

Summary
Aims
The proline‐rich Akt substrate of 40‐kDa (PRAS40) protein is a direct inhibitor of mTORC1 and an interactive linker between the Akt and mTOR pathways. The mammalian target of rapamycin (mTOR) is considered to be a central regulator of cell growth and metabolism. Several investigations have demonstrated that abnormal mTOR activity may contribute to the pathogenesis of several neurodegenerative disorders and lead to cognitive deficits.
Methods
Here, we used the PrP peptide 106‐126 (PrP106‐126) in a cell model of prion diseases (also known as transmissible spongiform encephalopathies, TSEs) to investigate the mechanisms of mTOR‐mediated cell death in prion diseases.
Results
We have shown that, upon stress caused by PrP106‐126, the mTOR pathway activates and contributes to cellular apoptosis. Moreover, we demonstrated that PRAS40 down‐regulates mTOR hyperactivity under stress conditions and alleviates neurotoxic prion peptide‐induced apoptosis. The effect of PRAS40 on apoptosis is likely due to an mTOR/Akt signaling.
Conclusion
PRAS40 inhibits mTORC1 hyperactivation and plays a key role in protecting cells against neurotoxic prion peptide‐induced apoptosis. Thus, PRAS40 is a potential therapeutic target for prion disease.
Keywords: apoptosis, mammalian target of rapamycin (mTOR), negative feedback mechanism, PI3K‐Akt‐mTOR signaling, prion diseases, proline‐rich Akt substrate of 40‐kDa (PRAS40)
1. Introduction
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are infectious, fatal neurodegenerative diseases caused by the induced conversion of normal host PrPC protein to an abnormal pathogenic PrPSc isoform characterized by protease resistance. TSEs affect humans and animals.1, 2 The pathological characteristics of TSEs are spongiform changes, microgliosis, astrocytosis, and neuronal death.3, 4, 5 Failure to clear the pathogenic PrPSc isoform from the brain induces neuronal dysfunction.6, 7, 8 There is no effective therapeutic or prophylactic strategy to control prion diseases. The prion fragment PrP106‐126 is used in cellular models of TSEs, because it is highly similar to PrPSc 9 PrP residues 106‐126 can polymerize to form amyloid‐like fibrils, which contribute to neurotoxicity and neuronal death.10, 11, 12, 13 These similarities make PrP106‐126 a useful model for in vitro studies of neurotoxicity and cell death in prion diseases.
The presence of excessive reactive oxygen species (ROS) in a cell induces oxidative stress. Oxidative stress is strongly linked to apoptotic neuronal cell death and neuronal dysfunction. In addition, oxidative stress has been implicated in the progression of several neurodegenerative disorders, including Parkinson's disease, Alzheimer's disease, and prion diseases.14, 15, 16, 17 Aggregation of misfolded proteins is a common etiology of oxidative stress related to neurodegenerative disorders.18, 19
Target of rapamycin (TOR) kinase was first identified in yeast as the target of rapamycin, an immunosuppressant.20, 21 Mammalian target of rapamycin (mTOR) is the mammalian orthologue of TOR22, 23 and a central regulator of cell growth and metabolism.24 Similar to TOR in yeast, mTOR functions in two multiprotein complexes: mTORC1 and mTORC2. mTORC1 is sensitive to rapamycin and regulates protein synthesis, nutrient responses, transcription, autophagy, and other cellular processes.25, 26, 27 mTORC2 contributes to regulation of cell growth, proliferation, and metabolism.28
Abnormal mTOR activity contributes to the pathogenesis of several neurodegenerative disorders.29, 30, 31 Experiments using an animal model of tuberous sclerosis (TSC) showed disinhibited mTOR signaling contributing to neuropsychiatric phenotypes, including cognitive deficits and seizures.32 Studies in AD patients and mouse models of AD have demonstrated activation of the mTOR pathway.33 Increased phosphorylation of ribosomal protein S6 kinase 1 (S6K1) has been observed in neurons from postmortem samples from patients with AD; moreover, these samples showed increased levels of phospho‐mTOR and phospho‐4E‐BP1, which were correlated with levels of total tau and phospho‐tau.34 [Correction added on 04 April 2017, after first online publication: The word ‘reducing’ has been deleted from ‘Increased phosphorylation of reducing ribosomal protein S6 kinase 1 (S6K1)’ in the statement above.] In mouse models of AD, rapamycin protected neurons against tau‐induced cell death,35 while reduced S6K1 expression improved spatial memory and synaptic plasticity.36 Aberrant mTOR signaling has also been observed in PD. Rapamycin‐induced autophagy prevents accumulation of ubiquitinated α‐synuclein in α‐synuclein overexpression models.37 The role of mTOR signaling in Huntington's disease (HD) is complex. Overexpression of huntingtin (Htt) protein in striatal neurons was associated with increased basal mTOR activity.38 In contrast, reduced phosphorylation of ribosomal protein S6 in striatal tissue samples collected from HD patients and HD mice indicated that impaired mTOR activity might contribute to neurodegeneration in HD.39
mTORC1 inhibitors, such as rapamycin and its analogs, have been used as mTOR‐targeted therapeutics in clinical practice. However, rapamycin and inhibitors of Akt (upstream of mTOR) show limited efficacy as inhibitors of mTORC1 signaling.40, 41 It is widely believed that the incomplete inhibition of mTORC1‐mediated phosphorylation following treatment with rapamycin and inhibitors of Akt may be a result of concomitant activation of AKT via the loss of a negative feedback mechanism.42, 43 Treatment with a combination of mTORC1 and Akt inhibitors dramatically repressed mTORC1 signaling.40 Moreover, these inhibitors inhibited phosphorylation of the proline‐rich Akt substrate of 40 kDa (PRAS40), which enhanced the repressive function of PRAS40 on mTORC1 signaling. This finding highlights the role of PRAS40 as a critical regulator of mTOR/Akt signaling. PRAS40 is a substrate of Akt,44 as well as an important substrate and component of mTORC1.45 In addition, PRAS40 is a direct inhibitor of mTORC1 and an interactive linker between the Akt and mTOR pathways. In this study, we report that mTORC1 inhibitor PRAS40 down‐regulated mTOR hyperactivity under stress and alleviated neurotoxic prion peptide‐induced apoptosis. This finding suggests that the mTOR pathway, especially PRAS40, may be considered a useful therapeutic target for prion diseases and other neurodegenerative disorders.
2. Materials and Methods
2.1. Reagents and antibodies
DMEM was purchased from Hyclone (Logan, UT, USA). FBS was purchased from Gibco (Grand Island, NY, USA). Prestained Protein Ladder (BD0027) was purchased from Bioworld Technology (Nanjing, China). WIP Tissue and Cell Lysis Reagent (C‐0013) was purchased from Beijing Biosynthesis Biotechnology (Beijing, China). SDS‐PAGE Sample Loading Buffer (P0015) was purchased from Beyotime Biotechnology (Shanghai, China). Peroxidase‐conjugated AffiniPure Goat Anti‐rabbit IgG (H+L; ZB‐2301) was purchased from ZSGB Biotechnology (Beijing, China). N‐Acetyl‐l‐cysteine (0108) was purchased from Amresco LLC (Solon, OH, USA). Rapamycin (S1842) and wortmannin (S1952) were purchased from Beyotime Biotechnology (Shanghai, China). Lipofectamine® 2000 Transfection Reagent (11668030) was purchased from Invitrogen, Thermo Fisher Scientific (Waltham, MA, USA). Opti‐MEM Reduced Serum Medium (31985062) was purchased from Gibco, Thermo Fisher Scientific. Phospho‐PRAS40 (Thr246) rabbit mAb (2997), PRAS40 Rabbit mAb (2691), phospho‐mTOR (Ser2448) rabbit mAb, phospho‐p70 S6 kinase (Thr389) rabbit mAb (9234), phospho‐4E‐BP1 (Thr37/46) rabbit mAb (2855), phospho‐Akt (Ser473) rabbit mAb (4060), phospho‐GSK‐3β (Ser9) rabbit polyclonal antibodies (9336), and phospho‐FoxO1 (Thr24)/FoxO3a (Thr32) rabbit polyclonal antibodies (9464) were purchased from Cell Signaling Technology (Danvers, MA, USA). [Correction added on 04 April 2017, after first online publication: ‘mTOR rabbit mAb (2983)’ has been changed to ‘PRAS40 Rabbit mAb (2691)’ in the statement above.] Rabbit anti‐caspase‐3, p17‐specific polyclonal antibodies (25546‐1‐AP), rabbit anti‐PARP1 polyclonal antibodies (13371‐1‐AP), rabbit anti‐beta tubulin polyclonal antibodies (10094‐1‐AP), and rabbit anti‐HA tag polyclonal antibodies (51064‐2‐AP) were purchased from Proteintech (Wuhan, China). The reagents and instruments used in the immunoblotting assays were purchased from Bio‐Rad (Hercules, CA, USA).
2.2. Cell culture
Mouse neuroblastoma N2a cells (Neuro2A cells) were grown in antibiotic‐free DMEM (Hyclone) supplemented with 10% FBS (Gibco). Cells were maintained in a humid incubator (37°C, 5% CO2). The experiments in which the cells were exposed to NAC, rapamycin or wortmannin utilized 1×105 cells per well. Cells were obtained from the Cell Culture Center of Xiehe Medical University (Beijing, China).
2.3. Prion protein peptide
The PrP106‐126 peptide (sequence KTNMKHMAGAAAA GAVVGGLG) was synthesized by Sangon Bio‐Tech (Shanghai, China). The purity of the prion peptide was >95% according to the data from the synthesizer. The peptide was dissolved in 0.1 mol/L PBS to a concentration of 1 mmol/L and shaken at 37°C for 1 week to facilitate peptide aggregation. All procedures were performed under sterile conditions. The experiments were conducted with a final peptide concentration of 100 μmol/L.
2.4. Assay of intracellular reactive oxygen species (ROS)
ROS abundance was measured using a ROS assay kit (Beyotime, China) according to the reagent protocol included by the manufacturer. Briefly, the growth medium was decanted from Neuro2A cells, after which the cells were washed with PBS, followed by incubation with fluorogenic probe 2′,7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA). DCFH‐DA was diluted in fresh DMEM to a final concentration of 10 μmol/L. Cells were incubated for 20 min at 37°C and washed three times with culture medium without serum. Finally, ROS abundance was assessed by measuring fluorescence at an excitation wavelength of 485 nm and an emission wavelength of 528 nm.
2.5. Plasmids and transfection
pRK5‐HA‐PRAS40 (Addgene plasmid #15481) and pLKO.1‐mPRAS40 (Addgene plasmid #15480) were gifts from Do‐Hyung Kim.46 Plasmid DNA was isolated using the Easy Pure® HiPure Plasmid MaxiPrep Kit (EM111, TransGen Biotech, Beijing, China). Opti‐MEM Reduced Serum Medium (Invitrogen, Carlsbad, CA, USA) and Lipofectamine 2000 Reagent (Invitrogen) were used for the transfection procedure. For the transfection, 2500 ng DNA was added to 1×106 adherent cells in a six‐well plate, after which 6 μL of Lipofectamine 2000 was diluted in 150 μL Opti‐MEM medium and incubated for 5 minutes with the diluted DNA at room temperature.
2.6. Measurement of apoptosis by flow cytometry
Apoptotic cell counting was conducted using the Annexin V‐FITC Apoptosis Detection Kit (A211‐01; Vazyme Biotech Co., Ltd., Nanjing, China). Apoptosis analysis was performed as described previously.47 Apoptotic cells, including Annexin V‐FITC‐positive cells, PI‐negative cells, and double‐positive cells, were counted.
2.7. Western blotting
Western blotting was performed as described previously.48 Briefly, equal amounts of protein (40 μg in each lane) were separated by SDS‐PAGE on 12% gels, after which the separated proteins were transferred onto a nitrocellulose membrane. Nonspecific binding sites were blocked by incubating the membrane with BSA Blocking Buffer (cw0054, pH 7.5; CWBIO, Beijing, China).
2.8. Statistical analysis
All assays were performed on three separate occasions. Data are expressed as mean±SD. All comparisons of parametric data were performed using Student's t test or one‐way ANOVA followed by Tukey's multiple comparison test. Statistical analysis was conducted using SPSS (version 13.0; IBM Co., Armonk, NY, USA), GraphPad Prism 5 (GraphPad Inc., La Jolla, CA, USA), and ImageJ (National Institutes of Health, Bethesda, MD, USA). The threshold for statistical significance was P<.05.
3. Results
3.1. Activation of mTOR in N2a cells upon exposure to PrP106‐126
Several investigations have shown that abnormal mTOR activity contributes to the pathogenesis of neurodegenerative disorders and leads to cognitive deficits.29, 30, 32, 33, 38 Moreover, hyperactive mTORC1 signaling has been reported to sensitize cells to apoptosis.49, 50 Hence, we decided to investigate the function of mTOR signaling in cells exposed to PrP106‐126. N2a cells were treated with 100 μmol/L PrP106‐126 for 6‐48 hours, after which the level of phosphorylated mTOR at Ser2448 was determined by Western blotting. Treatment of cells with PrP106‐126 induced mTOR phosphorylation over time. The mTOR phosphorylation level peaked at 24 hours post‐treatment (Figure 1A).
Figure 1.
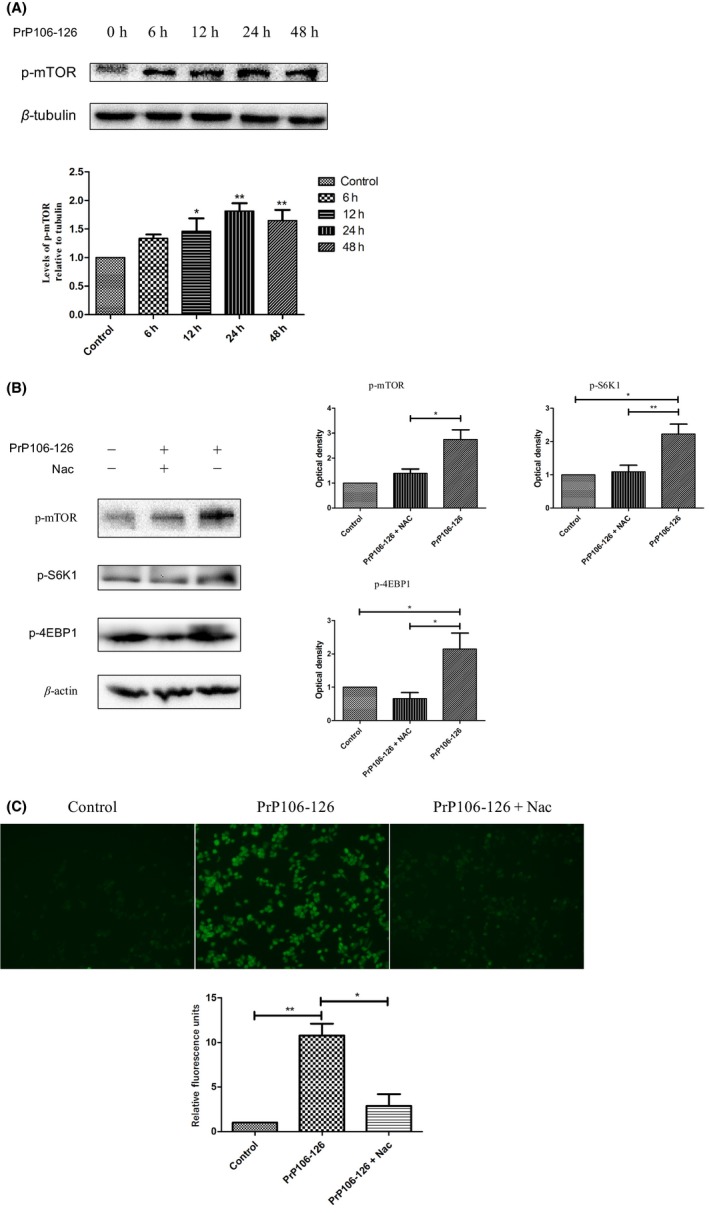
Activation of mTOR in N2a cells upon exposure to PrP106‐126. (A) N2a cells were treated with PrP106‐126 for the indicated time periods. Phosphorylation of mTOR at serine 2448 (p‐mTOR) was analyzed by Western blotting. The lower panel shows the densitometric quantification of mTOR phosphorylation levels. Values are presented as the mean±SD of triplicate experiments. *P<.05; **P<.01 (compared with the corresponding control group). (B) N2a cells were treated with PrP106‐126 in the presence or absence of NAC (5 mmol/L). Phospho‐mTOR, phospho‐S6K1, and phospho‐4EBP1 were analyzed by Western blotting. The histogram plot shows the densitometric quantification. Values are presented as the mean±SD of triplicate experiments. *P<.05; **P<.01. (C) Representative confocal microscopy images of reactive oxygen species (ROS). ROS are indicated by green DCF fluorescence
We examined whether induction of mTOR phosphorylation by PrP106‐126 is due to ROS production. Antioxidant N‐acetyl cysteine (NAC) was used as a scavenger to abolish induction of cellular ROS production. Pretreatment with NAC (5 mmol/L) for 2‐hour attenuated phosphorylation of mTOR and its downstream targets in N2a cells (Figure 1B). PrP106‐126‐treated cells showed significantly increased ROS production in comparison with that of PBS‐treated control cells. Cell treated with NAC (5 mmol/L) had significantly reduced ROS abundance in comparison with that of cells pretreated with the neurotoxic prion peptide (Figure 1C). These results indicate that oxidative stress was involved in the effects of PrP106‐126‐treatment. Activation of the mTOR pathway after exposure to PrP106‐126 was at least partially due to ROS production.
3.2. Overexpression of PRAS40 inhibited neuronal apoptosis induced by neurotoxic prion peptide
We hypothesized that mTOR hyperactivation contributes to neuronal apoptosis induced by aggregation of misfolded proteins. Thus, we overexpressed PRAS40 to determine whether its neuroprotective effect against neurotoxic prion peptide‐induced neuronal apoptosis is mediated by its direct inhibitory effect on TORC1.
Cells were transfected with the HA‐PRAS40 plasmid for 24 hours (or with the empty HA vector as a control treatment). First, we confirmed PRAS40 overexpression in vitro via Western blotting (Figure 2A). Knockdown of PRAS40 by shPRAS40 was also confirmed by Western blotting (Figure 2A).
Figure 2.
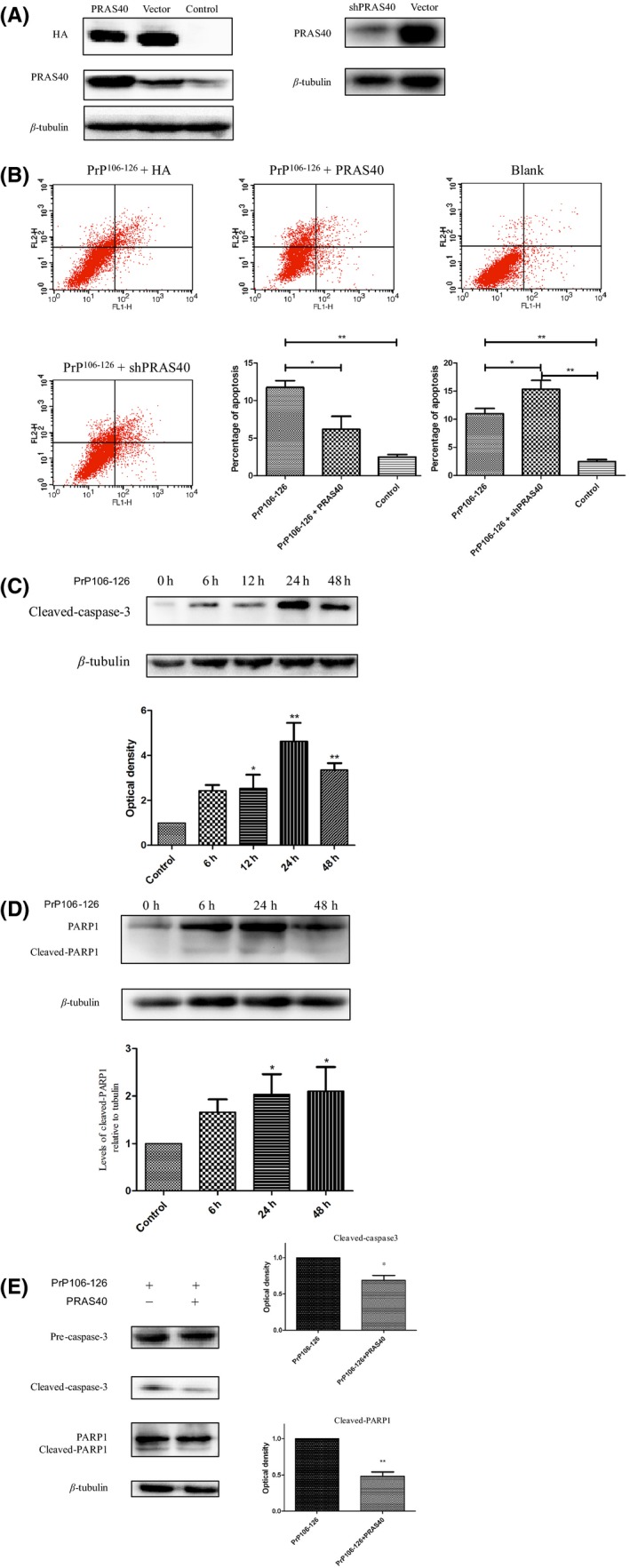
PRAS40 inhibited neurotoxic prion peptide‐induced neuronal apoptosis. (A) Western blot confirmation of the effects of overexpression or knockdown of PRAS40 in cells. Protein levels of PRAS40 were increased in cells transfected with PRAS40. Knockdown of PRAS40 reduced PRAS40 abundance in cells. (B) Cells transfected with the indicated plasmid were treated with PrP106‐126 for 24 h. Apoptotic cells were detected using an Annexin V‐FITC/PI apoptosis detection kit. Flow cytometry analysis showed that PRAS40 overexpression inhibited apoptosis induced by the prion peptide. Knockdown of PRAS40 enhanced PrP106‐126‐induced apoptosis in cells. (C and D) N2a cells were treated with PrP106‐126 for the indicated time periods. The abundance of cleaved caspase‐3 (C) and cleaved PARP1 (D) was analyzed by Western blotting. The histogram plots show the densitometric quantification. (E) Cells were transfected with the HA‐PRAS40 vector and treated with PrP106‐126 for 24 h, after which the abundance of cleaved caspase‐3 and cleaved PARP1 was analyzed by Western blotting. The histogram plots (right panel) show the densitometric quantification. Values are presented as the mean±SD of triplicate experiments. *P<.05; **P<.01
Cells transfected with HA‐PRAS40 were treated with PrP106‐126 for 24 hours. In comparison with the HA control cells, overexpression of PRAS40 clearly reduced the number of apoptotic cells following exposure to PrP106‐126 (Figure 2B). To confirm this result, we knocked down PRAS40 using shRNA, followed by treatment with PrP106‐126 for 24 hours. Knockdown of PRAS40 enhanced PrP106‐126‐induced apoptosis (Figure 2B).
To confirm the presence of PrP106‐126‐induced apoptosis, activation of apoptosis via caspase‐3 and PARP was assessed by immunoblotting. Protein levels of cleaved caspase‐3 and cleaved PARP were increased after PrP106‐126 treatment in a time‐dependent manner. The abundance of cleaved caspase‐3 was significantly increased at 12 hours post‐treatment and peaked at 24 hours post‐treatment (Figure 2C). The abundance of cleaved PARP was significantly increased at 24 hours post‐treatment (Figure 2D).
Cleaved caspase‐3 and cleaved PARP were also assessed by Western blotting following PRAS40 overexpression. The abundance of cleaved caspase‐3 and cleaved PARP was reduced in cells transfected with HA‐PRAS40 following exposure to PrP106‐126 (Figure 2E).
Collectively, these findings indicate that overexpression of PRAS40 attenuated neurotoxic prion peptide‐induced neuronal apoptosis.
3.3. PRAS40 alleviates PrP106‐126‐induced mTOR activation
PRAS40 overexpression attenuated mTOR activation by inhibiting mTORC1. Cells were transfected with the HA‐PRAS40 vector and treated with PrP106‐126 for 24 hours. S6K1 and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4EBP1; two well‐characterized substrates and downstream effector molecules of mTORC1) were analyzed by Western blotting. As expected, cells transfected with HA‐PRAS40 showed reduced phosphorylation of S6k1 and 4EBP1 (Figure 3A), while the abundance of PRAS40 and p‐PRAS40 was dramatically increased. The attenuated activation of these mTOR downstream effectors shows that PRAS40 inhibited the mTOR pathway.
Figure 3.
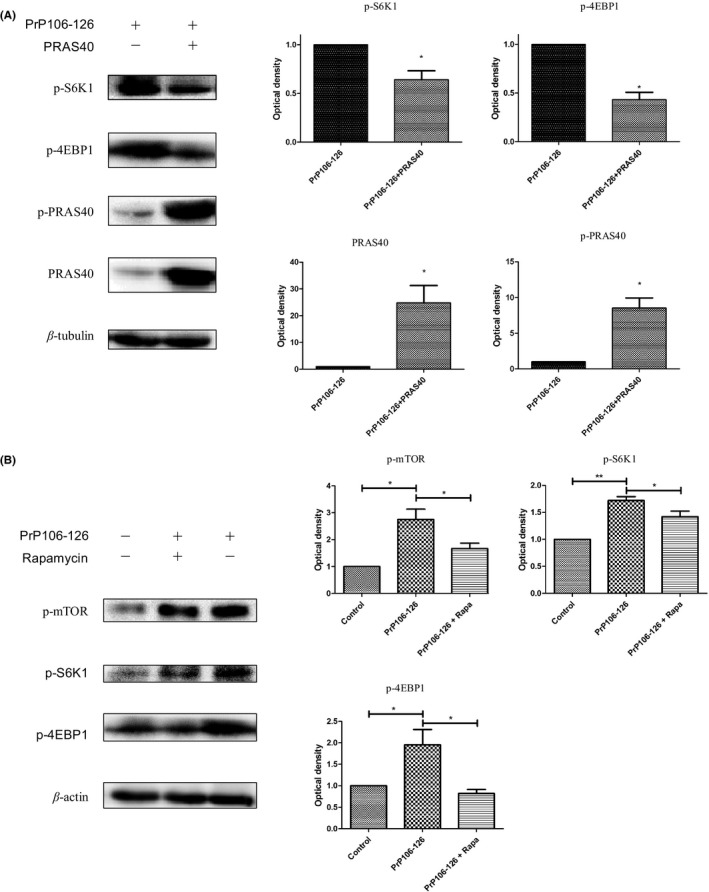
PRAS40 alleviates PrP106‐126‐induced mTOR activation. (A) Cells were transfected with the HA‐PRAS40 vector and treated with PrP106‐126 for 24 h. The abundance of phospho‐S6K1, phospho‐4EBP1, PRAS40, and phospho‐PRAS40 was analyzed by Western blotting. (B) N2a cells were treated with PrP106‐126 in the presence or absence of rapamycin (0.2 μg/mL), after which phospho‐mTOR, phospho‐S6K1, and phospho‐4EBP1 were analyzed by Western blotting. The histogram plot (right panel) shows the densitometric quantification. Values are presented as the mean±SD of triplicate experiments. *P<.05; **P<.01
N2a cells were exposed to mTOR inhibitor rapamycin (0.2 μg/mL) 2 hours before exposure to PrP106‐126 for 24 hours. S6K1 and 4EBP1 were significantly phosphorylated following exposure to PrP106‐126, which was consistent with phosphorylation of mTOR at Ser2448 (Figure 3B). [Correction added on 04 April 2017, after first online publication: The words ‘rapamycin and’ has been deleted and ‘Ser2248’ has been changed to ‘Ser2448’ in the statement above.] Rapamycin partially reduced phosphorylation of mTOR and its downstream effector S6K1 (Figure 3B), confirming that the mTOR pathway was activated by PrP106‐126. In summary, the mTOR pathway was activated after exposure to PrP106‐126, and this activation can be inhibited by PRAS40 overexpression or rapamycin treatment.
3.4. PRAS40 alleviates PrP106‐126‐induced neuronal apoptosis via mTOR‐AKT activation
Experiments were conducted to elucidate the manner in which mTOR promotes neuronal apoptosis and assess the link between apoptosis and mTOR. One likely mechanism for the link between apoptosis and mTOR is a negative feedback loop (NFL), in which mTORC1 activation inhibits PI3K‐Akt signaling;51, 52 this NFL is mediated via phosphorylation of the insulin receptor substrate (IRS) by S6K1. Furthermore, activation of Akt prevents apoptosis.53 In cancer cells, suppression of apoptosis via Akt inhibits the activity of transcription factors forkhead box O1 and forkhead box O3A (FoxO1/FoxO3A).54 As a key molecular link between mTORC1 and apoptosis, Akt activation was analyzed. Akt activation can be inhibited by mTORC1 activation, so we determined whether Akt was activated after PRAS40 overexpression. Following treatment with PrP106‐126 for 24 hours, Western blots showed that the abundance of phosphorylated Akt (a substrate of PI3K) and glycogen synthase kinase 3 beta (GSK3β; a substrate of Akt) was increased by PRAS40 overexpression (Figure 4A), indicating activation of PI3K‐Akt signaling. In addition, phosphorylation of FoxO1/FoxO3A was induced by PRAS40 overexpression (Figure 4A), indicating that the activity of these transcription factors was inhibited by Akt activation; this effect of PRAS40 may contribute to its inhibitory effect on oxidative stress‐induced apoptosis.
Figure 4.
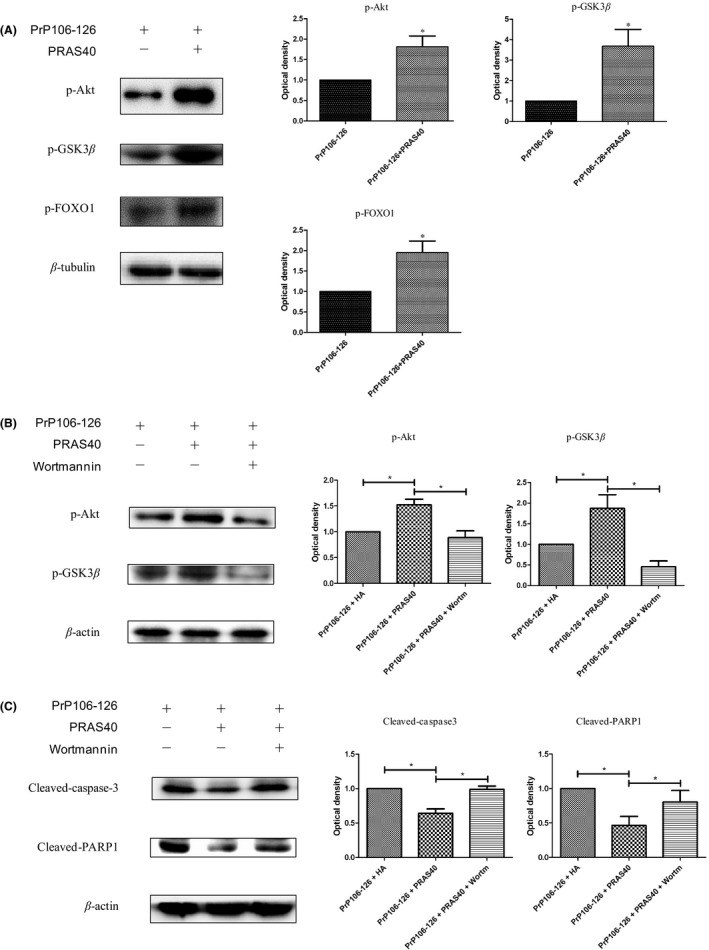
Overexpression of PRAS40 enhanced Akt activation. (A) Cells were transfected with the HAPRAS40 vector and treated with PrP106‐126 for 24 h. The phosphorylation levels of Akt (substrate of PI3K) and GSK3β (substrate of Akt) were increased, indicating activation of PI3K‐Akt signaling. FoxO1/O3A phosphorylation was induced, indicating that transcription factors FoxO1/O3A were inhibited by Akt activation. The histogram plots show the densitometric quantification. (B and C) Cells were transfected with the HA‐PRAS40 vector and treated with PrP106‐126 for 24 h in the presence or absence of wortmannin (100 nmol/L, added 2 h before PrP106‐126 treatment). Phospho‐Akt and phospho‐GSK3β were analyzed by Western blotting (B). The abundance of cleaved caspase‐3 and cleaved PARP1 was analyzed by Western blotting. The histogram plots (right panel) show the densitometric quantification (C). Values are presented as mean±SD of triplicate experiments. *P<.05
PRAS40‐transfected cells were treated with PrP106‐126 combined with wortmannin, a specific inhibitor of the PI3K/Akt pathway, for 24 hours, after which phosphorylated Akt was assessed by Western blotting. Wortmannin significantly attenuated the PRAS40‐mediated increase in the abundance of phosphorylated Akt (Figure 4B).
Furthermore, wortmannin abolished the inhibitory effect of PRAS40 overexpression on apoptosis. The abundance of cleaved caspase‐3 and cleaved PARP was reduced in cells transfected with PRAS40, but this effect was abolished by wortmannin (Figure 4C).
These results indicate that overexpression of PRAS40 alleviates PrP106‐126‐induced neuronal apoptosis by restoring Akt phosphorylation. Akt is at least partially involved in mTOR‐mediated inhibition of PrP106‐126‐induced neuronal apoptosis.
4. Discussion
In this study, we found that PRAS40‐mediated mTOR signaling is an important pathway in neuronal death in response to PrP106‐126 treatment. PrP106‐126 triggered oxidative stress and neuronal apoptosis; as a response, the mTOR pathway was activated. mTOR activation contributed to neuronal apoptosis through a negative feedback loop, which inhibited Akt activation. Overexpression of PRAS40 inhibited the mTOR pathway and alleviated neurotoxic prion peptide‐induced apoptosis.
4.1. Misfolded proteins, oxidative stress and apoptosis
Misfolded proteins evoke oxidative stress, which contributes to the development of apoptotic neuronal cell death and neuronal dysfunction.14, 15, 16, 17 PrP106‐126 induced mitochondrial dysfunction and apoptosis in N2a cells, consistent with our previous work.55, 56 Apoptosis is a complex process of programmed cell death and destruction that is involved in many diseases and disorders.57 In many types of cancer, resistance to apoptosis is a major driver of pathogenesis. Therefore, modulation of aberrant apoptosis is an attractive anticancer therapeutic strategy.58, 59 Under inflammatory conditions, a prolonged neutrophil lifespan due to delayed apoptotic death may be needed to protect the host from pathogens.60, 61, 62 However, neurons do not divide, so promotion of neuron survival is extremely important in patients with neurodegenerative disorders.
4.2. mTOR activation in neurodegenerative disorders
mTORC1 is activated under oxidative stress mainly because of inactivation of the TSC1‐TSC2 complex.63 Growth factors and hormones such as insulin stimulate the mTORC1 pathway. In addition, mTORC1 is activated by G‐protein Rheb and inhibited by TSC2‐mediated inhibition of Rheb.64 Under arsenite stress, TSC2 down‐regulation is associated with S6K1 phosphorylation.51 Therefore, oxidative stress activates mTORC1 mainly by inhibiting TSC2 activation, abolishing the inhibitory effect of TSC2 on mTORC1.
Abnormal mTOR activity contributes to the pathogenesis of neurodegenerative disorders and leads to cognitive deficits.29, 30, 32, 33, 38 Moreover, hyperactive mTORC1 signaling has been reported to sensitize cells to apoptosis.49, 50 In this study, neurotoxic prion peptide‐induced neuronal apoptosis was inhibited by abolishing ROS‐induced cellular stress or inhibiting mTORC1 activation. These results are consistent with the studies described above.
The reason why mTOR activation is needed under oxidative stress is interesting. We supposed that, as a central regulator of growth and metabolism, the mTOR pathway is required for synthesis and expression of stress response proteins. Several stress‐related proteins, including Hsp70, require mTORC1 activation for their expression during stress responses.65
Abnormal mTOR signaling pathway activity has been implicated in the pathogenesis of neurodegenerative disorders. It has been hypothesized that inhibiting mTOR signaling may attenuate neurodegeneration. However, some findings suggest that enhancing the activity of mTORC1 is neuroprotective.39 These results contradict the conventional view that inhibition of mTORC1 is neuroprotective.38, 66, 67 Davidson et al.39 showed that mTORC1 activity was reduced in their HD model, and found that reinstating aberrant mTORC1 activity improved disease phenotypes. However, in other studies,36 suppressing mTORC1 under conditions associated with hyperactive mTORC1 improved disease phenotypes. We believe that, as mentioned by Davidson et al., restoration of homeostatic mTORC1 function can improve the phenotypes of neurological diseases. In our study, hyperactive mTOR signaling was observed after exposure to PrP106‐126; in this case, inhibition of mTOR alleviated prion peptide‐induced neuronal toxicity.
4.3. PRAS40 and mTOR inhibition
PRAS40 is an important substrate and inhibitory component of mTORC1. Phosphorylation of PRAS40 results in disassociation of PRAS40 from mTORC1, which relieves its inhibitory constraint on mTORC1 activity.46, 68 We found that PRAS40 down‐regulated mTOR hyperactivity under prion peptide‐induced neuronal toxicity and alleviated neuronal apoptosis. Cells were exposed to rapamycin to compare its effects with those of PRAS40. Rapamycin is a classical inhibitor of mTORC1.40, 69, 70 Rapamycin consistently inhibited mTORC1 and attenuated apoptosis. These findings support the role of PRAS40 in protecting neurons from neurotoxic prion peptide‐induced apoptosis.
Inhibition of mTORC1 by PRAS40 releases the negative feedback loop, leading to activation of Akt. Consistent with this role, increased Akt phosphorylation has been observed frequently following treatment with rapamycin,71 but not following exposure to dual mTORC1/mTORC2 inhibitors.72 Moreover, impaired mTORC2 complex integrity may inhibit Akt signaling.73 Therefore, we used PRAS40 as a specific inhibitory protein to mediate mTORC1 activation.
4.4. FOXO transcription factors and Akt signaling
Forkhead box O (FOXO) transcription factors play a critical role in many cellular processes, including the cell cycle, metabolism, apoptosis, and resistance to oxidative stress.74, 75 The FOXO family (FOXO1, 3, 4, and 6) can respond to insulin/growth factors and is negatively regulated by PI3K‐Akt signaling.76 Akt‐mediated phosphorylation of FOXOs inhibits their transcriptional activity.74 In the presence of growth factors, FOXOs are sequestered by the PI3K‐Akt pathway in the cytoplasm. FOXOs translocate into the nucleus and transcribe target genes when Akt is inactive in the absence of growth factors.77 In our study, FoxO1/O3A phosphorylation was induced following PRAS40 overexpression (Figure 4A), which shows that the activity of these transcription factors was inhibited by Akt activation, thus facilitating neuronal survival under oxidative stress‐induced apoptosis.
Many studies have demonstrated that the Akt pathway supports neuronal survival.53, 78 In this study, PRAS40 down‐regulated mTOR hyperactivity and induced AKT activation via a negative feedback mechanism. PRAS40 overexpression inhibited apoptosis via Akt activation, but wortmannin abolished the reduction in apoptosis associated with PRAS40 overexpression. This finding confirmed the role of PRAS40 in Akt phosphorylation.
4.5. Negative feedback loop between mTOR and Akt signaling
Akt activation prevents apoptosis.53 mTORC1 is inhibited by TSC2‐mediated inhibition of Rheb. Upstream of Rheb, Akt phosphorylates and inactivates TSC2, allowing Rheb to activate mTORC1.79 In other words, Akt activation activates the mTOR pathway. However, upon mTORC1 activation, negative feedback loops inhibit PI3K‐Akt signaling.52 Phosphorylation of AKT is mediated by mTORC1/S6K1, mTORC2, and other pathways.73, 80 These negative feedback loops are the reason that mTORC1 hyperactivation induces apoptosis, although Akt is known to prevent apoptosis (Figure 5).
Figure 5.
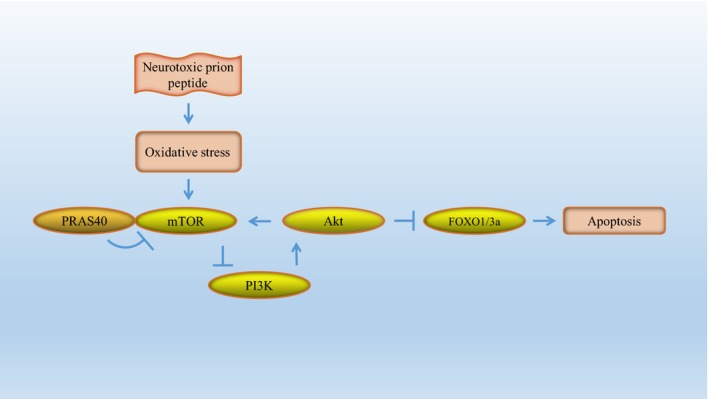
Schematic representation of PRAS40‐mediated mTORC1 inhibition and apoptosis suppression. mTORC1 is activated by neurotoxic prion peptide‐induced oxidative stress. Upstream of mTORC1 signaling, Akt can indirectly phosphorylate and activate mTORC1. Upon mTORC1 activation, negative‐feedback loops mediated by mTORC1/S6K1, mTORC2, or other pathways inhibit PI3K‐Akt signaling. Akt‐mediated phosphorylation of FOXOs results in inhibition of the transcriptional activity of FOXO. PRAS40, a direct inhibitor of mTORC1, attenuates mTOR hyperactivity under stress conditions and alleviates neurotoxic prion peptide‐induced apoptosis
Rapamycin induced activation of Akt via a feedback loop and weakened the effect of mTOR inhibition. Therefore, we focused on PRAS40 and the pivotal role of PRAS40 in the regulation of aberrant mTOR signaling.
We believe that the site where stimulation acts in the loop is important. Persistent stimulation by oxidative stress enhances mTORC1 hyperactivation and induces apoptotic neuronal signaling and neurotoxicity. Thus, PRAS40 inhibits mTORC1 hyperactivation and plays a key role in protecting neurons from neurotoxic prion peptide‐induced apoptosis. These findings suggest that the mTOR pathway, and PRAS40 in particular, should be exploited as a therapeutic target for prion diseases and other neurodegenerative disorders.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank Dr Chunfa Liu, Dr Ruichao Yue, Dr Guangyu Cheng, Dr Jie Wang, Dr Wei Wu, and Dr Haodi Dong for their valuable suggestions and critical reading of this manuscript.
Yang W, Yang L‐F, Song Z‐Q, et al. PRAS40 alleviates neurotoxic prion peptide‐induced apoptosis via mTOR‐AKT signaling. CNS Neurosci Ther. 2017;23:416–427. 10.1111/cns.12685
Funding information
This work was supported by the Natural Science Foundation of China (Project No. 31472166), the Ministry of Agriculture of China, 948 projects (2014‐S9), and the Foundation of the Chinese Ministry of Science and Technology (Project No. 2015BAI07B02).
The first two authors contributed equally to this work.
References
- 1. Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363‐13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeArmond SJ. Overview of the transmissible spongiform encephalopathies: prion protein disorders. Br Med Bull. 1993;49:725‐737. [DOI] [PubMed] [Google Scholar]
- 3. Fraser H. Diversity in the neuropathology of scrapie‐like diseases in animals. Br Med Bull. 1993;49:792‐809. [DOI] [PubMed] [Google Scholar]
- 4. Bell JE, Ironside JW. Neuropathology of spongiform encephalopathies in humans. Br Med Bull. 1993;49:738‐777. [DOI] [PubMed] [Google Scholar]
- 5. DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB. Changes in the localization of brain prion proteins during scrapie infection. Neurology. 1987;37:1271‐1280. [DOI] [PubMed] [Google Scholar]
- 6. Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet. 2004;363:51‐61. [DOI] [PubMed] [Google Scholar]
- 7. Fu Y, Zhao D, Pan B, et al. Proteomic analysis of protein expression throughout disease progression in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015;47:915‐926. [DOI] [PubMed] [Google Scholar]
- 8. Pan Y, Liu R, Terpstra E, et al. Dysregulation and diagnostic potential of microRNA in Alzheimer's disease. J Alzheimers Dis. 2016;49:1‐12. [DOI] [PubMed] [Google Scholar]
- 9. Pan Y, Sun L, Wang J, et al. STI571 protects neuronal cells from neurotoxic prion protein fragment‐induced apoptosis. Neuropharmacology. 2015;93:191‐198. [DOI] [PubMed] [Google Scholar]
- 10. Forloni G, Angeretti N, Chiesa R, et al. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543‐546. [DOI] [PubMed] [Google Scholar]
- 11. Brown DR, Herms J, Kretzschmar HA. Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. NeuroReport. 1994;5:2057‐2060. [DOI] [PubMed] [Google Scholar]
- 12. Selvaggini C, De Gioia L, Cantu L, et al. Molecular characteristics of a protease‐resistant, amyloidogenic and neurotoxic peptide homologous to residues 106‐126 of the prion protein. Biochem Biophys Res Commun. 1993;194:1380‐1386. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y, Zhao D, Pan B, et al. Death receptor 6 and caspase‐6 regulate prion peptide‐induced axonal degeneration in rat spinal neurons. J Mol Neurosci. 2015;56:966‐976. [DOI] [PubMed] [Google Scholar]
- 14. Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205‐214. [DOI] [PubMed] [Google Scholar]
- 15. Melo A, Monteiro L, Lima RM, Oliveira DM, Cerqueira MD, El‐Bacha RS. Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxid Med Cell Longev. 2011;2011:467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med. 2013;62:170‐185. [DOI] [PubMed] [Google Scholar]
- 17. Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49‐60. [DOI] [PubMed] [Google Scholar]
- 18. Bertoni A, Giuliano P, Galgani M, et al. Early and late events induced by polyQ‐expanded proteins: identification of a common pathogenic property of polYQ‐expanded proteins. J Biol Chem. 2011;286:4727‐4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hands S, Sajjad MU, Newton MJ, Wyttenbach A. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J Biol Chem. 2011;286:44512‐44520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905‐909. [DOI] [PubMed] [Google Scholar]
- 21. Kunz J, Henriquez R, Schneider U, Deuter‐Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585‐596. [DOI] [PubMed] [Google Scholar]
- 22. Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1‐arresting rapamycin‐receptor complex. Nature. 1994;369:756‐758. [DOI] [PubMed] [Google Scholar]
- 23. Sabatini DM, Erdjument‐Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin‐dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35‐43. [DOI] [PubMed] [Google Scholar]
- 24. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yun YS, Kim KH, Tschida B, et al. mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol Cell. 2016;61:625‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aylett CHS, Sauer E, Imseng S, et al. Architecture of human mTOR complex 1. Science. 2016;351:48‐52. [DOI] [PubMed] [Google Scholar]
- 28. Gaubitz C, Prouteau M, Kusmider B, Loewith R. TORC2 structure and function. Trends Biochem Sci. 2016;41:532‐545. [DOI] [PubMed] [Google Scholar]
- 29. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744‐755. [DOI] [PubMed] [Google Scholar]
- 30. Kassai H, Sugaya Y, Noda S, et al. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014;7:1626‐1639. [DOI] [PubMed] [Google Scholar]
- 31. Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379‐392. [DOI] [PubMed] [Google Scholar]
- 32. Ehninger D, de Vries PJ, Silva AJ. From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res. 2009;53:838‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed). 2012;4:941‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. An WL, Cowburn RF, Li L, et al. Up‐regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siman R, Cocca R, Dong Y. The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early‐stage Alzheimer‐type tauopathy. PLoS One. 2015;10:e142340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caccamo A, Branca C, Talboom JS, et al. Reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer's disease. J Neurosci. 2015;35:14042‐14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha‐synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578‐13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pryor WM, Biagioli M, Shahani N, et al. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington's disease. Sci Signal. 2014;7:a103. [DOI] [PubMed] [Google Scholar]
- 39. Lee JH, Tecedor L, Chen YH, et al. Reinstating aberrant mTORC1 activity in huntington's disease mice improves disease phenotypes. Neuron. 2015;85:303‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mi W, Ye Q, Liu S, She QB. AKT inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E‐BP1 axis. Oncotarget. 2015;6:13962‐13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grzmil M, Hemmings BA. Overcoming resistance to rapalogs in gliomas by combinatory therapies. Biochim Biophys Acta. 2013;1834:1371‐1380. [DOI] [PubMed] [Google Scholar]
- 42. O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun SY, Rosenberg LM, Wang X, et al. Activation of Akt and eIF4E survival pathways by rapamycin‐mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052‐7058. [DOI] [PubMed] [Google Scholar]
- 44. Kovacina KS, Park GY, Bae SS, et al. Identification of a proline‐rich Akt substrate as a 14‐3‐3 binding partner. J Biol Chem. 2003;278:10189‐10194. [DOI] [PubMed] [Google Scholar]
- 45. Wang L, Harris TE, Roth RA, Lawrence JJ. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036‐20044. [DOI] [PubMed] [Google Scholar]
- 46. Vander HE, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316‐323. [DOI] [PubMed] [Google Scholar]
- 47. Wang J, Zhou X, Pan B, et al. Investigation of the effect of Mycobacterium bovis infection on bovine neutrophils functions. Tuberculosis. 2013;93:675‐687. [DOI] [PubMed] [Google Scholar]
- 48. Song ZQ, Yang LF, Wang YS, et al. Overexpression of BAT3 alleviates prion protein fragment PrP106‐126‐induced neuronal apoptosis. CNS Neurosci Ther. 2014;20:737‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee CH, Inoki K, Karbowniczek M, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26:4812‐4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650‐1656. [DOI] [PubMed] [Google Scholar]
- 51. Thedieck K, Holzwarth B, Prentzell MT, et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell. 2013;154:859‐874. [DOI] [PubMed] [Google Scholar]
- 52. Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209‐218. [DOI] [PubMed] [Google Scholar]
- 53. Appenzeller‐Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274‐282. [DOI] [PubMed] [Google Scholar]
- 54. Myatt SS, Brosens JJ, Lam EW. Sense and sensitivity: FOXO and ROS in cancer development and treatment. Antioxid Redox Signal. 2011;14:675‐687. [DOI] [PubMed] [Google Scholar]
- 55. Pan B, Yang L, Wang J, et al. C‐Abl tyrosine kinase mediates neurotoxic prion peptide‐induced neuronal apoptosis via regulating mitochondrial homeostasis. Mol Neurobiol. 2014;49:1102‐1116. [DOI] [PubMed] [Google Scholar]
- 56. Song Z, Zhu T, Zhou X, et al. REST alleviates neurotoxic prion peptide‐induced synaptic abnormalities, neurofibrillary degeneration and neuronal death partially via LRP6‐mediated Wnt‐beta‐catenin signaling. Oncotarget. 2016;7:12035‐12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770‐776. [DOI] [PubMed] [Google Scholar]
- 58. Kumar D, Shankar S, Srivastava RK. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2014;343:179‐189. [DOI] [PubMed] [Google Scholar]
- 59. Li GY, Jung KH, Lee H, et al. A novel imidazopyridine derivative, HS‐106, induces apoptosis of breast cancer cells and represses angiogenesis by targeting the PI3K/mTOR pathway. Cancer Lett. 2013;329:59‐67. [DOI] [PubMed] [Google Scholar]
- 60. El KD, Jozsef L, Pan W, Filep JG. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res. 2008;103:352‐359. [DOI] [PubMed] [Google Scholar]
- 61. Wang J, Zhou X, Pan B, et al. Expression pattern of interferon‐inducible transcriptional genes in neutrophils during bovine tuberculosis infection. DNA Cell Biol. 2013;32:480‐486. [DOI] [PubMed] [Google Scholar]
- 62. Andina N, Conus S, Schneider EM, Fey MF, Simon HU. Induction of Bim limits cytokine‐mediated prolonged survival of neutrophils. Cell Death Differ. 2009;16:1248‐1255. [DOI] [PubMed] [Google Scholar]
- 63. Yoshida S, Hong S, Suzuki T, et al. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2‐Rheb GTPase pathway. J Biol Chem. 2011;286:32651‐32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E‐BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457‐1466. [DOI] [PubMed] [Google Scholar]
- 65. Chou SD, Prince T, Gong J, Calderwood SK. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One. 2012;7:e39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585‐595. [DOI] [PubMed] [Google Scholar]
- 67. Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119:398‐407. [DOI] [PubMed] [Google Scholar]
- 68. Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin‐regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903‐915. [DOI] [PubMed] [Google Scholar]
- 69. Bar‐Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cao K, Graziotto JJ, Blair CD, et al. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson‐Gilford progeria syndrome cells. Sci Transl Med. 2011;3:58r‐89r. [DOI] [PubMed] [Google Scholar]
- 71. Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin‐mediated resistance in diffuse large B‐cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114:2926‐2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP‐competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288‐298. [DOI] [PubMed] [Google Scholar]
- 73. Liu P, Gan W, Inuzuka H, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15:1340‐1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf). 2008;192:19‐28. [DOI] [PubMed] [Google Scholar]
- 75. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83‐97. [DOI] [PubMed] [Google Scholar]
- 76. Kloet DE, Burgering BM. The PKB/FOXO switch in aging and cancer. Biochim Biophys Acta. 2011;1813:1926‐1937. [DOI] [PubMed] [Google Scholar]
- 77. Sandri M. FOXOphagy path to inducing stress resistance and cell survival. Nat Cell Biol. 2012;14:786‐788. [DOI] [PubMed] [Google Scholar]
- 78. Xiong X, Xie R, Zhang H, et al. PRAS40 plays a pivotal role in protecting against stroke by linking the Akt and mTOR pathways. Neurobiol Dis. 2014;66:43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex‐2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3‐kinase/akt pathway. Mol Cell. 2002;10:151‐162. [DOI] [PubMed] [Google Scholar]
- 80. Rozengurt E, Soares HP, Sinnet‐Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 2014;13:2477‐2488. [DOI] [PMC free article] [PubMed] [Google Scholar]