

Summary
Aims
To investigate the roles of N‐myc downstream‐regulated gene 2 (NDRG2) in the pathology of aging and neurodegenerative disease such as Alzheimer's disease (AD).
Results
In this study, we confirmed the upregulation of NDRG2 in the brains of aging and AD animal models. To explore the role of NDRG2 in the pathology of AD at molecular level, we conducted a cell‐based assay of highly expressed wild‐type human APP695 SK‐N‐SH cells (SK‐N‐SH APPwt). By silencing and overexpressing gene of NDRG2, we demonstrated that NDRG2‐mediated increase in Aβ1‐42 was through the pathways of BACE1 and GGA3. NGRG2 improved tau phosphorylation via enhanced activity of CDK5 and decreased Pin1, but it was not affected by GSK3β pathway. NDRG2 might also induce cell apoptosis through the extrinsic (caspase 8) apoptotic pathway by interaction with STAT3.
Conclusion
Our study confirmed the upregulation of NDRG2 in AD animal models and demonstrated its important roles in AD pathology. NDRG2 might be a potential target for studying and treatment of AD.
Keywords: Alzheimer's disease, apoptosis, beta‐amyloid, NDRG2, tau phosphorylation
1. INTRODUCTION
Alzheimer's disease (AD) is one of the most common forms of neurodegenerative disease among the elder people, which brings a huge economic burden to the whole society. Currently, our knowledge about AD is incomplete and there is urgent demand for finding novel AD‐related proteins and characterizing these proteins in relation to AD pathology, which will provide new insight into the molecular changes occurring in AD.
N‐myc downstream‐regulated gene 2 (NDRG2) is a member of a new family of genes involved in cell proliferation, differentiation, and apoptosis. This family is composed of four members, NDRG1, NDRG2, NDRG3, NDRG4, and has been found to involve in various biological activities.1, 2 Among them, NDRG2 is a well‐characterized protein and has gained much attention in the past decades due to its studies of prodifferentiative and antiproliferative. It was first described by Deng et al.3 using subtractive hybridization approach from glioblastoma based on polymerase chain reaction in 2001. Since then, NDRG2 was found to be highly expressed in brain, muscle, and heart and moderately expressed in liver and kidney. Usually, NDRG2 acts as a potential tumor suppressor and the expression of which is negatively correlated with cancer progression. Reduced expression of NDRG2 has been reported in a range of human cancers including skin cancer,4 pancreatic cancer,5 breast cancer,6 lung cancer,7 liver cancer,8 thyroid cancer,9 and colon cancer.10
Using two‐dimensional electrophoresis technology, our previous study has demonstrated that the protein level of NDRG2 was elevated in 15‐month‐old senescence‐accelerated mouse (SAM) compared to 5‐month‐old SAM, indicating NDRG2 might be involved in the pathology of age‐related disease such as AD.11 Till now, only one report was found about the relation between NDRG2 and AD, which described both mRNA and protein levels of NDRG2 were significantly increased in patients with sporadic AD and it colocalized with senile plaques and NFTs in the brain regions, such as entorhinal cortex and CA1 subregion of the hippocampus known to be vulnerable to AD pathology.12 However, this initial observation did not explain how NDRG2 level was elevated during AD and what was the role of this protein in AD pathology. These questions remain to be elucidated.
In this study, we verified the abnormal expressions of NDRG2 in various AD animal models and investigated the molecular mechanisms of NDRG2 in AD pathology. Our study demonstrates that the protein NDRG2 affects on multiple pathways contributing the development of the AD‐like phenotype and indicates that NDRG2 might be a useful target in study of AD.
2. MATERIALS AND METHODS
2.1. Animals
APP/PS1 double‐transgenic mice and wild‐type littermates were purchased from The Jackson Laboratory. These mice express a mouse/human APP containing the K595 N/M596 L Swedish mutations and a mutant human PS1 carrying the exon 9‐deleted variant under the control of mouse prion promoter elements, directing transgene expression predominantly to CNS neurons. Male ICR mice and Sprague Dawley (SD) rats were obtained from Vital Rital Laboratories (Beijing, China). The SAMP8 and SAMR1 mice were supplied from the First Affiliated Hospital of Tianjing College of Traditional Chinese Medicine (Tianjing, China). The animals were group‐housed in a room maintained at 23±1°C with a 12‐hour light/dark cycle and free access to water and food. The process of intracerebroventricular‐injecting Aβ1‐42 in the rat and intraperitoneal injection of scopolamine in the mice has been described in previous report.13, 14 Briefly, Aβ1‐42 peptide was resuspended in DMSO at 5 mmol/L and diluted by cold F12 medium to yield a 100 μmol/L stock solution and then incubated at 4°C for 24 hours. The preparation was then centrifuged at 14 000 g for 10 minutes at 4°C. The supernatants were collected. All experiments were approved and performed in accordance with the institutional guidelines of the Experimental Animal Center of the Chinese Academy of Medical Science, Beijing, China.
2.2. Cell culture
SK‐N‐SH and SK‐N‐SH APPwt cells were grown in DMEM, plus 10% fetal bovine serum (Hyclone, Los Angeles, CA, USA), 100 U/mL penicillin/streptomycin. In addition, SK‐N‐SHAPPwt cells were supplemented with 200 μg/mL G418. Cell cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2 and passed every 2‐4 days based on 85% confluence.
2.3. RNA interference
For NDRG2 knockdown, specific siRNAs for human NDRG2 and negative control were all obtained from Invitrogen (Carlsbad, CA, USA). The short interference RNA sequence is (5′‐CCUGGCGAGAUAUGCUCUUTT‐3′). Mixtures of si‐NDRG2 and control siRNA were transfected into SK‐N‐SH APPwt cells by lipofectamine RNAiMAX (Invitrogen) under the forverse transfection protocol according to the manufacurer's specification. Briefly, 10 μL lipofectamine RNAiMAX and 5 μL siNDRG2 or siControl with DMEM (no FBS) were diluted in 250 μL DMEM (no FBS). They were incubated at the room temperature for 5 minutes. The siRNA and lipofectamine RNAiMAX were mixed and incubated at the room temperature for 20 minutes. Final concentration of siRNA was 70 nmol/L in DMEM, and 2 μL lipofectamine RNAiMAX was added per well. The mixture was incubated in a final volume of 500 μL for 20 minutes and then added to 80%‐90% confluent SK‐N‐SHAPPwt cells in six‐well plates for a final volume of 3 mL. Five hours after transfection, the medium was changed to the fresh complete medium for an additional 20, 44, or 68 hours before cells were harvested.
2.4. NDRG2 gene transfection
pcDNA4.0‐NDRG2 and pcDNA4.0‐lac Z were provided by professor Liu Xinping (Department of Biochemistry and Molecular Biology, The Fourth Military Medical University). The NDRG2 gene was amplified by PCR and digested by BamH I and EcoR I and then cloned into the expression vector pCNDA4.0 by Solution I ligase reaction system. The protein expression of vectors was detected by Western blotting. All transfections were performed using lipofectamine™2000, according to the manufacturer's instructions. Briefly, 4 μg pcDNA4.0‐lacZ or pcDNA4.0‐NDRG2 and 10 μL lipofectamine™2000 were transfected into SK‐N‐SH cells with a final volume of 2.5 mL per well. Cells were incubated for 5 hours with the lipofectmine/plasmid mixture in DMEM, and the medium was replaced by the fresh complete medium for an additional 20, 44, or 68 hours before cells were harvested.
2.5. Western blot analysis
Total protein was extracted using RIPA buffer and determined by the Bradford method (Bradford, 1976). Samples were run on polyacrylamide gel, transferred on to PVDF membranes, blocked with 5% skim milk, and incubated with primary antibodies overnight at 4°C. After washing with TBST, the membranes were incubated with secondary antibodies (1:10 000) at room temperature for 1 hour. The signals were detected using an enhanced chemiluminescence (ECL) kit, scanned using an LAS3000 Fujifilm imaging system (Fujifilm, Tokyo, Japan), and analyzed by densitometric evaluation using the Quantity‐One 4.31software (Bio‐Rad, Hercules, CA, USA). The primary antibodies used in this study were listed in Table 1.
Table 1.
Primary antibodies used in this study
| Antibody | Vendor | Application | Dilution |
|---|---|---|---|
| Mouse monoclonal anti‐NDRG2 | Santa Cruz biotechnology | WB, IP,IF | 1:200 |
| Rabbit polyclonal anti‐p‐tau (Ser199) | Invitrogen | WB | 1:1000 |
| Rabbit polyclonal anti‐p‐tau (Thr205) | Invitrogen | WB | 1:1000 |
| Rabbit polyclonal anti‐p‐tau (Ser396) | Abcam | WB | 1:1000 |
| Rabbit monoclonal anti‐p‐AKT | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anti‐AKT | Cell Signal Technology | WB | 1:1000 |
| Rabbit polyclonal anti total GSK3 | Abcam | WB | 1:1000 |
| Rabbit polyclonal anti‐GSK3β (Ser9) | Abcam | WB | 1:500 |
| Rabbit polyclonal anti‐p‐CDK5 | Santa Cruz biotechnology | WB | 1:200 |
| Mouse monoclonal anti‐CDK5 | Santa Cruz biotechnology | WB | 1:200 |
| Mouse monoclonal anti‐β‐actin | Sigma | WB | 1:10 000 |
| Rabbit monoclonal anti‐p35 | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anti‐BACE1 | Eptitomics | WB,IF | 1:1000, 1:100 |
| Rabbit monoclonal anti‐Aβ | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anti‐Bcl‐2 | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anti‐Bax | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anticaspase‐3 | Cell Signal Technology | WB | 1:1000 |
| Rabbit monoclonal anticaspase‐8 | Cell Signal Technology | WB | 1:1000 |
| Rabbi monoclonal anticaspase‐9 | Cell Signal Technology | WB | 1:1000 |
| Rabbi monoclonal anti‐p‐STAT3(Tyr705) | Cell Signal Technology | WB | 1:2000 |
| Rabbi monoclonal anti‐p‐STAT3(Ser727) | Cell Signal Technology | WB | 1:2000 |
| Rabbi monoclonal anti‐STAT3 | Cell Signal Technology | WB | 1:1000 |
IHC, immunohistochemistry; IF, immunofluorescent staining; WB, Western blot.
2.6. Immunostaining analysis
Culture cells were grown overnight. The medium was removed, and cells were washed three times with PBS and fixed in 4% paraformaldehyde for 30 minutes. After fixation, cells were thoroughly rinsed with PBS and incubated with goat serum in PBS containing 0.5% Triton X‐100 for 2 hours at room temperature. Then, the cells were incubated with an anti‐NDRG2 antibody (1:100), an anti‐β‐catenin antibody (1:100), or an anti‐STAT3 antibody (1:100) overnight at 4°C. Then, the cells were rinsed with PBS for 3 times, followed by incubating with secondary antibodies [Fluorescent‐labeling AlexaFluor 488 (anti‐mouse,1:200) and 594(anti‐rabbit, 1:200)](2 hours, RT). The immunostaing images were performed by fluorescence microscope (Nikon Eclipse Ti‐S) after treatment with Hoechst 33342 for 15 minutes to label nuclear DNA.
2.7. Cell viability assay
Cell viability was quantified by its ability to reduce tetrazolium salt 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) to a colored formazan products (Sigma, St. Louis, MO, USA) according to the manufacturer's instruction. Briefly, MTT reagent (5 mg/mL in PBS) was added to the cells at 1/10 volume of the medium. Cells were incubated at 37°C for 4 hours. Then, the supernatant was discarded followed by adding 150 μL DMSO and incubated at 37°C for 15 minutes. The absorbance was measured at 570 nm with MQX200 microplate reader (Bio‐Tek, Winooski, VT, USA). Cell viability was expressed as the percentage of absorbance obtained in control cultures.
2.8. RNA extraction, reverse transcription, and real‐time PCR
Total RNA was extracted from cells usingTRIzol reagent (Invitrogen). The reverse transcription PCR was performed using PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara, Dalian, China), and quantification of the target genes was performed with SYBR Premix Ex Taq™ in 7300 Real Time PCR System (Applied Biosystems, Warrington, UK), according to the manufacturer's instruction. The Ct value of BACE1 was normalized against those of 18sRNA from the same samples, and then, the absolute amount was determined according to the standard curve (the start point is 2.26*107). Primer sequences were as follows: BACE1 (5′‐GAGGTATCGACCACTCGC‐3′ and 5′‐GACTCCACCCGCACAAT‐3′) and 18sRNA(5′‐CGTCTGCCCTATCAACTTT‐3′ and 5′‐TTTCTCAGGCTCCCTCTC‐3′).
2.9. NDRG2 subcellular localization analysis
The protein sample isolated from SK‐N‐SH/SK‐N‐SH APP695 human neuroblastoma cell was subfractionated into cytosol and nuclear fraction by solubility‐based separation and sequential centrifugation using Nuclear‐Cytosol Extraction Kit (Applygen, Beijing, China) according to the manufacture's instruction. Briefly, cells (about 107) were incubated with 500 μL ice‐cold cytosol extraction buffer A for 10 minutes, followed by incubated with 30 μL buffer B for 1 minute. Then, the whole solution was centrifuged for 5 minutes with a speed of 12 000 g. The supernatant fraction was immediately transferred to a new tube. For nuclei extraction, the pellet was washed once with the CBE‐A, centrifuged for 5 minutes at 1000 g, and discarded the supernatant. The precipitate was added 100 μL ice‐cold nuclear extraction buffer, incubated for 5 minutes, and centrifuged at 12 000 g for 5 minutes. The supernatant fraction contained the protein extracted from the nuclei.
2.10. Co‐immunoprecipitation
Co‐immunoprecipitation was carried out using protein A/G PLUS‐agarose immunoprecipitation reagent (Santa Cruz biotechnology, Santa Cruz, CA, USA) according to the manufacture's instruction. Briefly, the lysates were precleared with 20 μL protein A/G PLUS‐agarose (Santa Cruz biotechnology) together with 1 μg species‐matched control IgG for 30 minutes at 4°C. To detect the proteins that could interact with NDRG2, the lysates (100‐500 μg) were incubated with 2 μg anti‐NDRG2 antibody for 1 hour with rocking at 4°C. Then, 20 μL protein A/G PLUS‐agarose was added and incubated overnight with rocking at 4°C. The imunnoprecipitates were pelleted and washed four times by RIPA buffer. The precipitates were resolved by 40 μL of 1× electrophoresis sample buffer and then subjected to Western blotting analysis.
2.11. Statistical analysis
Statistical analysis of the data was performed with one‐way ANOVA using SPSS version 13.0 (SPSS Inc., Chicago, IL, USA). The differences between the groups were analyzed by Bonferroni's post hoc test. For Western blot analysis, the control group was normalized to 100%. All data were shown as mean±SEM, and prism software (GraphPad Prism 5, La Jolla, CA, USA) was used to draw the picture. A value of P<.05 was considered to be statistically significant.
3. RESULTS
3.1. NDRG2 was upregulated in brain tissues of aging and AD animal models
Our previous proteomics study has demonstrated that NDRG2 level was significantly upregulated in the cortex and hippocampus of 15‐month‐old SAM compared with 5‐month‐old SAM.11 To confirm this result, the Western blot analysis was carried out using the tissues of cerebral cortex and hippocampus. A notable increase in NDRG2 level was observed in the hippocampus of 15‐month‐old SAMR1 as compared to 5‐month‐old SAMR1 (Figure 1A). At the meantime, we found NDRG2 level was also increased in both the cortex and hippocampus of 15‐month‐old SAMP8 mice compared to 5‐month‐old SAMP8 (Figure 1B). We further explored whether NDRG2 was involved in the process of aging. The result showed that NDRG2 level was significantly enhanced in the cortex of aged rats (24 months) compared to young rats (3 months), although only a slight increase in NDRG2 level was observed in the hippocampus (Figure 1C).
Figure 1.
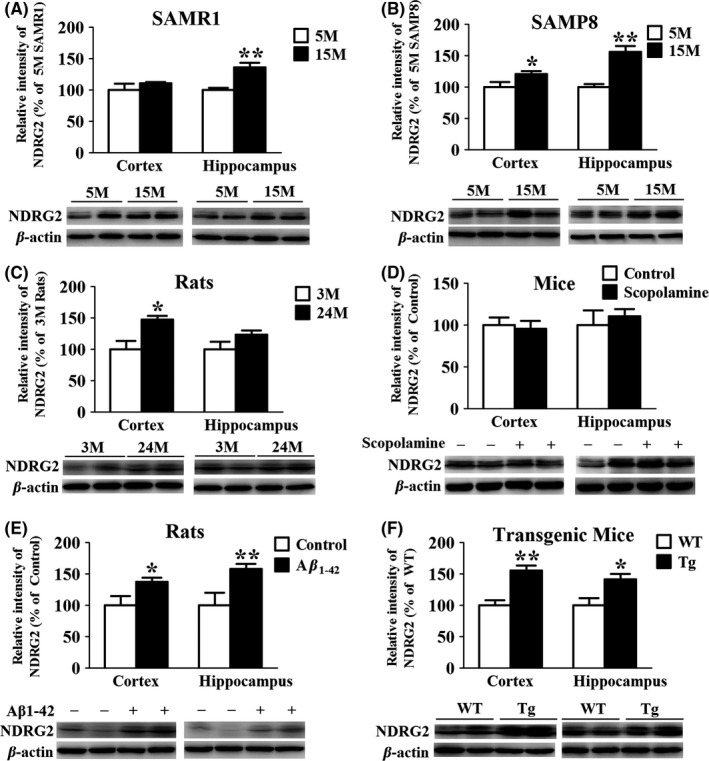
NDRG2 level is elevated in the brain tissues of 15‐mo‐old SAMR1, SAMP8, aged rats, Aβ1‐42 intracerebroventricular‐injected rats, and APP/PS1 double‐transgenic mice but not in scopolamine intraperitoneal‐injected mice. (A and B) Representative Western blots and quantitative analysis of NDRG2 in the cortical and hippocampal homogenates of SAMR1 and SAMP8. (C) Representative Western blots and quantitative analysis of NDRG2 in the cortical and hippocampal brain homogenates of aging rats. (D) Representative Western blots and quantitative analysis of NDRG2 in the cortical and hippocampal brain homogenates of scopolamine intraperitoneal‐injected mice. (E) Representative Western blots and quantitative analysis of NDRG2 in the cortical and hippocampal brain homogenates of Aβ1‐42 intracerebroventricular‐injected rats. (F) Representative Western blots and quantitative analysis of NDRG2 in the cortical and hippocampal brain homogenates of APP/PS1 mice. The results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4~7 per group. *P<0.05, **P<0.01 vs control group
The above results suggested that NDRG2 was an aging‐related protein, and we hypothesized that NDRG2 might also participate in the pathology of AD. The Aβ1‐42 intracerebroventricular‐injected AD rat model was used. NDRG2 level was found upregulated in the cortex and hippocampus of the rats (Figure 1E). In addition, similar results were also found in 14‐month‐old APP/PS1 transgenic mice (Figure 1F), which displayed distinct pathological characteristics of AD with obvious cognitive impairment (data are shown as Fig. S1 in Supporting Information). However, in scopolamine intraperitoneal‐injected mice model, no significant change of NDRG2 level was observed (Figure 1D).
3.2. NDRG2 promoted APP metabolism and beta‐amyloid production
It has been proposed that aberrant APP metabolism may result in the accumulation of Aβ peptides in vulnerable brain regions and initiate the events in pathophysiology of AD. Therefore, we employed RNA interference to assess the effect of NDRG2 knockdown on APP metabolism using neuroblastoma SK‐N‐SH APPwt cells. It showed that NDRG2 level was enhanced by 1.5‐fold compared to the control of SK‐N‐SH cells (Figure 2A), whereas NDRG2 expression was reduced by 85.7% in SK‐N‐SH APPwt cells when it was knocked down with siNDRG2 compared to the cells transfected with siControl (Figure 2B).
Figure 2.
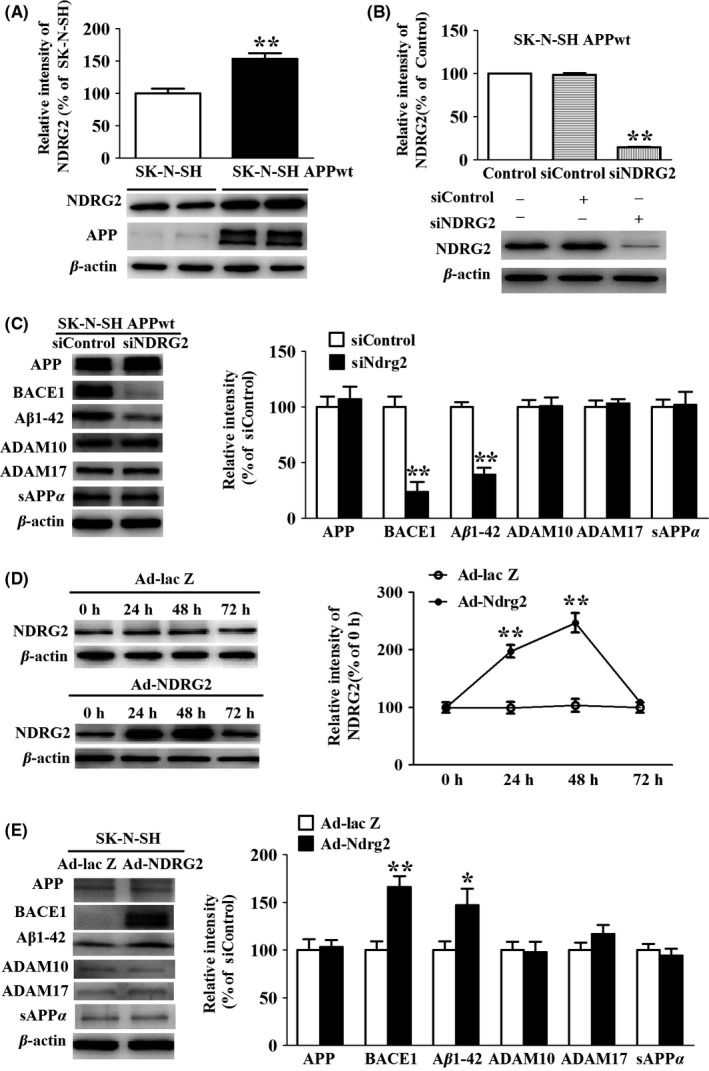
NDRG2 affects the amyloidogenic pathways of APP metabolism. (A) Representative Western blots and quantitative of NDRG2 in SK‐N‐SH cells and SK‐N‐SH APPwt cells. (B) Representative Western blots of NDRG2 after SK‐N‐SH APPwt transfected with siControl and siNDRG2. (C) Representative Western blots and quantitative of APP, ADAM10, ADAM17, BACE1, and Aβ1‐42 after SK‐N‐SH APPwt cells were transfected with siControl or siNDRG2. (D) Representative Western blots of NDRG2 after SK‐N‐SH cells transfected with Ad‐lacZ and Ad‐NDRG2. (E) Representative Western blots and quantitative of APP, ADAM10, ADAM17, BACE1, and Aβ1‐42. Representative Western blots and quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4 per group. *P<0.05, **P<0.01 vs control group
We then examined the effects of NDRG2 silencing on the level of APP, α‐secretase (including ADAM10 and ADAM17), and β‐secretase. We found a marked decrease in BACE1 and Aβ1‐42 levels in siNDRG2 group compared to siControl group, whereas there was no significant difference for the levels of APP, ADAM10, ADAM17, and sAPPα between the two groups (Figure 2C).
In the above study, we had demonstrated NDRG2 knockdown alleviating APP metabolism in SK‐N‐SH APPwt cells. We wondered whether the reciprocal effect could be achieved via NDRG2 overexpression in SK‐N‐SH cells. For this purpose, we performed a time course study of NDRG2 expression after SK‐N‐SH cells were transfected with Ad‐NDRG2 vector or Ad‐lac Z vector. We found that NDRG2 level increased slowly and reached to peak level at 48 hours in Ad‐NDRG2 vector transfected cells. At the meantime, we observed a marked increase in BACE1 and Aβ1‐42 in Ad‐NDRG2 group compared to Ad‐lac Z group, whereas there were no significant difference for the levels of APP, ADAM10, ADAM17, and sAPPα between the two groups (Figure 2E).
3.3. NDRG2 increases BACE1 levels and activity through GGA3‐mediated post‐translational stabilization
BACE1 cleaves APP at the β‐secretase site to initiate the production of Aβ peptides, which constitutes the rate‐limiting step of the amyloid production. As we detected a prominent decrease in BACE1 level after NDRG2 knockdown, we wondered whether this reduction was due to the inhibition of transcription. Absolute real‐time PCR showed the transcription of BACE1 was unchanged whether in NDRG2 knockdown SK‐N‐SH APPwt cells or in NDRG2 overexpresed SK‐N‐SH cells (Figure 3A, B).
Figure 3.
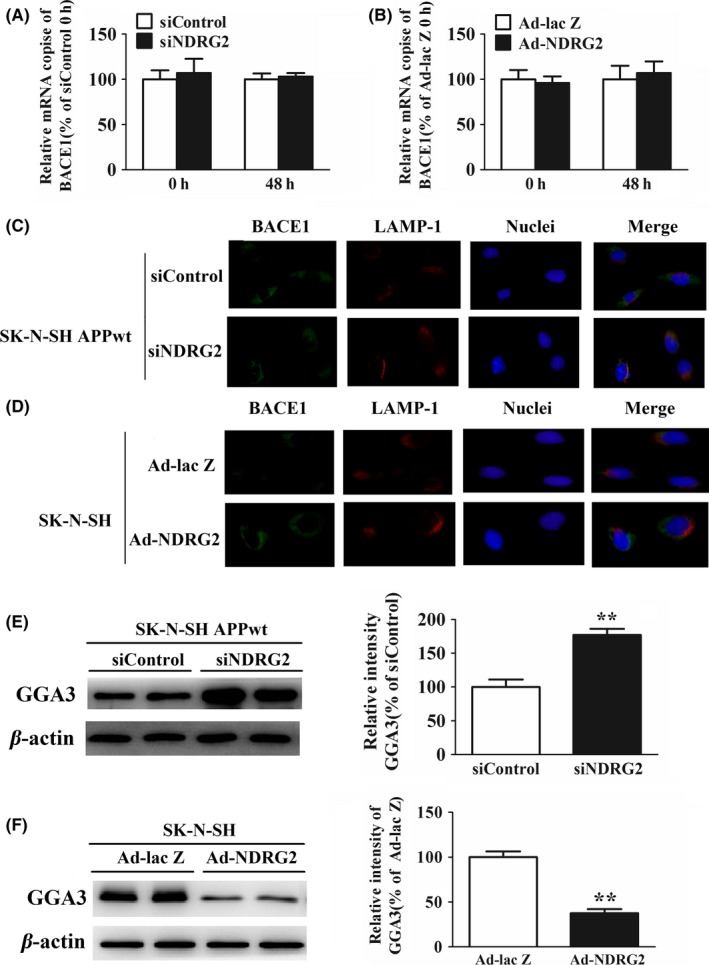
NDRG2 increased BACE1 level and activity through GGA3‐mediated post‐translatonal stabilization. (A) The mRNA expression of BACE1 after SK‐N‐SH APPwt cells were transfected with siControl or siNDRG2. (B) The mRNA expression of BACE1 after SK‐N‐SH cells were transfected with Ad‐NDRG2 or Ad‐lacZ. (C) Image of BACE1 and LAMP‐2A SK‐N‐SH APPwt cells after treated with siControl and siNDRG2. (D) Image of BACE1 and LAMP‐2A after SK‐N‐SH APPwt cells were transfected with Ad‐NDRG2 and Ad‐lacZ. (E) Representative Western blots and quantitative analysis of GGA3 after SK‐N‐SH APPwt cells after treated with siControl and siNDRG2. (F) Representative Western blots and quantitative analysis of GGA3 after SK‐N‐SH APPwt cells were transfected with Ad‐NDRG2 and Ad‐lacZ. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4~5 per group. *P<0.05, **P<0.01 vs control group
Some evidence has indicated an increase in BACE1 level in the brain of AD patients has been attributed to alterations of its intracellular trafficking,15 which might be a mechanism of NDRG2‐mediated BACE1 alteration. After transfection, cells were costained with BACE1 and LAMP1 antibody, a marker for lysosomes, to analyze subcellular localization of BACE1. Immunofluorescence analysis showed more BACE1 appeared to colocalize with LAMP1 in siNDRG2 group compared with siControl group in SK‐N‐SHAPPwt cells, which means siNDRG2 could promote BACE1 migrating to lysosomes for degradation (Figure 3C). On the contrary, more BACE1 seemed to departure from lysosomes in Ad‐NDRG2 group (NDRG2 overexpression) compared to Ad‐lac Z group in SK‐N‐SH cells, which was demonstrated by the apparent space of immunoflurescence between BACE1 and LAMP‐1 (Figure 3D).
GGA3 is an adaptor protein responsible for sorting of BACE1 to lysosomal for degradation as demonstrated by Vassar et al.16 After NDRG2 silencing in SK‐N‐SH APPwt cells, a robust increase in GGA3 protein expression was observed after NDRG2 knockdown in SK‐N‐SHAPPwt cells (Figure 3E), which could sort more BACE1 to the lysosomes for degradation. Alternatively, Western blot analysis revealed GGA3 level was largely reduced after NDRG2 overexpression in SK‐N‐SH cells (Figure 3F).
3.4. NDRG2 facilitates the hyperphosphorylation of tau
Previous study has demonstrated NDRG2 colocalizes with NFTs in AD cerebral cortex;12 therefore, whether there may be some correlation between NDRG2 and NFTs at the molecular level based on the colocalization. Immunostaining images showed the hyperphosphorylated tau appeared at high level in SK‐N‐SH APPwt cells but not in SK‐N‐SH cells (Figure 4A).
Figure 4.

NDRG2 facilitates the hyperphosphorylation of tau. (A) Image of PHF in SK‐N‐SH and SK‐N‐SH APPwt cells. (B) Representative Western blots and quantification of phosphorylated tau at Ser199, Thr205, and Ser396 after SK‐N‐SH APPwt cells were transfected with siControl or siNDRG2. (C) Representative Western blots and quantification of phosphorylated tau at Ser199, Thr205, and Ser396 after SK‐N‐SH cells were transfected with Ad‐NDRG2 or Ad‐lacZ. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4~5 per group. *P<0.05, **P<0.01 vs control group
We found that knockdown of NDRG2 engendered a dramatic reduction in tau phosphorylation in SK‐N‐SH APPwt cells. Quantitative analysis showed that the level of tau phosphorylation at Ser199, Thr205, Thr231, Ser396, and Ser404 reduced by 77.8%, 64.5%, 76.6%, 54.5%, and 78.3%, respectively, compared to siControl group (Figure 4B). Alternatively, these phosphorylation epitopes could be clearly observed after NDRG2 overexpression in SK‐N‐SH cells, whereas in Ad‐lacZ group, they were barely detectable (Figure 4C).
3.5. NDRG2 enhanced activity of CDK5 and inhibited the activity of Pin1
Further, we identified the molecular mechanism of NDRG2‐mediated tau phosphorylation. We examined the effect of siNDRG2 on the activity of GSK‐3β and CDK‐5,17, 18 two major kinases for phosphorylation of tau at abundant epitopes. The results showed the level of phosphor‐CDK‐5(activity form) was obviously reduced in siNDRG2 group compared to siControl group (P<.01) (Figure 5B). Conversely, phosphor‐GSK‐3β (Ser9) level remained relatively unchanged between the two groups (Figure 5A). Meanwhile, phosphor‐CDK‐5 expression was obviously enhanced in Ad‐NDRG2 group, whereas it was very low in Ad‐lac Z group (Figure 5C). In contrast, there was no significant difference in phosphor‐GSK‐3β (Ser9) between the two groups (data not shown). In addition, PP2A (measured by its inactive form phosphor‐PP2A Y307) was showed negatively regulated by NDRG2.
Figure 5.
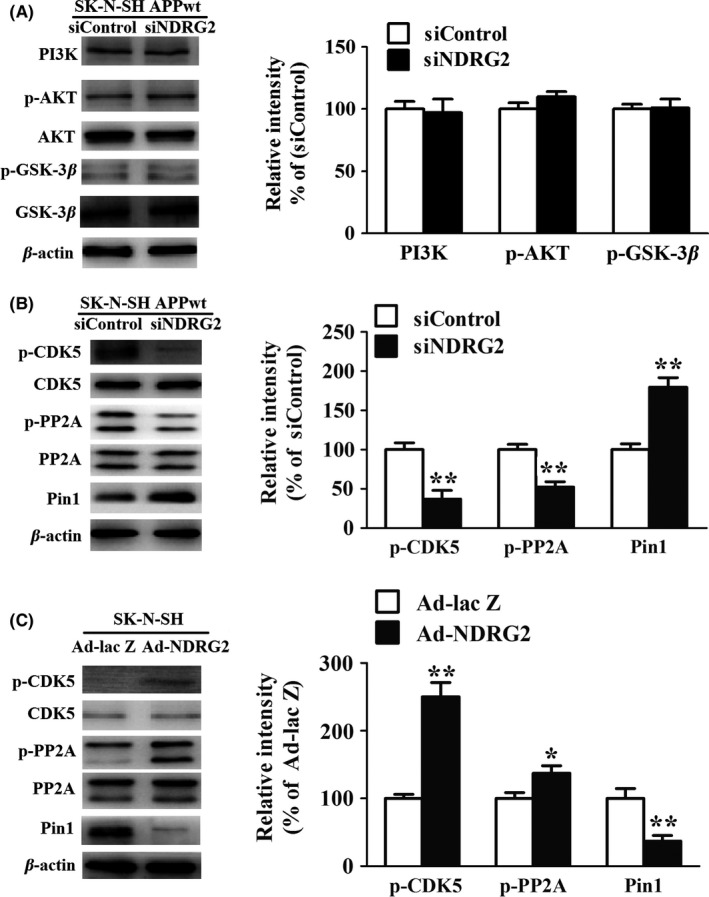
NDRG2 facilitates tau hyperphosphorylation by increasing CDK5 and inhibiting PP2A activity. (A) Representative Western blots and quantification of PI3K, p‐AKT (Ser473), AKT, phosphorylated GSK‐3β (Ser9), and total GSK‐3β after SK‐N‐SH APPwt cells were transfected with siControl and siNDRG2. (B) Representative Western blots and quantification of phosphorylated CDK‐5 (Ser159), total CDK‐5 after SK‐N‐SH APPwt cells were transfected with siControl and siNDRG2. (C) Representative Western blots and quantification of phosphorylated CDK‐5 (Ser159), total CDK‐5 after SK‐N‐SH cells were transfected with Ad‐lac Z and Ad‐NDRG2. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=3 per group. *P<0.05, **P<0.01 vs control group
The effect of NDRG2 on Pin1 was also studied. Silencing NDRG2 might increase Pin1 expression, and NDRG2 overexpression might inhibit it (Figure 5B, C). Our data indicated that CDK5, Pin1, and PP2A participated in the NDRG2‐mediated Tau hyperphosphorylation (Figure 5C).
3.6. NDRG2 promotes extrinsic caspase‐dependent apoptosis
NDRG2 was reported as a negative regulator of cell proliferation.19 Therefore, we examined the potential role of NDRG2 to induce cell apoptosis, one of the important pathological changes of AD.20 The MTT assays showed the cell viability dramatically increased by 55% in siNDRG2 group compared to siControl group (Figure 6A), and cell apoptosis decreased by 68.9% showed by Hoechst staining (Figure 6C). However, overexpression of NDRG2 might increase the apoptosis significantly (Figure 6D). The percentage of the apoptotic cells was calculated by the ratio of apoptotic cells to the total cells counted. At least 500 cells were counted from more than three random microscopic fields. Further, it was demonstrated that NDRG2 knockdown attenuating apoptosis was mainly due to a prominent reduction in cleaved caspase‐8 and caspase‐3 in the cells, indicating NDRG2‐mediated cell apoptosis is through the extrinsic apoptotic pathways (Figure 6E, F).
Figure 6.
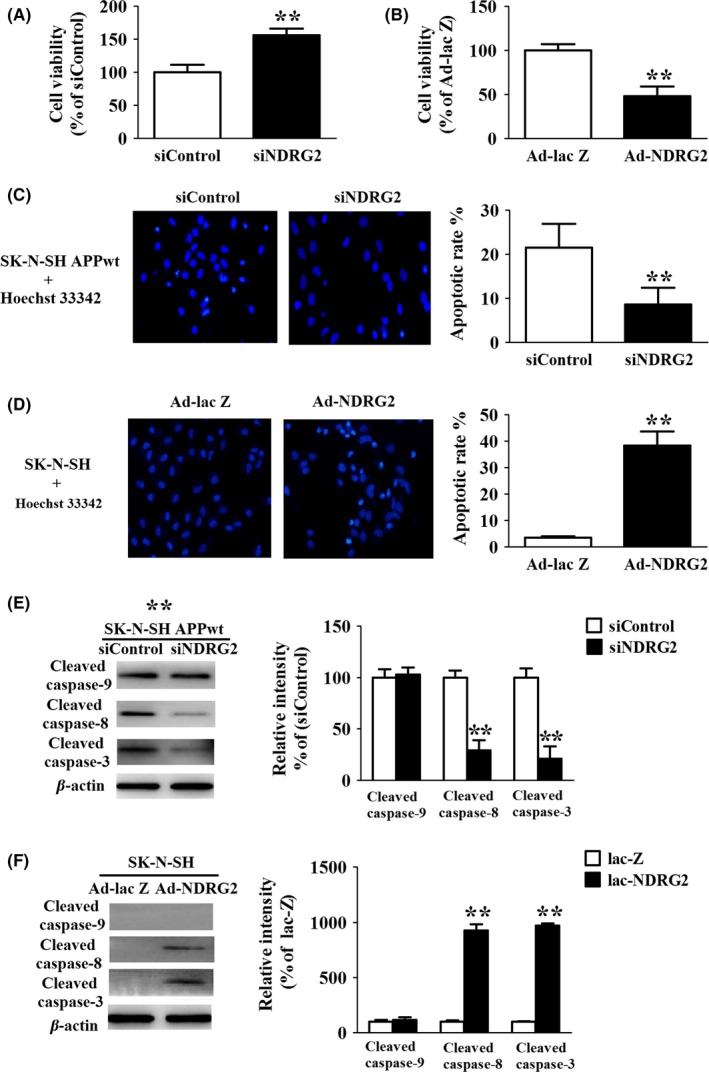
NDRG2 contributes to apoptosis via the extrinsic apoptotic pathways (A) MTT assay of SK‐N‐SH APPwt cells after cells were transfected with siControl and siNDRG2 for 48 h. (B) MTT assay of SK‐N‐SH cells after cells were transfected with Ad‐NDRG2 and Ad‐lacZ for 48 h. (C) Hoechst staining of SK‐N‐SH APPwt cells after treated with siControl and siNDRG2 for 48 h. (D) Hoechst staining of SK‐N‐SH APP cells after treated with Ad‐NDRG2 and Ad‐lacZ for 48 h. (E) Representative Western blots and quantitative analysis of cleaved caspase‐9, cleaved caspase‐8, cleaved caspase‐3 after SK‐N‐SH APPwt cells were transfected with siControl and siNDRG2. (F) Representative Western blots and quantitative analysis of cleaved caspase‐9, cleaved caspase‐8, cleaved caspase‐3 after SK‐N‐SH cells were transfected with Ad‐NDRG2 and Ad‐lacZ for 48 h. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4~5 per group. *P<0.05, **P<0.01 vs control group
3.7. NDRG2 inhibit the activity of STAT3, a mechanism to trig extrinsic apoptosis
As mentioned above, NDRG2‐mediated caspase‐3 activation was caspase‐8‐dependent, which is a well‐characterized downstream target of Fas ligand‐Fas system. Based on this notion, we tested the level of Fasl and Fas in our experiment, and we found both of them were largely downregulated in siNDRG2 group compared to siControl group (Figure 7A, B). Up to now, there are several factors that control the expression of Fas. Among them, STAT3 was the most deeply studied protein, which has a strong inhibitory effect on the fasl‐fas system.21 Contrary to Fasl and Fas, Western blot analysis demonstrated a robust increase in phosphorylation of STAT3 at Tyr 705 site upon SK‐N‐SH APPwt cells transfected with siNDRG2 and a markedly decrease in phosphorylation of STAT3 at Tyr 705 site upon SK‐N‐SH cells transfected with Ad‐NDRG2.
Figure 7.
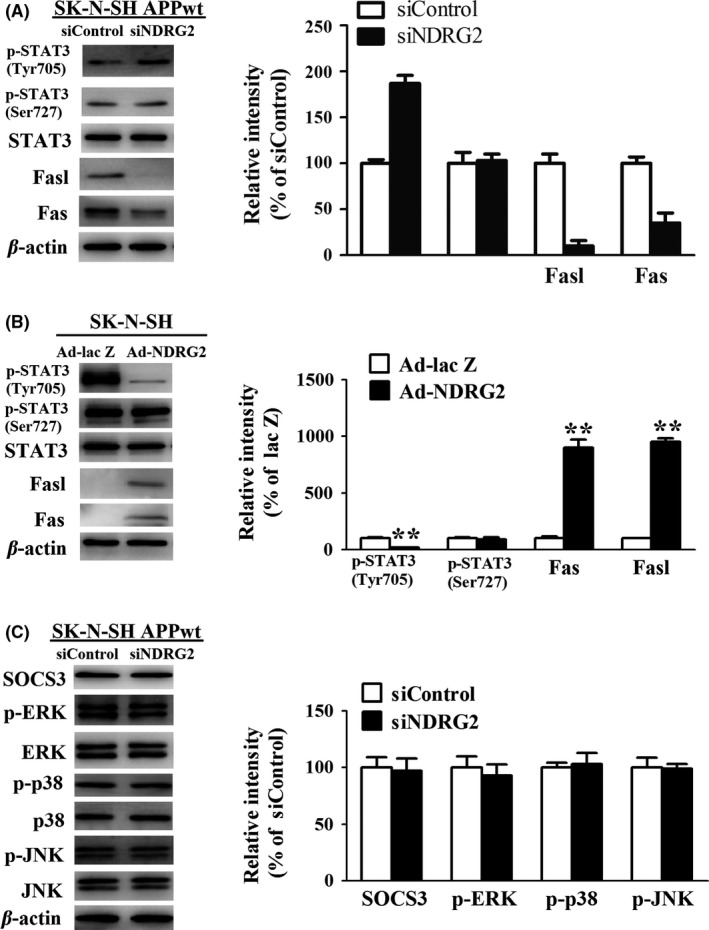
NDRG2 inhibits the activity of STAT3, triggering the extrinsic caspase‐dependent apoptosis. (A) Representative Western blots and Quantitative analysis of fasl, fas, p‐STAT3 (Tyr705), and p‐STAT3(Ser727) after SK‐N‐SH APPwt cells after treated with siControl, siNDRG2, and siNDRG2. (B) Representative Western blots and Quantitative analysis of fasl, fas, p‐STAT3(Tyr705), and p‐STAT3(Ser727) after SK‐N‐SH cells after treated with Ad‐NDRG2 and Ad‐lacZ. (C) Representative Western blots and quantitative analysis of SOCS3,p‐ERK, p‐p38,p‐JNK after SK‐N‐SH APPwt cells after treated with siControl, siNDRG2. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown. Values were expressed as percentages compared to the control group (set to 100%) and represented as group mean±SEM. n=4~5 per group. *P<0.05, **P<0.01 vs control group
To further elucidate the underlying mechanism of NDRG2‐mediated inhibition in STAT3 phosphorylation, we tested STAT3 endogenous inhibitor SOCS3 and MAPK signal pathways, which has been widely studied as a major positive regulator of STAT3 activity.22, 23 However, no significant differences were observed between siNDRG2 group and siControl group (Figure 7C), indicating there might be other mechanism that controlled the NDRG2‐medatied STAT3 phosphorylation.
3.8. NDRG2 translocates into the nuclear and interacted with STAT3
Previous studies on the function of NDRG2 in cancer have demonstrated that this protein might interact with other molecules and exert its biological function. Therefore, we observed whether NDRG2 interacted with STAT3. Immunoflureorescent images demonstrated a high level of colocalization between NDRG2 and STAT3 in SK‐N‐SH APPwt cells, whereas in SK‐N‐SH cells, this phenomenon is barely observed (Figure 8A). Co‐IP was used to verify the complex formation of the two proteins, and we found that NDRG2 indeed interacted with STAT3 in SK‐N‐SH APPwt cells with high affinity. Conversely, there was barely binding between NDRG2 and STAT3 in SK‐N‐SH cells (Figure 8B).
Figure 8.

NDRG2 translocates into the nuclear and interacted with STAT3. (A) Immunofluorescence colocalization analysis of cellular NDRG2 and STAT3 in SK‐N‐SH cells and SK‐N‐SH APPwt cells, NDRG2 antiboby(green), STAT3 antibody(red). (B) Representative Western blots of NDRG2 expression in nucleus and cytoplasm extraction. (C) Lysates from SK‐N‐SH and SK‐N‐SH APPwt cells were immunoprecipitated with mouse anti‐NDRG2 anti‐STAT3 or nonspecific mouse IgG antibodes. Cell lysates(imput) and immunoprecipitates were analyzed by Western blot for NDRG2 and STAT3. Quantified results were normalized to β‐actin expression. For all the results above, a representative experiment of three performed is shown.
At the meantime, we found a shift of NDRG2 expression from cytoplasm to nucleus after SK‐N‐SH cells transfected with APPwt. To further support the finding of NDRG2 nuclear translocation, a cell fraction assay was performed. The results showed that NDRG2 was expressed mainly in the cytoplasm and could hardly be detected in nucleus in SK‐N‐SH cells, whereas in SK‐N‐SH APPwt cells, the NDRG2 expression in both nucleus and cytoplasm was obviously increased. A possible explanation could be that NDRG2 translocated into the nuclear and interacted with STAT3 consequently inhibited the activity of STAT3. The details of NDRG2 translocation and interaction with STAT3 remain to be studied.
4. DISCUSSION
Our previous study demonstrated that NDRG2 protein expression was significantly increased in both cortex and hippocampus of 15‐month‐old SAM compared to that of 5‐month‐old SAM, which indicating NDRG2 might be associated with brain aging process.11 The SAM showed various characteristic age‐associated phenotypes, and with a short life span, integrated background information and high repetition rate, it was considered as an ideal model to study the mechanisms of aging or age‐related diseases such as AD.24 In the present study, we showed that NDRG2 protein level was upregulated in the brain tissues of different aging or AD animal models, indicating this protein involved indeed in the pathology of aging or AD. In above models, animals showed cognition deficiency; for example, there was an obvious cognition disorder in the step‐down tests of APP/PS1 mice (data are shown as Fig. S1 in Supporting Information). It should be mentioned that in scopolamine intraperitoneal‐injected mice model, no significant difference of NDRG2 level was observed, which means it has no relationship with short‐term acetylcholine dysfunction.
Early researches of NDRG2 were mainly concentrated on the area of cancer due to the natural prosperity of inhibition of cell proliferation.25 However, the role of NDRG2 in central nerve system (CNS) has also attracted more and more attention recently. NDRG2 was reported to be upregulated and colocalized with TUNEL‐positive cells in ischemic penumbra following transient focal cerebral ischemia, suggesting that NDRG2 may play an important role in cell apoptosis after stroke.26 In addition, NDRG2 is implicated to be involved in the synaptic transduction and in neurite sprouting, extension, and guidance, which offers novel insights into the physiological roles of NDRG2 in the CNS.27 Increase in both mRNA and protein levels of NDRG2 was found in the sporadic AD patient.12 Nevertheless, the mechanisms for upregulation of NDRG2 and its role in AD are still unclear. Recently, NDRG2 was reported to express in different type of cells, such as neurons and astrocytes. We also observed the expression of NDRG2 in astrocytes in the absence and presence of Aβ25‐35 in our study. NDRG2 was enhanced by Aβ25‐35 (see Fig. S2 in Supporting Information). However, our purpose of this study was mainly revealing the role of NDRG2 in neurons during the Alzheimer's disease, so we did not conduct additional studies on astrocytes. In this study, we used human SK‐N‐SH neuroblastoma cell line overexpressing wt APP695 (APPwt), which is a widely used cell line in the study of AD, and carried out relevant mechanism research of NDRG2.
In this study, we focused on the relationship between NDRG2 and AD pathology, especially the three main characteristics of AD at the molecular level: for example, accumulation of beta‐amyloid, abnormal tau phosphorylation, and neuronal apoptosis.28 Regarding the process of Aβ production, it has been demonstrated that the role of BACE1 is very important. It was showed in our study, silencing NDRG2 reduced expressions of BACE1 and Aβ1‐42 in SK‐N‐SH APPwt cells compared to control cells, whereas overexpressing NDRG2 might increase both BACE1 and Aβ1‐42, indicating changes in NDRG2 might relate to the generation of beta‐amyloid. Further, we investigated the mechanisms of BACE1 increase in pathology of AD. BACE1 is relatively stable with a half‐life of 16 hours, but once it is ubiquitinated at C‐terminal Lys501, it could be recognized by GGA3, a key protein for sorting BACE1 to the lysosome for degradation.29, 30 Our results revealed that NDRG2 was negatively correlated to GGA3 expression and could promote BACE1 trafficking to the endosome, which decreased the degradation of BACE1 and ultimately increased Aβ production in AD.
It is well known that in healthy subjects, tau is a component of microtubules, which represent the internal support structures for transport of nutrients, vesicles, mitochondria and chromosomes within the cell. In AD, tau protein is abnormally hyperphosphorylated, and it might reduce binding affinity to microtubules and result in aberrant tau aggregation into tangles.31 It has a profound effect on the progression of AD.32
Tau phosphorylation occurs mainly at proline‐directed Ser/Thr sites, which are targeted by peptidyl‐prolyl cis/trans isomerase 1(Pin1). Pin1 catalyzes cis to trans prolyl isomerisation of phosphorylated tau, facilitating its dephosphorylation by PP2A and apparently restores the ability to promote microtubules polymerization.33 Our study showed that the level of tau phosphorylation in SK‐N‐SH cells was positively correlated with the expression of NDRG2. Our data indicated that CDK5 strongly and positively responded to NDRG2 and PP2A also responded to it. But Pin1 negatively responded to NDRG2; therefore, the phosphorylation of tau was increased in NDRG2 overexpressed cells and decreased in NDRG2 knock‐down cells. It was also demonstrated in this study that the signaling pathway of AkT‐GSK‐3β was not affected by NDRG2, although it was showed to mediate tau phosphorylation in some other cases. Additionally, tau phosphorylation might be also induced by Aβ. Due to the lack of specific Aβ inhibitors, we cannot exclude this possibility in our study.
Neuronal loss by apoptosis is a pathological change during neurodegenerative disorders especially in AD.34 Therefore, explicating the mechanisms of neuronal apoptosis during AD is critical for elucidating the pathophysiology of the disease. In this study, we demonstrated that NDRG2 was strongly related to apoptosis of SK‐N‐SH APPwt cells as it was reported in cancer previously. It was found that NDRG2‐induced cell death was not through the mitochondria‐cytochrome c pathway, but through the death receptor apoptotic pathway by highly expressed Fas‐Fasl system and consequently boosting caspase‐8‐dependent caspase‐3 activation.
The Fas/Fasl expression is mainly modulated by STAT3. In our experiments, we observed NDRG2 primarily localized to the cytoplasm of SK‐N‐SH cells, whereas in SK‐N‐SH APPswe cells, NDRG2 significantly translocated into the nucleus. Furthermore, we found NDRG2 interacted with STAT3 in SK‐N‐SH APPwt nucleus, while in SK‐N‐SH cells, this interaction was barely observed. We speculate that the colocalization of NDRG2 and STAT3 in the nucleus may affect the transcription activity of STAT3 for the target genes like Fas. Our result is in line with a previous study, which elucidated NDRG2 moved from cytosol to nucleus to form the complex with MSP58 upon cell stress and inhibited MSP58‐induced cell proliferation.35 However, the mechanisms of APP‐mediated NDRG2 translocation into nucleus as well as the interaction between NDRG2 and STAT3 need to be studied in the future.
Our data suggested that although some clear pathological roles of NDRG2 were showed in this study, we cannot exclude the possibility that cell apoptosis and tau phosphorylation induced by NDRG2 might be attributed to increased Aβ production. It has been reported that Aβ induced cell apoptosis and tau phosphorylation.25, 36 Due to lack of specific Aβ inhibitor, the role of Aβ in NDRG2‐induced neuronal apoptosis and tau phosphorylation could not be ignored. In addition, we demonstrated that NDRG2 might directly affect the activities of CDK5, an important enzyme for phosphorylation of tau. However, the effects of Aβ on CDK5 could not be ruled out because Aβ can also increase the activity of CDK5.37 Therefore, the pathological effects of NDRG2 in AD might be indirect through the increased Aβ, whereas the distinct pathway of NDRG2‐induced cell apoptosis by enhancing expression and function of Fas/FasI system was found in this study.
In conclusion, we have investigated the pathophysiological roles and mechanisms of NDRG2 in AD animal and cell models. Increased level of NDRG2 in cortex or hippocampus may be a potential risk for AD. NDRG2 increased Aβ1‐42 might be related to the changes of BEAC1 and GGA3. It might also affect CDK5 and tau overphosphorylation. NDRG2 interacting with STAT3 and modulating Fas/Fasl signaling pathway plays important roles in cell death. Further studies of NDRG2 as a drug target in treatment of AD remain to be carried out.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by National Nature Science Foundation of China (No. 81573417) and Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation (No. BZ0150).
Rong X‐F, Sun Y‐N, Liu D‐M, et al. The pathological roles of NDRG2 in Alzheimer's disease, a study using animal models and APPwt‐overexpressed cells. CNS Neurosci Ther. 2017;23:667–679. 10.1111/cns.12716
REFERENCES
- 1. Qu X, Zhai Y, Wei H, et al. Characterization and expression of three novel differentiation‐related genes belong to the human NDRG gene family. Mol Cell Biochem. 2002;229:35‐44. [DOI] [PubMed] [Google Scholar]
- 2. Zhou RH, Kokame K, Tsukamoto Y, Yutani C, Kato H, Miyata T. Characterization of the human NDRG gene family: a newly identified member, NDRG4, is specifically expressed in brain and heart. Genomics. 2001;73:86‐97. [DOI] [PubMed] [Google Scholar]
- 3. Deng Y, Yao L, Liu X, et al. Exploring a new gene containing ACP like domain in human brain and expression it in E. coli . Prog Biochem Biophys. 2001;1:19. [Google Scholar]
- 4. Hummerich L, Muller R, Hess J, et al. Identification of novel tumour‐associated genes differentially expressed in the process of squamous cell cancer development. Oncogene. 2006;25:111‐121. [DOI] [PubMed] [Google Scholar]
- 5. Yamamura A, Miura K, Karasawa H, et al. Suppressed expression of NDRG2 correlates with poor prognosis in pancreatic cancer. Biochem Biophys Res Commun. 2013;441:102‐107. [DOI] [PubMed] [Google Scholar]
- 6. Park Y, Shon SK, Kim A, et al. SOCS1 induced by NDRG2 expression negatively regulates STAT3 activation in breast cancer cells. Biochem Biophys Res Commun. 2007;363:361‐367. [DOI] [PubMed] [Google Scholar]
- 7. Wang L, Liu N, Yao L, et al. NDRG2 is a new HIF‐1 target gene necessary for hypoxia‐induced apoptosis in A549 cells. Cell Physiol Biochem. 2008;21:239‐250. [DOI] [PubMed] [Google Scholar]
- 8. Lee DC, Kang YK, Kim WH, et al. Functional and clinical evidence for NDRG2 as a candidate suppressor of liver cancer metastasis. Cancer Res. 2008;68:4210‐4220. [DOI] [PubMed] [Google Scholar]
- 9. Zhao H, Zhang J, Lu J, et al. Reduced expression of N‐Myc downstream‐regulated gene 2 in human thyroid cancer. BMC Cancer. 2008;8:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim YJ, Yoon SY, Kim JT, et al. NDRG2 expression decreases with tumor stages and regulates TCF/beta‐catenin signaling in human colon carcinoma. Carcinogenesis. 2009;30:598‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu L, Yu J, Shi Q, et al. Strain‐ and age‐related alteration of proteins in the brain of SAMP8 and SAMR1 mice. J Alzheimers Dis. 2011;23:641‐654. [DOI] [PubMed] [Google Scholar]
- 12. Mitchelmore C, Buchmann‐Moller S, Rask L, West MJ, Troncoso JC, Jensen NA. NDRG2: a novel Alzheimer's disease associated protein. Neurobiol Dis. 2004;16:48‐58. [DOI] [PubMed] [Google Scholar]
- 13. Liu Z, Wang W, Feng N, Wang L, Shi J, Wang X. Parishin C's prevention of Abeta 1‐42‐induced inhibition of long‐term potentiation is related to NMDA receptors. Acta Pharm Sin B. 2016;6:189‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin J, Huang L, Yu J, et al. Fucoxanthin, a marine carotenoid, reverses scopolamine‐induced cognitive impairments in mice and inhibits acetylcholinesterase in vitro. Mar Drugs. 2016;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan J, Evin G. Beta‐site APP‐cleaving enzyme 1 trafficking and Alzheimer's disease pathogenesis. J Neurochem. 2012;120:869‐880. [DOI] [PubMed] [Google Scholar]
- 16. Vassar R. Caspase‐3 cleavage of GGA3 stabilizes BACE: implications for Alzheimer's disease. Neuron. 2007;54:671‐673. [DOI] [PubMed] [Google Scholar]
- 17. Sharma P, Sharma M, Amin ND, Albers RW, Pant HC. Regulation of cyclin‐dependent kinase 5 catalytic activity by phosphorylation. Proc Natl Acad Sci USA. 1999;96:11156‐11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang JZ, Wu Q, Smith A, Grundke‐Iqbal I, Iqbal K. Tau is phosphorylated by GSK‐3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A‐kinase. FEBS Lett. 1998;436:28‐34. [DOI] [PubMed] [Google Scholar]
- 19. Yao L, Zhang J, Liu X. NDRG2: a Myc‐repressed gene involved in cancer and cell stress. Acta Biochim Biophys Sin (Shanghai). 2008;40:625‐635. [DOI] [PubMed] [Google Scholar]
- 20. Zilkova M, Koson P, Zilka N. The hunt for dying neurons: insight into the neuronal loss in Alzheimer's disease. Bratisl Lek Listy. 2006;107:366‐373. [PubMed] [Google Scholar]
- 21. Ivanov VN, Bhoumik A, Krasilnikov M, et al. Cooperation between STAT3 and c‐jun suppresses Fas transcription. Mol Cell. 2001;7:517‐528. [DOI] [PubMed] [Google Scholar]
- 22. Gao H, Ward PA. STAT3 and suppressor of cytokine signaling 3: potential targets in lung inflammatory responses. Expert Opin Ther Targets. 2007;11:869‐880. [DOI] [PubMed] [Google Scholar]
- 23. Tarutani M, Nakajima K, Takaishi M, Ohko K, Sano S. Epidermal hyperplasia induced by Raf‐MAPK signaling requires Stat3 activation. J Dermatol Sci. 2013;72:110‐115. [DOI] [PubMed] [Google Scholar]
- 24. Chrisp C. Animal models in aging research. Fed Proc. 1986;45:43‐44. [PubMed] [Google Scholar]
- 25. LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21‐30. [DOI] [PubMed] [Google Scholar]
- 26. Li Y, Shen L, Cai L, et al. Spatial‐temporal expression of NDRG2 in rat brain after focal cerebral ischemia and reperfusion. Brain Res. 2011;1382:252‐258. [DOI] [PubMed] [Google Scholar]
- 27. Takahashi K, Yamada M, Ohata H, Honda K. Ndrg2 promotes neurite outgrowth of NGF‐differentiated PC12 cells. Neurosci Lett. 2005;388:157‐162. [DOI] [PubMed] [Google Scholar]
- 28. Hardy J, Duff K, Hardy KG, Perez‐Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355‐358. [DOI] [PubMed] [Google Scholar]
- 29. Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin‐proteasome pathway. FASEB J. 2004;18:1571‐1573. [DOI] [PubMed] [Google Scholar]
- 30. Tesco G, Koh YH, Kang EL, et al. Depletion of GGA3 stabilizes BACE and enhances beta‐secretase activity. Neuron. 2007;54:721‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kosik KS, Joachim CL, Selkoe DJ. Microtubule‐associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044‐4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blurton‐Jones M, Laferla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3:437‐448. [DOI] [PubMed] [Google Scholar]
- 33. Butterfield DA, Abdul HM, Opii W, et al. Pin1 in Alzheimer's disease. J Neurochem. 2006;98:1697‐1706. [DOI] [PubMed] [Google Scholar]
- 34. Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol. 1996;6:493‐506. [DOI] [PubMed] [Google Scholar]
- 35. Zhang J, Liu J, Li X, et al. The physical and functional interaction of NDRG2 with MSP58 in cells. Biochem Biophys Res Commun. 2007;352:6‐11. [DOI] [PubMed] [Google Scholar]
- 36. Garwood CJ, Pooler AM, Atherton J, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Aβ‐induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Aβ‐induced model of Alzheimer's disease: the role of Cdk5. Aging Cell. 2010;9:64‐77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials