐D1R antibody and anti‐GSK‐3β antibody, respectively. D1R was visualized by Alexa Fluor® 488 (green), whereas GSK‐3β was visualized by Cy3 red secondary antibody. (C) Colocalization of GSK‐3β (red) and exogenously expressed D1R‐YFP (green) in primary‐cultured striatal neurons (panel a). An amplification of image (panel b). (D) D1R interacted with GSK‐3β in stable HEK‐293‐D1R cells. Cells were lysed and immunoprecipitated using anti‐HA‐tag antibody. Western blot analysis was carried out with anti‐GSK‐3β antibody to detect the presence of GSK‐3β in immunoprecipitates (lane 5). (E) D1R was associated with GSK‐3β in the native brain tissue. Rat prefrontal cortex (PFC) tissues were prepared as described in Materials and Methods and immunoprecipitated with anti‐D1R antibody, and the blots were revealed by GSK‐3β antibody (lane 5). Multiple controls for the immunoprecipitation assays. Lane 2: anti‐HA (or anti‐D1R) antibody without cell lysate (or rat tissue); Lane 3: cell lysate (or rat tissue) without anti‐HA (or anti‐D1R) antibody; Lane 4: cell lysate (or rat tissue) + rabbit IgG. All experiments have been repeated at least three times.
Modulation of GSK‐3β Activity Alters the Association between D1R and GSK‐3β
We next explored whether alteration in GSK‐3β activity could affect the D1R and GSK‐3β interaction. Two kinds of inhibitors for GSK‐3β were employed. LiCl (5 mM for 4 h), which inhibits GSK‐3β activation via elevating the phosphorylation of Ser‐9 of GSK‐3β (Figure 2A,B), resulted in a decreased association between D1R and GSK‐3β. SB216763 (5 μM for 20 min), a competitive GSK‐3β inhibitor, also decreased GSK‐3β in D1R precipitates (Figure 2A,B), indicating that GSK‐3β enzymatic activity is required for the receptor–protein interaction. This was further confirmed in experiments in which a dominant active plasmid of Akt (Akt‐myr) was transfected into stable HEK‐293‐D1R cells. In these experiments, the constitutive activated Akt resulted in the inactivation of GSK‐3β and decreased the association between D1R and GSK‐3β, thereby mimicking the effect of a GSK‐3β inhibitor. In contrast, transfection with dominant negative mutant Akt (Akt‐K179M), or wild‐type Akt produced no effect on the Ser‐9 phosphorylation of GSK‐3β and on D1R‐GSK‐3β association (Figure 2C,D). These data showed that inhibition of GSK‐3β activity decreased the interaction between D1R and GSK‐3β.
Figure 2.
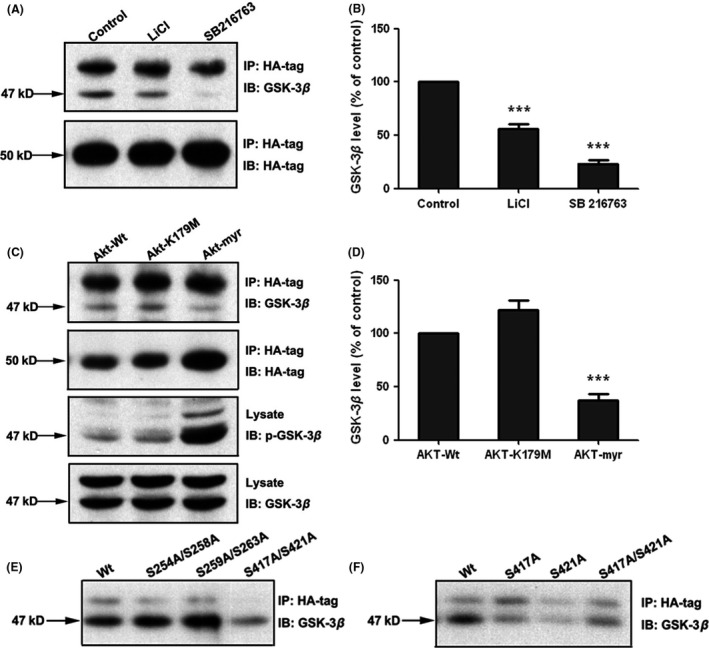
Inhibition of GSK‐3β decreases the association between D1R and GSK‐3β and D1R interacts with GSK‐3β through the C‐terminus of D1R. (A) GSK‐3β inhibitors decreased the association between GSK‐3β and D1R. Immunoblots were performed on HA‐D1R immunoprecipitates. Stable D1R‐expressing HEK‐293 cells were treated with either LiCl (5 mM for 4 h) or SB216763 (5μΜ for 20 min) or saline vehicle before cells were collected for immunoprecipitation and immunoblot assay. Representative immunoblots were shown. (C) Akt‐myr inhibited the association between D1R and GSK‐3β. Stable D1R‐expressing HEK‐293 cells were transiently transfected with cDNA encoding Akt‐wt, Akt‐myr or Akt‐K179M. Forty‐eight hours after transfection, cells were lysed and the immunoprecipitation assay was performed using anti‐HA‐tag antibody. Akt‐myr increased the GSK‐3β phosphorylation of Ser9 and decreased the association of D1R and GSK‐3β. (B) and (D) The quantification of optical density ratios for A and C, respectively. (E) and (F) Co‐immunoprecipitation analysis for mutated D1R and GSK‐3β. HEK‐293 cells were transiently transfected with wild‐type or mutated D1R (HA‐tagged) and prepared for co‐immunoprecipitation assays. After immunoprecipitated with anti‐HA antibody, the blots were revealed by GSK‐3β antibody. Site mutation on C‐terminus of D1R significantly decreased the interaction between D1R and GSK‐3β. Histograms show mean ± SEM. for at least three experiments. (***P < 0.001, LiCl/SB216763 vs. control or Akt‐mutant vs. Akt‐wt).
Interaction between D1R and GSK‐3β through the C‐terminus of D1R
The GSK‐3β activity‐sensitive association with D1R implied two possibilities: Either the interacting site of D1R with the kinase contains a structure motif that can be recognized as a substrate of GSK‐3β phosphorylation, or the activity change in GSK‐3β results in the conformation alteration that interrupts the interaction. As the canonical phosphorylation sequence recognized by GSK‐3β is SXXXS, it contains two serine residues separated by three residues, which is called primed phosphorylation (by another protein kinase) 21, 22. There are three SXXXS motifs in the cytoplasmic part of full‐length human D1R cDNA: S(254)QPES(258) and S(259)FKMS(263) in the third intracellular loop, and S(417)PALS(421) in the carboxyl tail. To identify whether GSK‐3β interacts with D1R through its phosphorylation sequence, we made progressive substitutive mutants by replacing serine with alanine. After verification by DNA sequencing, plasmids of wild‐type or respective mutated D1R were transfected into HEK‐293 cells and subsequently prepared for a co‐immunoprecipitation study. As shown in Figure 2E, the S417A/S421A mutation significantly decreased the interaction between D1R and GSK‐3β, while wild‐type or S254A/S258A‐ and S259A/S263A‐mutated D1R displayed a similar association with GSK‐3β, indicating that GSK‐3β binds to the S(417)PALS(421) motif in the carboxyl tail of D1R. This result is further confirmed by co‐immunoprecipitation in HEK‐293 cells expressing S417A or S421A site‐mutated D1R (Figure 2F), and both S(417) and S(421) mutations inhibited the interaction between D1R and GSK‐3β. Therefore, we concluded that D1R interacts with GSK‐3β through the C‐terminus of D1R.
Inhibition of GSK‐3β Attenuates D1R‐Mediated Gαs Protein Activation
To explore the functional consequence of alterations in the interaction between D1R and GSK‐3β, stable HEK‐293‐D1R cells were pretreated with either LiCl or SB216763 to inhibit the activity of GSK‐3β prior to DA stimulation. Inhibition of GSK‐3β by either LiCl or SB216763 attenuated the maximal cAMP production stimulated by DA (Emax value: 72.1 ± 13.1% for LiCl; 87.7 ± 9.09% for SB216763) without changing the EC50 value (Figure 3A). To further confirm this observation, we performed [35S]GTPγS‐binding assays in a membrane preparation to detect DA‐stimulated Gαs protein activation. There was a significant decrease in the Emax value for Gαs activation in response to DA in D1R‐expressing cells that were pretreated with LiCl or SB216763 (53.0 ± 14.0% of control for LiCl; 75.6 ± 21.6% of control for SB216763). Again, EC50 values were not altered by GSK‐3β inhibition (Figure 3B). These data demonstrated that inhibition of GSK‐3β which disrupts D1R and GSK‐3β interaction attenuated the D1R‐mediated Gαs protein activation.
Figure 3.

Inhibition of GSK3β attenuates D1R‐mediated Gαs protein activation. (A) Effects of GSK‐3β inhibition by LiCl or SB216763 on agonist‐stimulated cAMP accumulation in stable HEK‐293‐D1R cells. The cells were pretreated with either 5 mM LiCl (4 h), 10 μM SB216763 (20 min), or vehicle prior to and during the stimulation by DA (from 10−11 to 10−5M). The cAMP assay was conducted as described in Materials and Methods. The data are expressed as a percentage of the maximal cAMP response produced by the control group. (B) Effects of GSK3β inhibition by LiCl or SB216763 on D1R‐mediated Gαs protein activation in stable HEK‐293‐D1R cells in response to dopamine stimulation. Cells were pretreated with or without LiCl (4 h) or SB216763 (20 min). Cells were harvested and membranes were then prepared. [35S]GTP γS‐binding assays were performed. The data are expressed as a percentage of the maximal Gαs protein activation over the control group. (C) SB216763 inhibited the D1R‐mediated Gαs protein activation in rat PFC tissues. Tissues were prepared and treated with 10 μM SB216763 or vehicle for 30 min, and [35S]GTP γS‐binding assays were then performed. Histograms show mean ± SEM for at least three experiments. (*P < 0.05, ** P < 0.01, *** P < 0.001, LiCl/SB216763 vs. control). (D) SB216763 decreased D1R‐GSK‐3β association in rat PFC tissues. Tissues were prepared and treated as described in (C) and then were lysed, and immunoprecipitation was performed using anti‐D1R antibody. Western blot analysis was carried out with anti‐GSK‐3β antibody to detect the presence of GSK‐3β in immunoprecipitates.
Inhibition of Prefrontal Cortical GSK‐3β Activity Results in the Dysfunction of Prefrontal D1R
As we have shown that inhibition of GSK‐3β attenuated D1R activation in vitro (Figure 3A and 3B), we wonder whether inhibition of GSK‐3β will also impair D1R activation in native brain tissue. Rat PFC tissue blocks were prepared as described in Materials and Methods. After incubation with SB216763 for 30 min, tissues were lysed for membrane preparations. SB216763 pretreatment significantly reduced the SKF38393‐stimulated Gαs activation (Figure 3C), implicating that inhibition of GSK‐3β impaired PFC D1R function. As expected, the association between D1R and GSK‐3β was also decreased (Figure 3D).
Inhibition of GSK‐3β Alters the Membrane D1R Expression
To check whether GSK‐3β inhibition alters membrane D1R expression that may contribute to the decrease in D1R activation, we employed a radioligand saturation binding assay to evaluate the membrane D1R expression in response to LiCl‐ or SB216763‐mediated inhibition of GSK‐3β. As shown in Figure S1, inhibition of GSK‐3β produced a significant decrease in membrane D1R density as assessed by saturation binding assay (Bmax value: 4.96 ± 0.29pmol/mg for control; 3.74 ± 0.28pmol/mg for LiCl treatment, P < 0.001; 3.84 ± 0.61pmol/mg for SB216763 treatment, P < 0.001). Moreover, SB216763 altered the Kd value of D1R binding from 1.36 ± 0.16 nM (control) to 2.09 ± 0.14 nM (P < 0.001). The results clearly demonstrated that inhibition of GSK‐3β decreased membrane D1R density, which is in agreement with the GSK‐3β inhibition‐attenuated D1R activation.
Inhibition of GSK‐3β Interrupts D1R Endocytosis in HEK‐293 Cells and Primary‐Cultured Striatal Neurons
Decreased D1R membrane density could result from decreased receptor insertion into plasma membrane due to either disrupted receptor recycling or decreased receptor synthesis. However, an acute GSK‐3β inhibition‐mediated decrease in D1R membrane distribution as showed above is unlikely due to a change in receptor synthesis, but rather may result from the disruption of D1R recycling.
It is known that internalization of desensitized receptors via endocytosis, a process in which the activated receptors are removed from the cell surface into intracellular vesicles, is a common feature of GPCR recycling 31, 32. Receptor recycling is critical for making receptors available in plasma membranes for subsequent receptor stimulation. To elucidate the involvement of GSK‐3β in D1R recycling, we first investigated whether GSK‐3β can co‐internalize with D1R. Stable HEK‐293‐D1R cells were transiently transfected with GSK‐3β‐YFP. The D1R and GSK‐3β internalization were visualized by application of anti‐HA antibodies to living cells as described in Materials and Methods. As shown in Figure 4A, the co‐internalization of D1R and GSK‐3β was evidenced in response to DA stimulation. Further, we employed a fluorescent intracellular accumulation assay to measure DA‐induced D1R internalization. As shown in Figure 4B,C, DA application induced a robust D1R internalization in the absence of GSK‐3β inhibitors, whereas LiCl or SB216763 treatment attenuated the receptor internalization significantly (P < 0.001, compared with control), indicating that GSK‐3β inhibition may interrupt D1R endocytosis. To further confirm this observation, GSK‐3β specific siRNAs were designed and synthesized (P259 and P1223). Stable HEK‐293‐D1R cells were transfected with siRNA to knock down GSK‐3β. Both siRNA for GSK‐3β (P259, P1223) reduced the endogenous GSK‐3β expression (Figure 5C). Knocking down GSK‐3β also resulted in a significant decrease in DA stimulation‐induced D1R internalization (Figure 5A,B, P < 0.001). Experiments were conducted in primary‐cultured striatal neurons in which the D1R‐selective agonist SKF81297 induced a dramatic internalization of D1R in primary‐cultured neurons in the absence of GSK‐3β inhibitors. In contrast, application of a GSK‐3β inhibitor significantly attenuated the D1R internalization (Figure 4D,E). These results demonstrate that GSK‐3β regulates D1R endocytosis.
Figure 4.
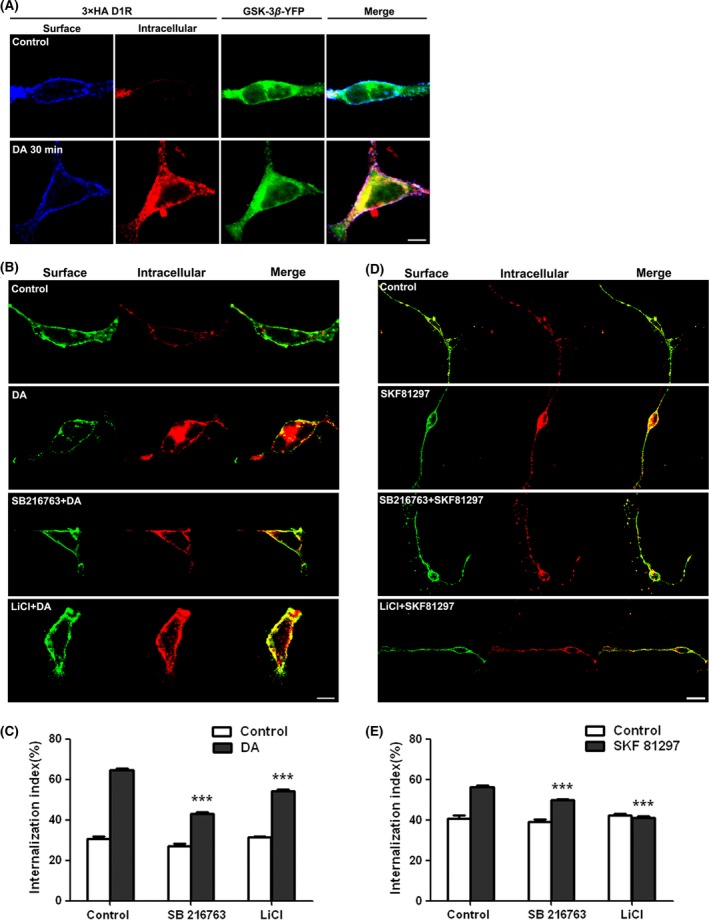
Inhibition of D1R internalization in HEK‐293‐D1R cells and in primary‐cultured striatal neurons by GSK‐3β inhibitors. (A) Co‐internalization of GSK‐3β with D1R. Stable HEK‐293‐D1R (HA‐tagged) cells were transiently transfected with GSK‐3β‐YFP. Thirty‐six hours after transfection, surface receptors were live‐conjugated with an anti‐HA antibody and were allowed for endocytosis (37°C, 30 min) in the absence or presence of DA (10 μM). (B) A representative image for DA‐stimulated D1R endocytosis in the presence or absence of GSK‐3β inhibitors 5 mM LiCI or 5 μM SB216763 in HEK‐293 cells stably expressing D1R. (D) The quantification for SKF81297‐stimulated D1R endocytosis in the presence or absence of GSK‐3β inhibitors in primary‐cultured striatal neurons transfected with HA‐D1R. (C) and (E) quantitation of the intracellular accumulation assays, measured as the ratio of internalized/total fluorescence (internalization index). Histograms show mean ± SEM (n = 30–35 for each condition). (***P < 0.001, LiCl/SB216763 vs. control).
Figure 5.

GSK‐3β siRNA inhibits the agonist‐induced D1R internalization. Stable HEK‐293‐D1R was transfected with GAPDH siRNA or GSK‐3β siRNA P259 or P1223 or mock. 48 h after transfection, cells were treated with 10 μM DA for 30 min. Fluorescence intracellular accumulation assay was used to test the receptor internalization. (A) Representative images of D1R internalization in stable HEK‐293‐D1R cells with GSK‐3β knockdown in response to DA stimulation. (B) Quantification of constitutive and agonist‐induced D1R internalization. Histograms show mean ± SEM (n = 30–35 for each condition). (***P < 0.001, siRNA vs. control). (C) GSK‐3β expression was decreased after the transfection of GSK‐3β siRNA. Cells were lysed, and expression of endogenous GSK‐3β and GSK‐3α was assessed using immunoblotting. Upper panel shows the GSK‐3α, and lower panel shows the GSK‐3β. The experiments have been repeated at least three times.
GSK‐3β is Required for D1R Stimulation‐Mediated Translocation of β‐arrestin‐2
β‐arrestins play a central role in mediating GPCR endocytosis. Binding of β‐arrestins to the receptor is a key regulatory step in receptor resensitization 33, 34. It was shown that β‐arrestin‐2 underwent a rapid membrane translocation in response to D1R activation. We investigated whether the GSK‐3β inhibition‐mediated reduction in agonist‐stimulated D1R endocytosis is associated with changes in translocation of β‐arrestin‐2 to plasma membrane. The GFP‐tagged β‐arrestin‐2 was transiently transfected into stable D1R‐expressing HEK‐293 cells. In agreement with previous observation, the GFP fluorescence of β‐arrestin‐2 was evenly distributed throughout the cytoplasm and exhibited no apparent enhanced plasma membrane localization in the absence of D1R activation. Upon D1R agonist stimulation, a time‐dependent rapid redistribution of GFP‐tagged β‐arrestin‐2 to the plasma membrane occurred (Figure 6A). However, agonist‐induced translocation of β‐arrestin‐2 to plasma membrane was attenuated in the presence of GSK‐3β inhibitors (LiCl and SB216763, Figure 6A). We measured the time‐dependent loss of β‐arrestin‐2 fluorescence from the cytoplasm in response to agonist treatment with or without a GSK‐3β inhibitor to quantify the translocation of β‐arrestin‐2 (Figure 6B). The data were then analyzed using a plateau with exponential decay nonlinear regression function (GraphPad Prism). The result indicated that LiCl and SB216763 reduced the redistribution of β‐arrestin‐2 toward plasma membrane in response to DA stimulation. This was further confirmed by immunoblot using a specific anti‐β‐arrestin‐2 antibody in a plasma membrane preparation (Figure 6C). Taken together, these data indicate that GSK‐3β is required for D1R stimulation‐mediated translocation of β‐arrestin‐2. GSK‐3β inhibition interrupted the translocation of β‐arrestin‐2 to plasma membrane, which, in turn, attenuated D1R recycling and resensitization and, consequently, resulted in the impairment of D1R function.
Figure 6.
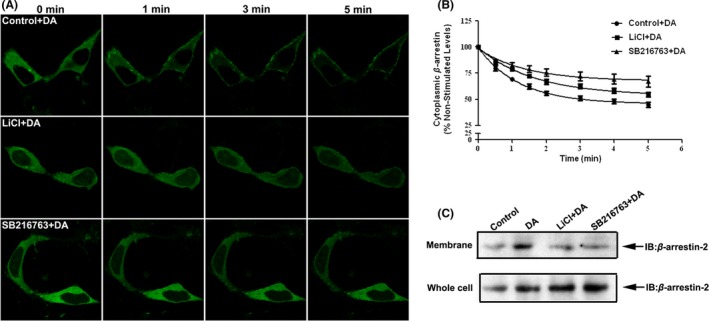
Translocation of β‐arrestin‐2 to plasma membrane is reduced by GSK‐3β inhibition. (A) Representative confocal microscope images of GFP‐β‐arrestin‐2 fluorescence at 0, 1, 3, and 5 min followed by DA treatment. HEK‐293 cells were transiently transfected with the HA‐D1R and GFP‐β‐arrestin‐2. Twenty‐four hours posttransfection, cells were plated on glass bottom culture dishes and analyzed with confocal microscope. Images were collected sequentially every 30 s after agonist stimulation with 20 μM DA using single‐line excitation (488 nm). (B) Quantitation of GFP‐β‐arrestin‐2 fluorescence was measured in the cytoplasm of cells before and after treatment with DA. Data represent the mean ± SEM of at least three independent experiments (n = 20 cells). (C) Agonist‐induced translocation of β‐arrestin‐2 in stable D1R‐expressing HEK‐293 cells was monitored by Western blotting using β‐arrestin‐2 antibody (Santa Cruz 1:1000; Santa Cruz Biotechnology). Cells were treated with DA or vehicle for 5 min, and reaction was terminated by placing the cells on ice. The cells were collected, plasma membranes were then prepared, and levels of endogenous β‐arrestin‐2 was assessed using immunoblotting using β‐arrestin‐2 antibody.
Inhibition of GSK‐3β Attenuates D1R Desensitization
As β‐arrestin‐2 translocation is a key step for agonist‐stimulated D1R internalization of the desensitized receptor, we next tested the functional consequence for GSK‐3β inhibition‐attenuated β‐arrestin‐2 translocation. Stable D1R‐expressing cells were pretreated with either DA or vehicle for 1 h, and the cell membrane was prepared for [35S]GTPγS‐binding assay to assess the Gαs protein activation in response to subsequent DA challenge. As shown in Figure S2, DA pretreatment induced significant desensitization of D1R (43.8 ± 6.32% reduction in the maximal Gαs activation relative to that of vehicle pretreatment). As expected, application of LiCl or SB216763 resulted in more dramatic reduction in D1R desensitization (29.2 ± 6.44% for LiCl and 23.8 ± 10.3% for SB216763, in relation to that of vehicle control). This clearly indicated that inhibition of GSK‐3β, which disrupted the β‐arrestin‐2 translocation attenuated D1R desensitization.
Decreased GSK‐3β Activity and D1R‐GSK‐3β Interaction are Associated with Prefrontal D1R Dysfunction in an Animal Model of Schizophrenia
Administration of PCP to animals or humans produces behavioral responses that closely resemble schizophrenia symptoms and, thus, has been widely used as a model for experimental schizophrenia research 28. Hypofunction of PFC in chronic PCP‐treated animals has been associated with the impairment of learning and memory and social withdrawal. In this regard, impaired D1R function in PFC may play critical role in the behavioral consequence of PFC hypofunction in schizophrenia. In agreement with our previous report 35, chronic PCP treatment resulted in impaired D1R activation in PFC. Coordinately, social interaction and novel objection recognition were also significantly impaired (Figure S3). We next checked whether PFC GSK‐3β activity was decreased in chronic PCP‐treated animals. As shown in Figure 7A,B, chronic PCP treatment induced a significant increase in Ser 9‐phosphorylated GSK‐3β, while total GSK‐3β expression was not altered, indicating decreased GSK‐3β activation in PFC of schizophrenia‐like animals. Inhibition of GSK‐3β resulted in decreased interaction between D1R and GSK‐3β, both in vitro and in native brain tissue, and is associated with dysfunction of D1R (Figure 3). Similarly, a significantly decreased D1R and GSK‐3β interaction was observed in PFC of PCP‐treated rats, in agreement with impaired D1R activation (Figure 7C). The present data indicate that the decreased GSK‐3β activity may be responsible for D1R dysfunction in schizophrenia.
Figure 7.
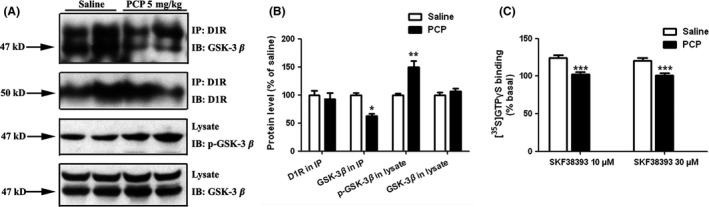
Decreased D1R‐GSK‐3β interaction in prefrontal cortex (PFC) of schizophrenia‐like animals. (A) Immunoblotting was performed on D1R immunoprecipitates in tissues with either saline or chronic PCP‐treated rats. The decreased interaction between D1R and GSK‐3β was evidenced in rat PFC with chronic PCP (5 mg/kg, i.p.) treatment. (B) The quantification of optical density ratios for A. Histograms show mean ± SEM for at least three experiments. (*P < 0.05, **P < 0.01, PCP vs. saline). (C) Prefrontal D1R activation was attenuated in schizophrenia‐like animals. PCP (5 mg/kg, i.p.) or saline was given once daily for 2 weeks. Then, rat brain PFC tissues were prepared as described in Materials and Methods. [35S]GTP γS‐binding assays were performed as described previously. D1R‐mediated Gs protein activation significantly decreased in PCP treatment. (***P < 0.001, PCP vs. saline).
Discussion
In this study, we showed that GSK‐3β kinase interacts physically with dopamine D1 receptors both in vitro and in vivo. We further found that the interaction occurred between the S(417)PALS(421) motif at the C‐terminus of D1R. Inhibition of GSK‐3β reduced the D1R‐GSK‐3β association and D1R activation. We further discovered that GSK‐3β inhibition induced the impairment in D1R function and is associated with the disruption of D1R recycling to plasma membrane. Decreased translocation of β‐arrestin‐2 appears to be responsible for the defects in D1R endocytosis. Importantly, we found that inhibition of GSK‐3β in rat PFC also resulted in impaired D1R activation. Moreover, in an animal model of schizophrenia, we detected a decrease in prefrontal GSK‐3β activity and D1R‐GSK‐3β association, along with impaired D1R activation. The present work reveals a novel mechanism for GSK‐3β‐regulated D1R function which may underlie prefrontal D1R dysfunction in schizophrenia (Figure 8).
Figure 8.

Proposed mechanism by which GSK‐3β regulates D1R cell signaling pathway. (A) When D1R is activated, colocalization or interaction with GSK‐3β accelerates β‐arrestin‐mediated D1R recycling and increases D1R signaling. (B) Disruption of GSK‐3β and D1R interaction results in that phosphorylated D1R cannot recover in time and D1R signaling is decreased. Dotted lines indicate signaling pathway is decreased. Red and blue colors indicate agonist of D1R and inhibitor of GSK‐3β, respectively.
GPCR functional regulation in response to agonist stimulation is a dynamic and complex process 1. Intracellular enzymes and some adaptor proteins were shown to play a critical role in the regulation of receptor traffic and function 26. For instance, G protein regulatory kinase (GRK) and protein kinase A (PKA) can phosphorylate the β‐adrenergic receptor and lead to functional uncoupling of the receptor with its respective G protein. Subsequently, the binding of β‐arrestin to the receptor results in the receptor internalization 34. Accumulated evidence revealed a similar regulatory role for PKA and GRK in D1R desensitization 32, 33, 34. GSK‐3β is a fascinating enzyme with broad actions in the regulation of cellular function. GSK‐3β activity is regulated by serine (inhibitory) and tyrosine (stimulatory) phosphorylation. The present study demonstrated, for the first time, that GSK‐3β is physically associated with D1R and participates in receptor functional regulation via affecting D1R recycling. In an effort to elucidate the details of this binding property, we performed progressive site‐mutation assays and found that GSK‐3β interacts with the C‐terminal of D1R. Interestingly, inhibition of GSK‐3β failed to produce any significant change in D1R phosphorylation (data not shown), indicating that the kinase most likely does not phosphorylate D1R directly. Given the facts that D1R can be phosphorylated by other protein kinases such as PKA and GRK upon receptor activation, it is, therefore, conceivable that D1R activation‐mediated phosphorylation of the receptor, in turn, modulates the D1R and GSK‐3β interaction.
DA exerts its effects through the activation of DA receptors and their subsequent coupling to downstream molecules. Disturbances in dopaminergic signaling are implicated in a number of neuropsychiatric disorders 1, 2, 3. It is believed that receptor endocytosis of desensitized GPCR is an initial step in the resensitization process that requires ligand–receptor dissociation and receptor dephosphorylation. Subsequently, the receptor may recycle back to the membrane or be targeted for degradation 1, 6, 9. The recovery at the membrane of internalized receptors has been attributed to the synthesis or insertion of more receptors, or to the recycling of endocytosed receptors. A few DRIPs have been shown to regulate this process including PSD‐95, β‐arrestin, GRK 6, 9, 11, 32, 33, 34. The present data demonstrated that agonist‐stimulated D1R endocytosis was reduced when GSK‐3β was inhibited, which was associated with the inhibition of membrane translocation of β‐arrestin‐2. We found that GSK‐3β is colocalized with D1R on the plasma membrane and co‐internalized with D1R in response to agonist stimulation. Inhibition of GSK‐3β activity resulted in decreased association with D1R and the attenuation in D1R desensitization and resensitization ultimately leads to impairment of receptor function. The finding that GSK‐3β‐modulated D1R function via affecting receptor recycling reveals a novel mechanism in D1R functional regulation (Figure 8).
Alterations in dopaminergic neurotransmission and dysregulation of GSK‐3β have been implicated in a variety of neurological and neuropsychiatric conditions including schizophrenia, mood disorders, and Alzheimer's disease 21, 22, 23, 24. Systemic administration of GSK‐3β inhibitor was reported to attenuate D1R agonist‐induced ambulatory and stereotypic activity 27. D1R is enriched in the PFC region and plays critical role in PFC function. In schizophrenia, prefrontal D1R dysfunction is known to contribute to the impaired cognitive function and negative symptoms 35, 36, 37, 38, 39, 40, 41. However, the underlying mechanism is poorly understood. Although previous reports have shown decreased GSK‐3β activity in PFC of schizophrenic patients or experimental animal models of schizophrenia 42, 43, 44, the functional consequence of this deficit is largely unknown. The present study reveals that the decreased prefrontal GSK‐3β activity in schizophrenia results in decreased D1R‐GSK‐3β association, consequently disrupting receptor recycling and ultimately impairing PFC D1R function. This finding not only provides a new mechanism for D1R impairment in schizophrenia but may also identify a novel means to treat schizophrenia.
It is also important to note that D1R and NMDA receptors play critical roles in the modulation of PFC function. Given the well‐established facts that D1R and NMDA receptors are functionally coupled through physical interaction, altered D1R function results in a change in NMDA function 6, 29, 30. Impaired D1R function due to decreased prefrontal GSK‐3β activity in schizophrenia could alter NMDA receptor function, which may be a critically important underlying mechanism of PFC dysfunction in schizophrenia.
Supporting information
Figure S1. Inhibition of GSK‐3β alters the membrane D1R expression.
Figure S2. Inhibition of GSK‐3β attenuates D1R desensitization.
Figure S3. Administration of phencyclidine (PCP) effects rat behavioral responses.
Acknowledgments
This work was financially supported by grants from The National Science Foundation of China (81130023, 81373382), The National Basic Research Plan (973) of the Ministry of Science and Technology of China (2011CB504403), The Specialized Research Fund for the Doctoral Program of Higher Education of China (20133201110017), The Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).
Conflict of Interest
The authors declare no conflict of interest.
The first two authors contributed equally to this work.
References
- 1. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 2011;63:182–217. [DOI] [PubMed] [Google Scholar]
- 2. Ye N, Neumeyer JL, Baldessarini RJ, et al. Update 1 of: Recent progress in development of dopamine receptor subtype‐selective agents: Potential therapeutics for neurological and psychiatric disorders. Chem Rev 2013;113:PR123–PR178. [DOI] [PubMed] [Google Scholar]
- 3. Ng GY, Trogadis J, Stevens J, et al. Agonist‐induced desensitization of dopamine D1 receptor‐stimulated adenylyl cyclase activity is temporally and biochemically separated from D1 receptor internalization. Proc Natl Acad Sci USA 1995;92:10157–10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 2007;69:451–482. [DOI] [PubMed] [Google Scholar]
- 5. Saab BJ, Georgiou J, Nath A, et al. NCS‐1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 2009;63:643–656. [DOI] [PubMed] [Google Scholar]
- 6. Lee FJ, Xue S, Pei L, et al. Dual regulation of NMDA receptor functions by direct protein‐protein interactions with the dopamine D1 receptor. Cell 2002;111:219–230. [DOI] [PubMed] [Google Scholar]
- 7. Liu F, Wan Q, Pristupa ZB, et al. Direct protein‐protein coupling enables cross‐talk between dopamine D5 and gamma‐aminobutyric acid A receptors. Nature 2000;403:274–280. [DOI] [PubMed] [Google Scholar]
- 8. Bermak JC, Li M, Bullock C, Zhou QY. Regulation of transport of the dopamine D1 receptor by a new membrane‐associated ER protein. Nat Cell Biol 2001;3:492–498. [DOI] [PubMed] [Google Scholar]
- 9. Sun P, Wang J, Gu W, et al. PSD‐95 regulates D1 dopamine receptor resensitization, but not receptor‐mediated Gs‐protein activation. Cell Res 2009;19:612–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhen X, Torres C, Wang HY, Friedman E. Prenatal exposure to cocaine disrupts D1A dopamine receptor function via selective inhibition of protein phosphatase 1 pathway in rabbit frontal cortex. J Neurosci 2001;21:9160–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurice P, Guillaume JL, Benleulmi‐Chaachoua A, et al. GPCR‐interacting proteins, major players of GPCR function. Adv Pharmacol 2011;62:349–380. [DOI] [PubMed] [Google Scholar]
- 12. Porras G, Berthet A, Dehay B, et al. PSD‐95 expression controls L‐DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest 2012;122:3977–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clinton SM, Meador‐Woodruff JH. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology 2004;29:1353–1362. [DOI] [PubMed] [Google Scholar]
- 14. Heinz A, Schlagenhauf F. Dopaminergic dysfunction in schizophrenia: Salience attribution revisited. Schizophr Bull 2010;36:472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 2004;74:1–58. [DOI] [PubMed] [Google Scholar]
- 16. Miyamoto S, Miyake N, Jarskog LF, et al. Pharmacological treatment of schizophrenia: A critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry 2012;17:1206–1227. [DOI] [PubMed] [Google Scholar]
- 17. Dallerac GM, Vatsavayai SC, Cummings DM, et al. Impaired long‐term potentiation in the prefrontal cortex of Huntington's disease mouse models: Rescue by D1 dopamine receptor activation. Neurodegener Dis 2011;8:230–239. [DOI] [PubMed] [Google Scholar]
- 18. Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK‐3 dysregulation disorder. Trends Neurosci 2007;30:142–149. [DOI] [PubMed] [Google Scholar]
- 19. Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci 2010;11:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim WY, Snider WD. Functions of GSK‐3 signaling in development of the nervous system. Front Mol Neurosci 2011;4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol 2009;49:327–347. [DOI] [PubMed] [Google Scholar]
- 22. Jope RS, Roh MS. Glycogen synthase kinase‐3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets 2006;7:1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beaulieu JM. A role for Akt and glycogen synthase kinase‐3 as integrators of dopamine and serotonin neurotransmission in mental health. J Psychiatry Neurosci 2012;37:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li YC, Gao WJ. GSK‐3beta activity and hyperdopamine‐dependent behaviors. Neurosci Biobehav Rev 2011;35:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sillivan SE, Black YD, Naydenov AV, et al. Binge cocaine administration in adolescent rats affects amygdalar gene expression patterns and alters anxiety‐related behavior in adulthood. Biol Psychiatry 2011;70:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beaulieu JM, Sotnikova TD, Marion S, et al. An Akt/beta‐arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005;122:261–273. [DOI] [PubMed] [Google Scholar]
- 27. Miller JS, Tallarida RJ, Unterwald EM. Inhibition of GSK3 attenuates dopamine D1 receptor agonist‐induced hyperactivity in mice. Brain Res Bull 2010;82:184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neill JC, Barnes S, Cook S, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: Focus on NMDA receptor antagonism. Pharmacol Ther 2010;128:419–432. [DOI] [PubMed] [Google Scholar]
- 29. Fiorentini C, Gardoni F, Spano P, et al. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N‐methyl‐D‐aspartate receptors. J Biol Chem 2003;278:20196–20202. [DOI] [PubMed] [Google Scholar]
- 30. Lin JW, Ju W, Foster K, et al. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 2000;3:1282–1290. [DOI] [PubMed] [Google Scholar]
- 31. Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR‐interacting proteins. Br J Pharmacol 2012;165:1717–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Zastrow M, Williams JT. Modulating neuromodulation by receptor membrane traffic in the endocytic pathway. Neuron 2012;76:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walther C, Ferguson SS. Arrestins: Role in the desensitization, sequestration, and vesicular trafficking of G protein‐coupled receptors. Prog Mol Biol Transl Sci 2013;118:93–113. [DOI] [PubMed] [Google Scholar]
- 34. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta‐arrestins. Science 2005;308:512–517. [DOI] [PubMed] [Google Scholar]
- 35. Guo Y, Zhang H, Chen X, et al. Evaluation of the antipsychotic effect of bi‐acetylated l‐stepholidine (l‐SPD‐A), a novel dopamine and serotonin receptor dual ligand. Schizophr Res 2009;115:41–49. [DOI] [PubMed] [Google Scholar]
- 36. Sawaguchi T, Goldman‐Rakic PS. D1 dopamine receptors in prefrontal cortex: Involvement in working memory. Science 1991;251:947–950. [DOI] [PubMed] [Google Scholar]
- 37. Arnsten AF. Prefrontal cortical network connections: Key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci 2011;29:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: Version III–the final common pathway. Schizophr Bull 2009;35:549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Castner SA, Williams GV, Goldman‐Rakic PS. Reversal of antipsychotic‐induced working memory deficits by short‐term dopamine D1 receptor stimulation. Science 2000;287:2020–2022. [DOI] [PubMed] [Google Scholar]
- 40. Okubo Y, Suhara T, Suzuki K, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997;385:634–636. [DOI] [PubMed] [Google Scholar]
- 41. Abi‐Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 2002;22:3708–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kozlovsky N, Belmaker RH, Agam G. Low GSK‐3beta immunoreactivity in postmortem frontal cortex of schizophrenic patients. Am J Psychiatry 2000;157:831–833. [DOI] [PubMed] [Google Scholar]
- 43. Nadri C, Lipska BK, Kozlovsky N, et al. Glycogen synthase kinase (GSK)‐3beta levels and activity in a neurodevelopmental rat model of schizophrenia. Brain Res Dev Brain Res 2003;141:33–37. [DOI] [PubMed] [Google Scholar]
- 44. Kozlovsky N, Belmaker RH, Agam G. Low GSK‐3 activity in frontal cortex of schizophrenic patients. Schizophr Res 2001;52:101–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Inhibition of GSK‐3β alters the membrane D1R expression.
Figure S2. Inhibition of GSK‐3β attenuates D1R desensitization.
Figure S3. Administration of phencyclidine (PCP) effects rat behavioral responses.