

Summary
Aims
Pseudoginsenoside‐F11 (PF11), an ocotillol‐type ginsenoside, has been reported to exert wide‐ranging neuroprotective properties. The aim of this study was to investigate the effect and potential mechanisms of PF11 on the autophagic/lysosomal pathway following ischemic stroke.
Methods
Male Sprague‐Dawley rats underwent permanent middle cerebral artery occlusion (pMCAO). Cerebral ischemia outcome, TUNEL staining, Fluoro‐Jade B staining were carried out 24 hours poststroke. The autophagic/lysosomal‐related proteins were measured.
Results
A single administration of PF11 significantly decreased the infarct area, reduced the brain water content, and improved neurological functions, even 4 hours after the onset of pMCAO. Meanwhile, PF11 lessened the ischemic insult‐mediated loss of neurons and activation of astrocytes and microglia. Furthermore, PF11 attenuated pMCAO‐induced accumulations of autophagosomes and apoptosis. We further observed a remarkable effect of PF11 in reversing the ischemic insult‐induced accumulation of autophagosomes (LC3‐II) and abnormal aggregation of autophagic proteins (SQSTM1 and ubiquitin). Furthermore, PF11 was capable of improving lysosomal function and lysosome/autophagosome fusion following pMCAO, and this change was reversed by the lysosomal inhibitor chloroquine. Also, the improvement of ischemic outcome and the antiapoptotic effect induced by PF11 was reversed by CQ.
Conclusion
These findings indicate that the autophagic flux is impaired in a rat model of pMCAO, and that PF11 exerts an excellent protective effect against ischemic stroke by alleviating autophagic/lysosomal defects.
Keywords: apoptosis, autophagic/lysosomal defects, permanent cerebral ischemia, PF11
1. Introduction
Stroke is one of the leading causes of adult death and disability worldwide. Until now, tissue plasminogen activator is the only drug approved for the treatment of ischemic stroke with a limited therapeutic time window and a high risk of hemorrhagic side effects.1, 2 Meanwhile, many neuroprotective interventions have failed in clinical trials because of a lack of efficacious curative effects.3 Autophagy has only recently been identified as an important mechanism that may be a key regulator of ischemic stroke. Increased induction of autophagy is relatively frequent in ischemic stroke,4 including hypoxia‐ischemia,5 global ischemia,6 and focal ischemia.7, 8 Stimulation of autophagy is thought to reduce neurodegenerative damage after focal ischemic insult.8, 9, 10 However, excessive autophagy may contribute to cell death.7, 11 A recent study has indicated that autophagy plays different roles in cerebral ischemia and the subsequent reperfusion phase.12 Thus, the contribution of autophagy to ischemic stroke needs to be reconsidered.
Pseudoginsenoside‐F11 (PF11), isolated from leaves of Panax pseudoginseng subsp. Himalaicus HARA (Himalayan Panax), is a form of ocotillol‐type ginsenoside contained in Panax quinquefolium L.13 Our studies have strongly suggested that PF11 plays an extensive role in treating many disorders of the central nervous system. For example, PF11 significantly improves AD‐like cognitive impairment.14 Also, PF11 exerts excellent antineuroinflammatory effects on LPS‐activated microglia.15 Furthermore, PF11 has a marked antagonistic effect on Parkinson's disease.16 Additionally, PF11 decreases morphine‐induced behavioral sensitization in mice.17, 18 Therefore, PF11, through its diverse pharmacological activities, may also have potential neuroprotective effects in the context of ischemic stroke.
Here, we focus on the effect and potential mechanisms of PF11 on the autophagic/lysosomal pathway in a rat model of pMCAO. The data show that one single administration of PF11 (3‐48 mg/kg, i.v.) significantly attenuates infarction and brain edema and improves neurological functions, even 4 hours after the onset of pMCAO, and the protective effect of PF11 may depend on restoration of autophagic flux by improving lysosomal function and autophagosome/lysosome fusion. What is more, a autophagic/lysosomal defect was detected in a rat pMCAO stroke model.
2. Materials and Methods
2.1. Administration of drugs and chemicals
PF11 was isolated according to previously described methods.19 The purity of PF11 detected with HPLC was more than 98%. PF11 (3, 6, 12, 24, 48 mg/kg, i.v.) was dissolved in normal saline and administered at 0.5 hour after the onset of pMCAO by the intravenous (i.v.) route. Edaravone (5 mg/kg; China National Medicines Guorui Pharmaceutical Co., Ltd., Beijing, China) was dissolved in normal saline and administered at 0.5 hour after the onset of pMCAO20 by the i.v. route. Five hour before, the animals were killed, treatment with chloroquine (25 mg/kg; Sigma, St. Louis, MO, USA, C6628) was carried out by a single intracerebroventricular (i.c.v) injection with a needle (26‐gauge) at the following stereotaxic coordinates: 0.8 mm anterior to the bregma, 1.5 mm lateral to the midline, and 3.6 mm ventral to the skull surface.21
2.2. Animals and pMCAO rat model
Male Sprague‐Dawley (SD) rats weighing 270‐300 g (age 8 weeks approximately), supplied by the Experimental Animal Centre of Shenyang Pharmaceutical University, were maintained under standard housing conditions with food and water available ad libitum. Rats were anesthetized with chloral hydrate (350 mg/kg, i.p.; Tianjin Chemical Reagent Co., Ltd., Tianjin, China). A ventral midline incision was made, and the right common carotid artery (CCA) identified. The external carotid artery (ECA) and internal carotid artery (ICA) were carefully divided and isolated. A 50 mm length of monofilament nylon suture (Φ0.26 mm), with its tip embedded in paraffin, was inserted from the right CCA to the ICA through a small incision in the CCA and then advanced to approximately 18 mm from the carotid artery bifurcation until a faint resistance was felt.22 Sham‐operated rats experienced the same procedures except for suture insertion. All experiments and procedures were in accordance with the guidelines for the Care and Use of Laboratory Animals from the Ethics Committee of Shenyang Pharmaceutical University.
2.3. Measurement of cerebral infarct volume, cerebral edema, and neurological function
Rats were killed at 0 hour (sham‐operated rats, sham) or 24 hours post‐pMCAO, and their brains were quickly removed, weighed accurately, and then sliced into six coronal sections, each of 2 mm thickness. The slices were incubated in a 1% solution of TTC (Sigma, T8877) at 37°C for 20 minutes, and then photographic images were taken. Afterward the slices were dried at 100°C for 24 hours and then weighed accurately. The brain water content was determined as an indicator of cerebral edema using a wet/dry method as previously described.23 The cross‐sectional areas, with or without infarction, in each brain slice were measured using Image J analysis software (version 1.6, National Institutes of Health, Bethesda, MD, USA). The total mean infarct area of each section was examined by the change in coloration.24 Neurological function was evaluated with the modified Neurological Severity Score (mNSS) test.25 Each function is graded on a scale of 0‐18 (normal score, 0; maximal deficit score, 18). Higher scores indicate more severe behavioral deficits. The mNSS test was performed in the saline‐ and PF11‐treated rats at 24 hours after the pMCAO procedure.
2.4. Fluoro‐Jade B staining
Staining was performed according to the manufacturer's protocol (Histo‐Chem Inc., Jefferson, AR, 2FJB). Sections were dried on slides at 50°C for 30 minutes, immersed in a solution containing 1% sodium hydroxide in 80% alcohol, followed by 2 minutes in 70% alcohol and 2 minutes in distilled water. Slides were transferred to a solution of 0.06% potassium permanganate for 10 minutes on a shaker table and, then, washed in distilled water. After 20 minutes in Fluoro‐Jade B staining solution, the slides were rinsed for 1 minute in each of three distilled water washes and, then, placed on a slide warmer at 50°C in the dark until they were completely dry. Sections were cleared by immersion in xylene for 1 minute before cover slipping with DPX (Sigma, 317616).
2.5. Immunoblotting
At 24 hours after pMCAO, brain tissues from the ischemic cortex and the corresponding area of sham‐operated rats were rigorously homogenized in RIPA buffer. An aliquot of 25 μg‐30 μg of total protein from each sample was separated using 10%‐12% SDS‐polyacrylamide gel electrophoresis using constant current. Proteins were subsequently transferred to nitrocellulose membrane or polyvinylidene fluoride membrane, which were subsequently incubated with 5% skimmed milk. Afterward, the membranes were incubated with corresponding primary antibodies against β‐actin (1:1000; Santa Cruz Biotechnology, Dallas, TX, USA, sc‐47778), LC3 (1:1000; Medical & Biological Laboratories Co., Ltd., MBL, Nagoya, Japan, PM036), SQSTM1 (1:1000; abcam, Cambridge, UK, ab56416), ATG7 (1:1000; Cell Signaling Technology, Danvers, MA, USA, 2361S), LAMP2 (1:2000; Sigma, L0668), Ubiquitin (1:1000; CST, 3936S), HSP70 (1:500; Santa Cruz, sc‐32239), VAMP7 (1:1000; CST, 13920), V‐ATPase (1:500; Santa Cruz, sc‐28801), cathepsin B (1:500; Santa Cruz, sc‐6493), Cleaved Caspase‐3 (1:1000; CST, 9661), and Parp (1:1000; CST, 9542) at 4°C overnight, followed by horseradish peroxidase‐conjugated secondary antibody (1:5000, Beyotime, Nantong, China). Band patterns were analyzed with ImageJ software and normalized to the loading control.
2.6. Immunofluorescence
Rats were perfused with PBS (pH 7.4) followed by 4% paraformaldehyde (PFA) (vol/vol). Brains were postfixed in 4% PFA, immersed in 20% and 30% sucrose. For immunofluorescence, sections (20 μm) were washed in phosphate‐buffered saline (PBS) and in 10 mmol L−1 sodium citrate buffer. Afterward, the sections were blocked in 10% normal goat serum containing 0.3% Triton X‐100. Then they were incubated overnight at 4°C with primary antibody against LC3 (1:200; MBL, PM036), NeuN (1:500; Millipore, Kenilworth, NJ, USA, MAB377), Iba‐1 (1:500; Millipore, MABN92), GFAP (1:500, Sigma, G3893), SQSTM1 (1:200; abcam, ab56416), LAMP2 (1:200; Sigma, L0668), and cathepsin B (1:50; Santa Cruz, sc‐6493) in PBS. The slices were then incubated with fluorochrome‐coupled secondary antibody (1:200; Beyotime, Alexa Fluor 488, Alexa Fluor 555). The slices were incubated with DAPI (Sigma, D8417), and then, the confocal images were taken using a Nikon C2 Plus with a 60× objective. Fluorescence measurement was conducted with Nikon Instruments Software (NIS‐Elements Advanced Research). The integrated optical density was analyzed by Image‐Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD).
2.7. TUNEL staining
To identify the apoptosis rate in cells expressing LC3, double staining of LC3 and the terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) were performed. Immunofluorescence staining of LC3 was performed according to the above procedure. TUNEL staining was applied using an in situ Cell Death Detection Kit (Roche, Basel, Switzerland, 11684795910). Staining was performed according to the manufacturer's protocol.
2.8. Statistical analysis
Statistical analysis was carried out using SPSS 19.0 software for Windows (SPSS Inc, Chicago, IL, USA). Western blot and immunofluorescence results were analyzed with one‐way analysis of variance (ANOVA), followed by Tukey's test. Data are expressed as means±SEM. Infarct area and brain water content data are expressed as mean±SEM and were statistically analyzed using one‐way ANOVA followed by Bonferroni's post hoc test. mNSS scores are expressed as the mean±SEM and were analyzed with Kruskal‐Wallis one‐way ANOVA on ranks, followed by Dunn's method. P<.05 was considered statistically significant.
3. Results
3.1. PF11 reduced the infarct volume and brain water content, and improved the behavioral outcome
The results showed that one single treatment of PF11 (3, 6, 12, 24, 48 mg/kg, i.v) at 0.5 hour in rats challenged by ischemic insult significantly reduced the cerebral infarct volume. PF11 (6 and 12 mg/kg) significantly reduced the infarct size by 24.8% (23.06±1.95; P<.01) and 25.7% (22.78±1.26; P<.01), respectively, when compared with saline vehicle (30.66±0.97) (Figure 1A and B). Treatment with PF11 at 6 and 12 mg/kg also produced a significant reduction in brain water content by 0.86% (79.40±0.36; P<.01) and 1.73% (79.34±0.32; P<.01), respectively, when compared with saline vehicle (80.74±0.20) (Figure 1C). PF11 also significantly increased the neurological outcome compared with the saline group (Figure 1D). To determine the therapeutic time window, rats were administered with PF11 (12 mg/kg) at 0.5, 2, 4, 8 hours after the onset of ischemia. As illustrated in Figure 1E, PF11 significantly decreased the infarct volume up to 2 hours after pMCAO. The brain water content was also significantly reduced by PF11 even 4 hours following ischemia (Figure 1F). All these results indicated that one single injection of PF11 attenuated the ischemic damage to the brain and had a relatively wide therapeutic time window.
Figure 1.
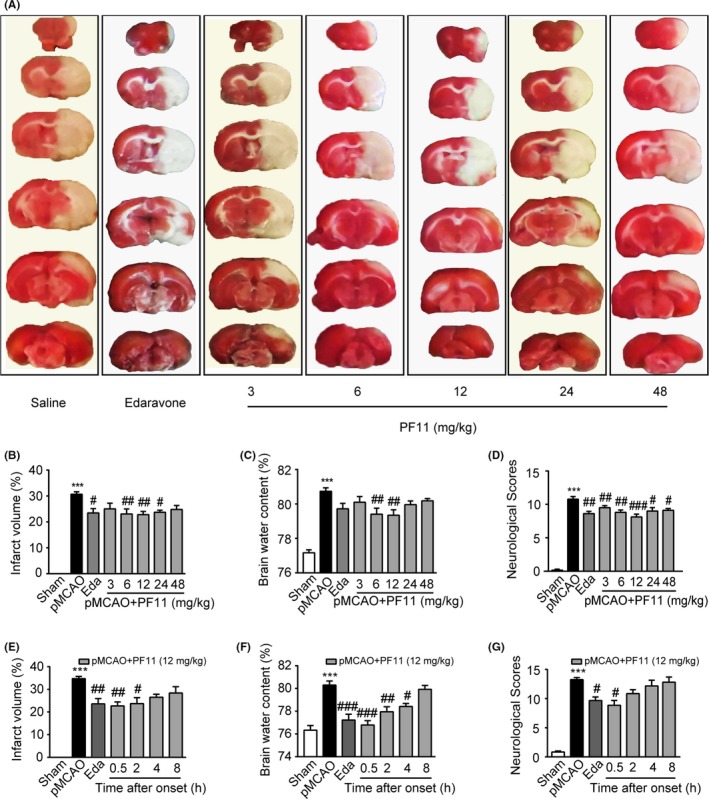
PF11 reduces infarct volume and brain water content and improves behavioral outcome after pMCAO. (A, B) Effect of PF11 on the cerebral infarct volume, examined by 2,3,5‐triphenyltetrazolium chloride staining of serial coronal brain sections at 24 h after pMCAO. PF11 (3, 6, 12, 24, 48 mg/kg) and edaravone (5 mg/kg) were administered (i.v.) at 0.5 h after the onset of pMCAO. The neuroprotective agent edaravone (Eda) was used as a positive control. Statistical analysis was performed with one‐way ANOVA, followed by the Bonferroni's test (the same statistical tests were used for C, E and F). Data are presented as mean±SEM from 8 to 10 rats/group (the same number in C and D). ***P<.001 vs sham group; ##P<.01, #P<.05 vs pMCAO group. (C) Brain water content measured at 24 h after pMCAO. ***P<.001 vs sham group; ##P<.01 vs pMCAO group. (D) Neurological scores of sham‐operated rats and pMCAO rats treated with PF11, edaravone, or saline. Statistical analysis was performed with Kruskal‐Wallis one‐way ANOVA on ranks, followed by Dunn's method (the same tests were used for G). ***P<.001 vs sham group; ##P<.01, #P<.05 vs pMCAO group. (E) Quantifications of the infarct volume 24 h after administration of PF11 (12 mg/kg) at 0.5 h, 2 h, 4 h, 8 h, and edaravone (5 mg/kg) at 0.5 h following pMCAO. Data are presented as mean±SEM from 8 to 10 rats/group (same number in F and G). ***P<.001 vs sham group; ##P<.01, #P<.05 vs pMCAO group. (F) Brain water content measured at 24 h after pMCAO. ***P<.001 vs sham group; ###P<.001, ##P<.01, #P<.05 vs pMCAO group. (G) Neurological scores measured at 24 h after pMCAO. ***P<.001 vs sham group; #P<.05 vs pMCAO group
3.2. PF11 rescued pMCAO‐induced neural cell death and glial cell activation
As shown in Figure 2A, in pMCAO‐treated rats, many neurons (identified by labeling with NeuN) were damaged as evidenced by irregularly aligned and shrunken somata, and this was accompanied by a marked loss of neurons compared to the sham group (95.71±4.11 vs 170.83±2.37; P<.01). PF11 (6, 12 mg/kg) rescued cell morphology and significantly reduced neuronal loss in a dose‐dependent manner (97.75±4.73, P<.001; 123.00±5.45, P<.001) in the corresponding area compared to the pMCAO group (95.71±4.11) (Figure 2A, E, and G). Immunofluorescence examination revealed prominent activation of microglia in the ipsilateral cortex 24 hours after ischemia, as shown by the changed morphology and Iba‐1‐positive staining,26 which was obviously attenuated by PF11 (Figure 2B). Compared with sham‐operated cortex, more GFAP‐reactive astrocytes were seen at 24 hours following pMCAO.27, 28 PF11 reversed the abnormal astrocyte activation induced by ischemic insult (Figure 2C). To measure the extent of neuronal degeneration in the pMCAO rats, neurons were stained with Fluoro‐Jade B (FJB). As shown in Figure 2D, few dying neurons were observed in sections from sham‐operated rats, while pMCAO induced a marked increase in neuronal death in the cortex region as evidenced by an obvious increase in the total number of FJB‐positive cells compared to the sham group. In contrast, PF11 (3, 6, 12 mg/kg) treatment resulted in a significant reduction in the number of FJB‐positive cells (102.00±2.86, P<.001; 84.17±2.50, P<.001; 77.33±3.45, P<.001) in the cortex region compared to the pMCAO group (133.88±3.54) (Figure 2F).
Figure 2.
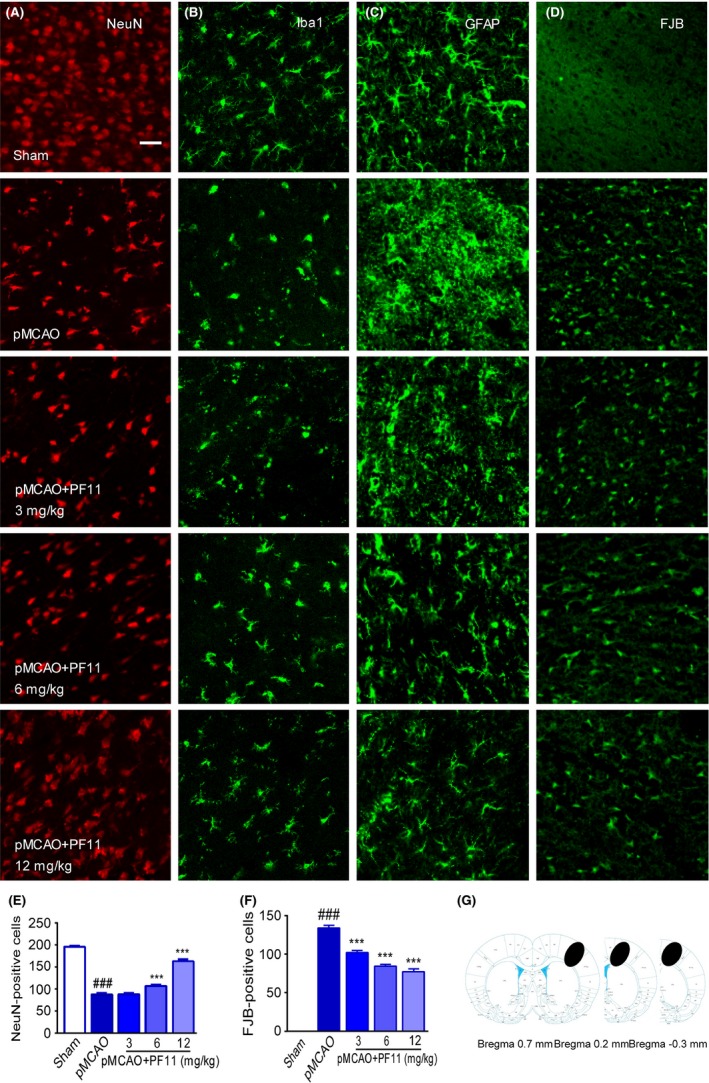
PF11 rescues pMCAO‐induced neural cell death and glial cell activation. Immunofluorescence images showing that PF11 significantly reversed the pMCAO‐induced neuron death (A), microglial activation (B), and astrocyte activation (C) in the ipsilateral cortex, as detected by laser confocal microscopy using antibodies against NeuN (red), Iba‐1 (green), and GFAP (green), respectively. Fluoro‐Jade B (FJB) staining was used to identify degenerating neuronal cells (D, green). Scale bar=20 μm. (E, F) Statistical analysis of surviving neurons (NeuN‐positive) and degenerating neurons (FJB‐positive) was conducted using Image‐Pro Plus 6.0. The values are presented as means±SEM (n=18). Statistical comparisons were carried out with ANOVA followed by Tukey's test. ###P<.001 vs sham group, ***P<.001 vs pMCAO group. pMCAO denotes rats subjected to pMCAO for 24 h; pMCAO+PF11 (3, 6, 12 mg/kg, iv) denotes rats that were injected with PF11 at 0.5 h after the onset of pMCAO. Rats were perfused with 4% paraformaldehyde, and 20 μm brain cortex sections were processed for immunofluorescence examination. (G) The black ovals indicate the regions selected for immunofluorescence detection
3.3. PF11 attenuated pMCAO‐induced accumulations of autophagosomes and apoptosis
LC3 and TUNEL costaining were performed on the histological sections from the pMCAO‐treated rat cortex (Figure 3). The right panel of Figure 3 shows cells that are positive for both TUNEL and LC3 staining, which indicate that apoptotic and autophagic pathways can occur in the same cells. In the pMCAO‐treated group, the majority of TUNEL‐positive cells also showed immunoreactivity for LC3 puncta, suggesting the concurrence of ischemic stroke‐induced cell death and autophagosome accumulation.5, 29 Further, PF11 at a dose of 12 mg/kg partly reduced the proportion of LC3/TUNEL‐positive cells, suggesting that PF11 increases the resistance of cells to ischemic stroke insult (Figure 3).
Figure 3.
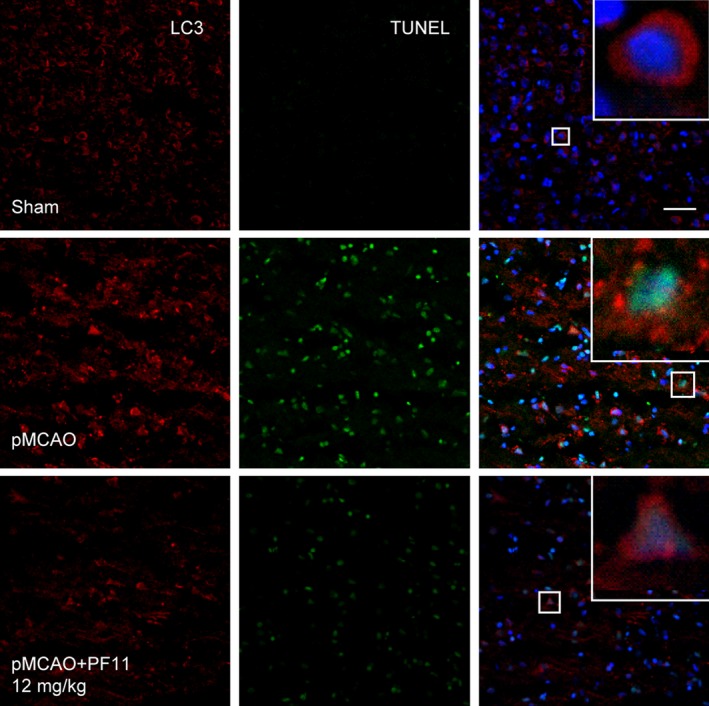
PF11 attenuates pMCAO‐induced accumulations of autophasosomes and apoptosis. Representative immunofluorescence staining of double‐labeled brain cortex sections showing LC3‐positive cells (red) and TUNEL‐positive cells (green) in sham‐operated rats and pMCAO rats treated with saline or PF11 (12 mg/kg) at 0.5 h after the onset of pMCAO. Nuclei are labeled with DAPI (blue). High‐magnification images of the boxed areas are shown in the inserts. Scale bar=20 μm
3.4. PF11 attenuated the pMCAO‐induced accumulations of autophagosomes and autophagic substrates
Although mounting evidence has suggested the existence of autophagy in ischemic stroke,7, 11, 12, 30 the functions of autophagy in this process remain unclear. To address this question, we conducted immunoblot analyses using a panel of autophagic pathway markers that allowed us to interrogate multiple steps in the autophagic pathway and the potential effect of PF11. As shown in Figure 4A and B, a dramatic enhancement in the ratio of LC3‐II/β‐actin was observed in the rat cortex region at 24 hours following pMCAO, which was in accordance with previous studies in multiple ischemic stroke models.7, 12, 21 Expression of SQSTM1, a well‐known autophagic substrate also named p62,31 was slightly but not significantly decreased by pMCAO (Figure 4A and C). This suggested an abnormal accumulation of protein substrate, in contrast to the majority of previous studies which showed that SQSTM1 is significantly decreased following ischemic stroke insult.12 Meanwhile, in response to ischemic insult, a considerable amount of insoluble cytoplasmic inclusions consisting of ubiquitinated proteins were detected (Figure 4A and E). The level of ATG7, which is involved in the stage of autophagosome formation, was increased following ischemic insult (Figure 4A and D). Next, we examined the effect of PF11 (3, 6, 12 mg/kg) on the pMCAO‐induced accumulation of autophagosomes. The results showed that PF11 significantly reduced the LC3‐II level (Figure 4A and B) and accelerated the digestion of the substrate SQSTM1 in a dose‐dependent manner (Figure 4A and C). The expression of ubiquitinated proteins was decreased by PF11 at a dose of 3 mg/kg (Figure 4A and E). The abundance of autophagosomes was also evaluated by immunofluorescence analysis of the colocalization of LC3 (red) and SQSTM1 (green) in the rat cortex (Figure 4F). The right panel of Figure 4F shows high‐magnification images which indicate that the accumulation of autophagosomes and the abnormal accumulation of protein substrates occurred in the same cells. Compared with the sham group, there was an obvious induction of LC3 and SQSTM1 puncta at 24 hours following pMCAO, which was reduced by PF11 treatment in a dose‐dependent manner. The accumulation of autophagosomes may be due to an increase in the formation of upstream autophagosomal precursors or impaired downstream autophagosome/lysosome fusion. To further investigate this, we analyzed the upstream mTOR signaling pathway, a well‐known regulator of autophagy.32 We used immunoblotting to examine the phosphorylation status of mTOR and its substrate, p70S6K. The data showed no significant difference in the p‐mTOR/t‐mTOR ratio or the p‐p70S6K/t‐p70S6K ratio 24 hours after pMCAO (Fig. S1A and 1B), suggesting that the accumulation of autophagosomes was independent of the mTOR/p70S6K pathway.
Figure 4.
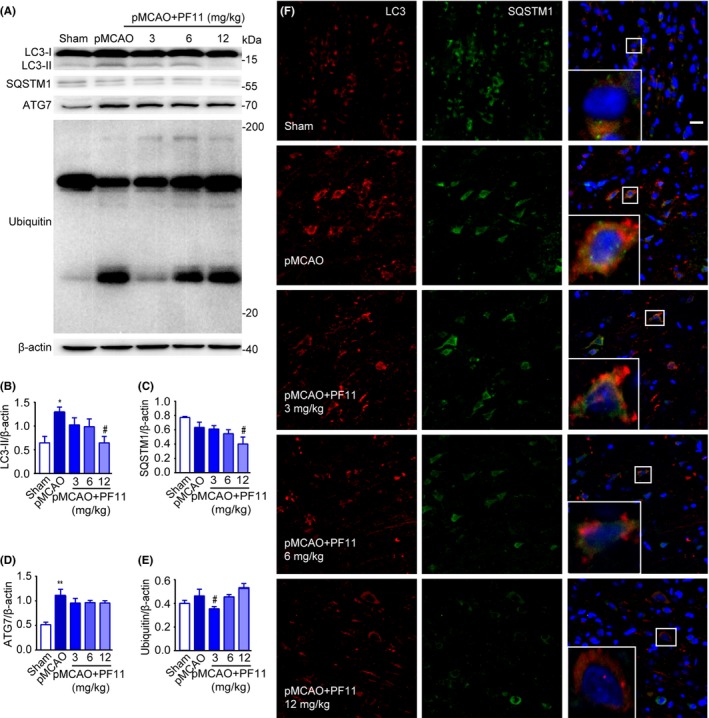
PF11 attenuates the pMCAO‐induced accumulations of autophagosomes and autophagic substrates. (A) Western blot analysis of LC3‐II, SQSTM1, ATG7, and Ubiquitin levels in cortex extracts 24 h after ischemic stroke. Rats were treated with PF11 (3, 6, 12 mg/kg) at 0.5 h after the onset of pMCAO. β‐actin was used as a loading control. (B‐E) Quantitative analysis of the immunoblotted proteins was performed with ImageJ. Statistical comparisons were carried out with ANOVA followed by Tukey's test. Data are presented as means±SEM. **P<.01, *P<.05 vs sham group; #P<.05 vs pMCAO group. (F) Immunofluorescence images showing double‐labeling of LC3‐positive vesicular “puncta” (red) and SQSTM1 (green) in the cortex of sham‐operated rats and of pMCAO rats 24 h after treatment with saline or PF11. Nuclei are labeled with DAPI (blue). High‐magnification images of the boxed areas are shown in the inserts. Bar=10 μm
3.5. PF11 attenuated pMCAO‐induced lysosome dysfunction and defective autophagosome/lysosome fusion
To further substantiate whether pMCAO induces autophagosome accumulation by increasing the formation or decreasing the degradation of autophagosomes, we investigated the function of lysosomes, which are responsible for the digestion of autophagic substrates. Western blot analysis of cortex extracts showed that expression of lysosome‐associated membrane protein‐2 (LAMP2) and the mature lysosomal protease cathepsin B was significantly decreased at 24 hours following pMCAO (Figure 5B‐D). We also assessed lysosomal function by using confocal microscopy to examine the colocalization of the lysosomal markers LAMP2 and cathepsin B (see high‐magnification images in the right panels of Figure 5A). Consistent with the immunoblotting results, immunofluorescence analysis of LAMP2 and cathepsin B in cortical cells revealed that the signals from both proteins were strongly reduced by pMCAO, which is suggestive of marked lysosomal dysfunction (Figure 5A). Moreover, the expression of V‐ATPase, a proton pump that establishes and maintains the acidic environment of lysosomes,33 was significantly decreased as determined by immunoblotting (Figure 5B and E). A significant decline in the level of VAMP7, a member of the SNARE complex which is responsible for the fusion of lysosomes with autophagosomes,34 was also observed, suggesting that the fusion process may be affected by ischemic insult (Figure 5B and F). All these data suggested that there was a selective impairment in lysosome function and autophagosome/lysosome fusion following pMCAO.
Figure 5.
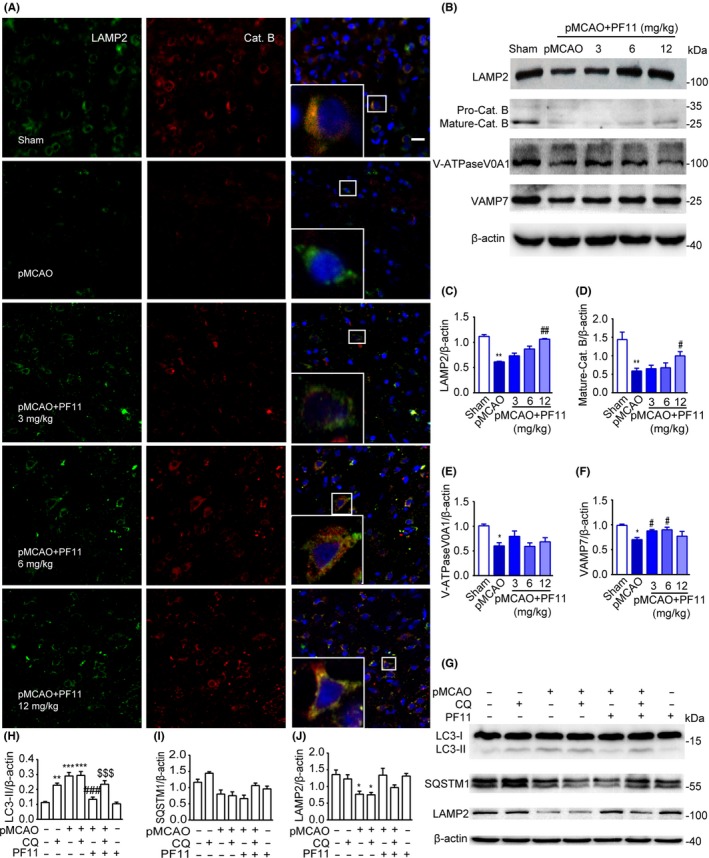
PF11 attenuates pMCAO‐induced lysosome dysfunction and promotes the fusion of lysosomes with autophagosomes. (A) Immunofluorescence studies showing the colocalization of LAMP2‐positive lysosomes (green) with the lysosomal enzyme CatB (red) in the cortex of sham‐operated rats and of pMCAO rats 24 h after treatment with saline or PF11 (3, 6, 12 mg/kg) at 0.5 h after the onset of pMCAO. Nuclei are stained with DAPI (blue). High‐magnification images of the boxed areas are shown in the inserts. Scale bar=10 μm. (B) Immunoblots showing the levels of LAMP2, mature‐CatB, V‐ATPase V0A1, and VAMP7 in brain cortex extracts from sham‐operated rats and of pMCAO rats 24 h after treatment with saline or PF11 (3, 6, 12 mg/kg) at 0.5 h after the onset of pMCAO. β‐actin was used as a loading control. (C‐F) Densitometric analysis of the immunoblots. Statistical comparisons were carried out with ANOVA followed by Tukey's test. Data are presented as means±SEM. **P<.01, *P<.05 vs sham group; ##P<.01, #P<.05 vs pMCAO group; n=4. (G‐J) Effects of CQ treatment on proteins in the autophagic‐lysosomal pathway. Brain cortex samples were obtained from sham‐operated rats with or without CQ, pMCAO‐operated rats with or without CQ, pMCAO+PF11‐treated rats with or without CQ, and rats treated with PF11 alone. Western blot analysis showing the levels of LC3, SQSTM1, and LAMP2 (G) and their ratio normalized to the β‐actin loading control (H‐J). CQ was administered 5 h before the rats were killed. Statistical comparisons were carried out with ANOVA followed by Tukey's test. Data are presented as means±SEM. ***P<.001, **P<.01, *P<.05 vs sham group; ###P<.001 vs pMCAO group; $$$P<.001 vs pMCAO+PF11 group; n=4
Next, the effect of PF11 (3, 6, 12 mg/kg) on lysosomal function was examined. The data showed that PF11 markedly increased the LAMP2 and V‐ATPase levels after the challenge by pMCAO. Similarly, confocal analysis showed that PF11 dramatically increased the expression and colocalization of LAMP2 and CatB in a dose‐dependent manner (Figure 5A). PF11 also partly reversed the ischemia‐induced decline in VAMP7, as judged by immunoblotting (Figure 5F). These data suggested that PF11 was capable of improving lysosomal function and lysosome/autophagosome fusion. To further clarify whether the effect of PF11 on reducing the abundance of autophagosomes could be attributed to improved lysosomal function, we assayed the levels of autophagic‐related proteins in animals treated with or without chloroquine (CQ), which inhibits lysosomal proteases or blocks downstream autophagosome/lysosome fusion.35 Immunoblots showed that an intracerebral ventricular injection of CQ (25 mg/kg) led to significant upregulation of LC3‐II as compared with the sham group (Figure 5G and H), suggesting intact autophagic flux. Ischemic insult caused a 2‐fold increase in autophagosomes as compared with the sham group, and this change was not altered in the presence of CQ, suggesting that autophagic flux was markedly impaired by pMCAO. Moreover, PF11 (12 mg/kg) significantly reduced the abundance of autophagosomes as compared with the ischemia group, and this decrease was reversed by CQ (Figure 5G and H). The variation in the levels of SQSTM1 and LAMP2 was shown in Figure 5G, I, and J. These results strongly indicated that treatment with a high concentration of PF11 can restore the autophagic flux by improving lysosomal function.
3.6. The protective effect of PF11 on ischemic stroke was abolished by chloroquine (CQ)
To further clarify whether the protective effect of PF11 on ischemic insult was dependent on alleviating the autophagic/lysosomal defects, we investigated the ischemic stroke outcome and the apoptosis‐related proteins in animals treated with or without CQ as described in the previous paragraph. Immunoblots showed that the apoptosis‐related proteins Cleaved Parp and Cleaved Caspase‐3, which are the key executioners of apoptosis, were significantly upregulated in the pMCAO group as compared to the sham group (Figure 6A‐C). PF11 (12 mg/kg) significantly reduced Cleaved Parp and Cleaved Caspase‐3 expression as compared to the pMCAO group, and this change was markedly reversed by CQ (25 mg/kg) (Figure 6A‐C). Also, the TTC staining, brain water content, and neurological scores indicated that the protective effect of PF11 on permanent ischemic insult was abolished by CQ (Figure 6D‐G). Additionally, confocal analysis showed that PF11 increased the expression of NeuN (Figure 6H) and decreased the colocalization of TUNEL and LC3 (Figure 6I) following ischemic insult, and these changes were reversed by CQ (right panels of Figure 6H and I). These results strongly indicated that the protective effect of PF11 against ischemic stroke may depend on alleviating autophagic/lysosomal defects.
Figure 6.
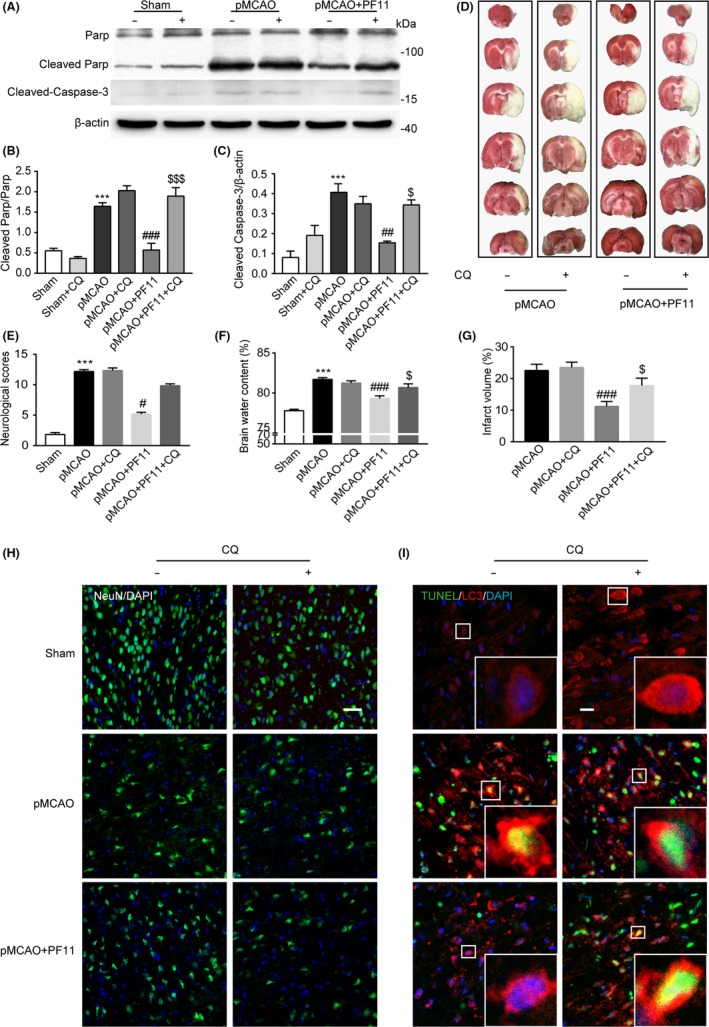
The protective effect of PF11 on ischemic stroke is abolished by CQ. (A‐C) Western blot analysis showing the expression of the apoptosis‐related proteins Cleaved Parp and Cleaved Caspase‐3 (A) and their ratio normalized to the loading control (B and C) in the sham‐operated rats with or without CQ, pMCAO‐operated rats with or without CQ, and pMCAO+PF11‐treated rats with or without CQ. Statistical comparisons were carried out with ANOVA followed by Tukey's test. Data are presented as means±SEM. ***P<.001 vs sham group; ###P<.001, ##P<.01 vs pMCAO group; $$$P<.001, $P<.05 vs pMCAO+PF11 group; n=4. (D and G) TTC staining showing the cerebral infarct volume in rats treated with PF11 with or without CQ. (E) Brain water content measured at 24 h following ischemic insult in rats treated with PF11 with or without CQ. (F) Neurological scores of sham‐operated rats and pMCAO rats treated with PF11 with or without CQ. Statistical comparisons were carried out with ANOVA followed by Tukey's test. Data are presented as means±SEM. ***P<.001 vs sham group; ###P<.001, #P<.05 vs pMCAO group; $P<.05 vs pMCAO+PF11 group; n=6. (H) Immunofluorescence images showing the NeuN immunoreactivity of cells in the cortex of sham‐operated rats and pMCAO rats treated with PF11 with or without of CQ. Scale bar=20 μm. (I) Immunofluorescence studies showing TUNEL‐positive nuclei (green) and LC3‐positive vesicular “puncta” (red) in cells in the cortex of sham‐operated and pMCAO‐operated rats treated with PF11 with or without CQ. High‐magnification images of the boxed areas are shown in the inserts. Scale bar=10 μm. PF11 (12 mg/kg, i.v.) was administered at 0.5 h after the onset of pMCAO. CQ (25 mg/, i.c.v.) was administered at 5 h before the animals were killed
4. Discussion
Our previous studies have demonstrated that PF11 exerts extensive protective effects when used to treat disorders of the central nervous system.14, 15, 16, 17, 18 The present study suggests that PF11 (3‐12 mg/kg) provides excellent protection against pMCAO‐mediated ischemic insult. However, the highest dose of PF11 (48 mg/kg) had no effect on infarct volume, but had a significant effect on neurological function. There is no strict parallel relationship between infarct volume and neurological scores.36 Although TTC staining allowed us to delineate clearly the boundaries of the necrotic tissues, this staining did not allow fine evaluation of the morphological status of the dying or intact neurons.36 Thus, it is possible that the high dose of PF11 (48 mg/kg) had a protective effect on dying or intact neurons, leading to a significant improvement in neurological function, but had little effect on necrotic neurons, so that no significant changes were found in infarct volume. Furthermore, it is particularly noteworthy that PF11 (12 mg/kg) exhibited significant neuroprotection. In addition, these findings indicate that the autophagic flux is impaired in a rat model of pMCAO. Our findings suggest a potential therapeutic role for PF11.
It is well known that apoptosis and necrosis contribute to the development of ischemia‐induced delayed cell death.37 In recent years, it has been shown that autophagic cell death, which is distinct from apoptosis and is also called type II programmed cell death,38 is involved in cerebral ischemia‐mediated cell death.29, 39, 40 Our data also showed that dying cells displayed large‐scale accumulation of autophagosomes following ischemic insult, which was reversed by PF11 at a dose of 12 mg/kg. These results support the idea that PF11 protects against cell death and has a potential relationship with autophagy.
Mounting evidence has suggested that the autophagic flux is stimulated, and autophagosomes accumulate in several models of ischemic stroke.7, 12 In the present study, the level of LC3‐II also strongly increased following pMCAO. Interestingly, in our study, the levels of the autophagic substrate SQSTM1 did not significantly decrease 24 hours after ischemic insult, a result which differed from other experimental stroke models.8, 12 However, in the latest study, using human postmortem tissue, increased expression of SQSTM1 was observed, suggesting an accumulation of autophagic substrates after human stroke.30 This increased level of SQSTM1 may be indicative of inhibition of lysosomal function. Furthermore, the increased oxidative stress that is known to occur in stroke is also associated with increased levels of SQSTM1.41 In addition, caspase‐8, which is activated after cerebral ischemic insult, recruits SQSTM1 to further regulate caspase‐8 activation.42 In a recent study, it was reported that self‐oligomerization of SQSTM1 is responsible for its localization to the autophagosome formation site, which is associated with the endoplasmic reticulum (ER), and this process occurs independently of LC3.43 Thus, there is not necessarily an inverse correlation between LC3 and SQSTM1. However, the detection of SQSTM1 levels alone is not sufficient to confirm autophagic removal, and for this reason, the level of polyubiquitinated protein aggregates has often been determined to confirm the efficiency of autophagic clearance in multiple diseases.44 Our data showed that ubiquitin levels tended to increase following ischemic insult, suggesting a low rate of autophagic clearance. Furthermore, PF11 reversed the accumulation of autophagic substrates, suggesting a high efficiency of autophagic clearance.
Because autophagosomes fuse with and are then degraded by lysosomes, the accumulation of autophagosomes or LC3‐II protein after brain ischemia can be due to either increased formation or decreased lysosomal degradation of autophagosomes. Therefore, we examined the steps involved in both the formation and degradation of autophagosomes following ischemic stroke. Our data showed that no significant change was detected in the expression of p‐mTOR and p‐p70S6K as compared to the sham group (Fig. S1A‐C), suggesting that the accumulation of autophagosomes was independent of the mTOR/p70S6K pathway. Next, we investigated whether lysosomal dysfunction and defective autophagosome/lysosome fusion could be detected following ischemic insult. Up to now, the function of lysosomes following ischemic stroke is still an area of widespread controversy. Although in some studies lysosomes and lysosomal hydrolases are activated to remove autophagic aggregates after ischemic stroke,7 it is noteworthy that ischemic injury can trigger cytosolic acidification and rupture/permeabilization of lysosomes, thereby leading to cathepsin‐mediated cell death.45, 46 We observed that PF11 had remarkable effects in reversing pMCAO‐mediated lysosomal dysfunction. Thus, we supposed that PF11 reduced the accumulation of autophagosomes and autophagic substrates by improving lysosomal function and accelerating autophagosome/lysosome fusion. To further confirm this hypothesis, a lysosomal inhibitor CQ was used to test the reason of accumulation of autophagosomes after brain ischemia, as judged by assaying the level of the autophagosomal marker protein LC3‐II.35 The results showed that the influence of PF11 on the abundance of autophagosomes and lysosomal dysfunction induced by pMCAO could be reduced by CQ to a level similar to that in the pMCAO group. Thus, PF11 had a relatively selective effect in reversing the pMCAO‐driven accumulation of autophagic vesicles by improving lysosomal function without affecting the normal baseline autophagic pathway proteins in the sham group.
Lysosomal dysfunction has been viewed as a pathogenic factor in several neurodegenerative diseases47, 48 and myocardial ischemia49 and is also a potential therapeutic target. It is now accepted that the magnitude of lysosomal rupture may influence the rate of cell death.50 However, the detailed mechanisms underlying lysosomal destabilization still remain poorly understood. Heat‐shock protein‐70 (HSP70) is known to stabilize lysosomal membranes and protect cells from oxidative stress and apoptotic stimuli.51 Interestingly, in the present study, the expression of HSP70 was significantly decreased after ischemic stroke, and PF11 could reverse this effect (Fig. S2A‐B), suggesting that the favorable effect of PF11 on lysosomes might be related to enhancement of the HSP70 protein level.
In conclusion, we have shown that the autophagic flux is impaired, and PF11 exerts an excellent protective effect by alleviating autophagic/lysosomal defects in a rat ischemic stroke model.
Conflict of Interest
The authors have no disclosure of conflict of interests related to this investigation.
Supporting information
Acknowledgments
The Project is sponsored by Liaoning BaiQianWan Talents Program (2014921036) and National Basic Scientific Personnel Training Funds (J1103606).
Liu Y‐Y, Zhang T‐Y, Xue X, et al. Pseudoginsenoside‐F11 attenuates cerebral ischemic injury by alleviating autophagic/lysosomal defects. CNS Neurosci Ther. 2017;23:567–579. 10.1111/cns.12702
Contributor Information
Chun‐Fu Wu, Email: wucf@syphu.edu.cn.
Jing‐Yu Yang, Email: yangjingyu2006@gmail.com.
References
- 1. Majid A. Neuroprotection in stroke: past, present, and future. ISRN Neurol. 2014;2014:515716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reed SD, Cramer SC, Blough DK, Meyer K, Jarvik JG. Treatment with tissue plasminogen activator and inpatient mortality rates for patients with ischemic stroke treated in community hospitals. Stroke. 2001;32:1832‐1840. [DOI] [PubMed] [Google Scholar]
- 3. Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann Neurol. 2007;61:396‐402. [DOI] [PubMed] [Google Scholar]
- 4. Adhami F. The Roles of Autophagy in Cerebral Ischemia. Autophagy. 2007;3:42‐44. [DOI] [PubMed] [Google Scholar]
- 5. Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia‐ischemia induced brain injury. Neurobiol Dis. 2008;32:329‐339. [DOI] [PubMed] [Google Scholar]
- 6. Wang JY, Xia Q, Chu KT, et al. Severe global cerebral ischemia‐induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3‐methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011;70:314‐322. [DOI] [PubMed] [Google Scholar]
- 7. Wen YD, Sheng R, Zhang LS, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762‐769. [DOI] [PubMed] [Google Scholar]
- 8. Viscomi MT, D'Amelio M, Cavallucci V, et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2014;8:222‐235. [DOI] [PubMed] [Google Scholar]
- 9. Buckley KM, Hess DL, Sazonova IY, et al. Rapamycin up‐regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Q, Zhang T, Wang J, et al. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun. 2014;444:182‐188. [DOI] [PubMed] [Google Scholar]
- 11. Xing S, Zhang Y, Li J, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012;8:63‐76. [DOI] [PubMed] [Google Scholar]
- 12. Zhang X, Yan H, Yuan Y, et al. Cerebral ischemia‐reperfusion‐induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2014;9:1321‐1333. [DOI] [PubMed] [Google Scholar]
- 13. Wang ZJ, Sun L, Peng W, et al. Ginseng derivative ocotillol enhances neuronal activity through increased glutamate release: a possible mechanism underlying increased spontaneous locomotor activity of mice. Neuroscience. 2011;195:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang CM, Liu MY, Wang F, et al. Anti‐amnesic effect of pseudoginsenoside‐F11 in two mouse models of Alzheimer's disease. Pharmacol Biochem Behav. 2013;106:57‐67. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Wang C, Wang J, et al. Pseudoginsenoside‐F11 (PF11) exerts anti‐neuroinflammatory effects on LPS‐activated microglial cells by inhibiting TLR4‐mediated TAK1/IKK/NF‐kappaB, MAPKs and Akt signaling pathways. Neuropharmacology. 2014;79:642‐656. [DOI] [PubMed] [Google Scholar]
- 16. Wang JY, Yang JY, Wang F, et al. Neuroprotective effect of pseudoginsenoside‐f11 on a rat model of Parkinson's disease induced by 6‐hydroxydopamine. Evid Based Complement Alternat Med. 2013;2013:152798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hao Y, Yang JY, Wu CF, Wu MF. Pseudoginsenoside‐F11 decreases morphine‐induced behavioral sensitization and extracellular glutamate levels in the medial prefrontal cortex in mice. Pharmacol Biochem Behav. 2007;86:660‐666. [DOI] [PubMed] [Google Scholar]
- 18. Li Z, Wu CF, Pei G, Guo YY, Li X. Antagonistic effect of pseudoginsenoside‐F11 on the behavioral actions of morphine in mice. Pharmacol Biochem Behav. 2000;66:595‐601. [DOI] [PubMed] [Google Scholar]
- 19. Besso H. Further Studies on Darnmarane‐saponins of American ginseng, roots of Panax quinquefblium L. Chem Pharm Bull. 1982;30:4534‐4538. [Google Scholar]
- 20. Amemiya S, Kamiya T, Nito C, et al. Anti‐apoptotic and neuroprotective effects of edaravone following transient focal ischemia in rats. Eur J Pharmacol. 2005;516:125‐130. [DOI] [PubMed] [Google Scholar]
- 21. Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84‐91. [DOI] [PubMed] [Google Scholar]
- 23. Vink R, Young A, Bennett CJ, et al. Neuropeptide release influences brain edema formation after diffuse traumatic brain injury. Acta Neurochir Suppl. 2003;86:257‐260. [DOI] [PubMed] [Google Scholar]
- 24. Jiang W, Zhang S, Fu F, Zhu H, Hou J. Inhibition of nuclear factor‐kappaB by 6‐O‐acetyl shanzhiside methyl ester protects brain against injury in a rat model of ischemia and reperfusion. J Neuroinflammation. 2010;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005‐1011. [DOI] [PubMed] [Google Scholar]
- 26. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312‐318. [DOI] [PubMed] [Google Scholar]
- 27. Petito CK, Halaby IA. Relationship between ischemia and ischemic neuronal necrosis to astrocyte expression of glial fibrillary acidic protein. Int J Dev Neurosci. 1993;11:239‐247. [DOI] [PubMed] [Google Scholar]
- 28. Schmidt‐Kastner R, Wietasch K, Weigel H, Eysel UT. Immunohistochemical staining for glial fibrillary acidic protein (GFAP) after deafferentation or ischemic infarction in rat visual system: features of reactive and damaged astrocytes. Int J Dev Neurosci. 1993;11:157‐174. [DOI] [PubMed] [Google Scholar]
- 29. Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy‐like cell death. Neurobiol Dis. 2008;29:132‐141. [DOI] [PubMed] [Google Scholar]
- 30. Frugier T, Taylor JM, McLean C, et al. Evidence for the recruitment of autophagic vesicles in human brain after stroke. Neurochem Int. 2016;96:62‐68. [DOI] [PubMed] [Google Scholar]
- 31. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131‐24145. [DOI] [PubMed] [Google Scholar]
- 32. Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121‐129. [DOI] [PubMed] [Google Scholar]
- 33. Marshansky V, Futai M. The V‐type H+‐ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI‐VAMP/VAMP7 and VAMP3/cellubrevin: two v‐SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901‐1916. [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wahl F, Allix M, Plotkine M, Boulu RG. Neurological and behavioral outcomes of focal cerebral ischemia in rats. Stroke. 1992;23:267‐272. [DOI] [PubMed] [Google Scholar]
- 37. Pandya RS, Mao L, Zhou H, et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11:81‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sheng R, Liu XQ, Zhang LS, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310‐325. [DOI] [PubMed] [Google Scholar]
- 40. Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012;18:250‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nezis IP, Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid Redox Signal. 2012;17:786‐793. [DOI] [PubMed] [Google Scholar]
- 42. Jin Z, Li Y, Pitti R, et al. Cullin3‐based polyubiquitination and p62‐dependent aggregation of caspase‐8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721‐735. [DOI] [PubMed] [Google Scholar]
- 43. Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self‐oligomerization but not LC3 binding. J Cell Biol. 2011;192:17‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273‐295. [DOI] [PubMed] [Google Scholar]
- 46. Yamashima T, Tonchev AB, Tsukada T, et al. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791‐800. [DOI] [PubMed] [Google Scholar]
- 47. Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8:108‐117. [DOI] [PubMed] [Google Scholar]
- 48. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170‐3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kilinc M, Gursoy‐Ozdemir Y, Gurer G, et al. Lysosomal rupture, necroapoptotic interactions and potential crosstalk between cysteine proteases in neurons shortly after focal ischemia. Neurobiol Dis. 2010;40:293‐302. [DOI] [PubMed] [Google Scholar]
- 51. Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann‐Pick disease‐associated lysosomal pathology. Nature. 2010;463:549‐553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials