

Summary
Aims
To investigate the roles of Claudin‐5 (CLDN5) in regulating the permeability of the blood‐brain barrier (BBB) during lung cancer brain metastasis.
Results
By silencing and overexpressing the CLDN5 gene in human brain vascular endothelial (hCMEC/D3) cells, we demonstrated the attenuation of cell migration ability and CLDN5's significant positive role in cell proliferation in CLDN5‐overexpressing hCMEC/D3 cells and observed the opposite result in the CLDN5 knockdown group. The reinforced CLDN5 expression reduced the paracellular permeability of hCMEC/D3 cells and decreased the invasion of lung adenocarcinoma A549 cells. Overall, 1685 genes were found to be differentially expressed between the CLDN5‐overexpressing cells and the control cells using the Affymetrix Human Transcriptome Array 2.0 (HTA 2.0), and the function of these genes was determined by Gene Ontology and pathway analyses. The possible biological functions of the 1685 genes include cell proliferation, adhesion molecules, and the Jak‐STAT, PI3K‐Akt, Wnt, and Notch signaling pathways. The identified sets of mRNAs that were specific to CLDN5‐overexpressing hCMEC/D3 cells were verified by a qRT‐PCR experiment.
Conclusion
CLDN5 regulates the permeability of BBB by regulating the proliferation, migration, and permeability of hCMEC/D3 cells, especially through the cell adhesion molecule signaling pathway, to enhance the function of the tight junctions, which was involved in reducing the formation of lung cancer brain metastasis.
Keywords: blood‐brain barrier, brain metastasis, cell adhesion molecules signaling, claudin‐5, permeability
1. INTRODUCTION
Metastasis is the presence of disease at distant sites due to the spread of cancer cells, which results in an overwhelming mortality in patients with cancer and accounts for almost 90% of all cancer‐related deaths.1 Major solid cancers, such as lung and breast cancers, produce a high incidence of central nervous system (CNS) metastases.2, 3 Brain metastases (BM) has reached an epidemic prevalence in the elderly and remains the worldwide leading cause of the death,4 outnumbering primary gliomas by 10:1.5 Thus, the underlying mechanisms in the process of brain metastasis need to be explored.
The human blood‐brain barrier (BBB) is formed by brain endothelial cells (ECs) and is an important mechanism for protecting the brain from fluctuations in plasma composition, neurotransmitters, and xenobiotics that are capable of disturbing neural function.6 The barrier also plays an important role in the homeostatic regulation of the brain microenvironment necessary for the stable and coordinated activity of neurons. Its poor permeability and penetrability to certain materials, especially BM, make the CNS “The Last Continent.” In vivo, the barrier is formed by specialized brain ECs that have continuous tight junctions (TJs), low pinocytotic activity, and a set of enzymes and ABC transporters that metabolize or exclude many xenobiotics.7, 8 Many studies have demonstrated that CLDNs are the major proteins involved in the TJs,9, 10 which are essential for maintaining the BBB structure. It was also reported that the members of this superfamily are involved not only in the barrier function of the ECs but also in cellular growth and the epithelial‐mesenchymal transition.11, 12
CLDN5 is an EC‐specific member of the CLDN family that is indispensable for the correct organization of TJs and maintenance of brain microvascular ECs integrity13 and is considered to contribute to the “sealing” of the TJs in modulating BBB permeability.14 CLDN5 closes the intercellular cleft by the interaction of its extracellular loops with the corresponding loops of CLDN5 of the adjacent EC. Recent literature has begun to highlight a potential role for CLDN5 in a number of disease pathologies, including Alzheimer's disease, edema, hypoglycemia, inflammation, toxic damage, trauma, and tumors.13, 15, 16, 17 Some studies have also revealed that a major role for CLDN5 is to selectively increase permeability to ions and macromolecules.18 In addition, CLDN‐5‐deficient mice displayed a higher permeability selective for small molecules (<800 Da).19 We also discovered that if a tumor was poised to permeate the BBB, there might be some changes in the expression of CLDN5.9 To elucidate the underlying mechanism of CLDN5 in regulating BBB permeability and the potential role of CLDN5 in cell proliferation and migration, we established an in vitro model using an immortalized human cerebral microvascular EC line (hCMEC/D3) to evaluate the function of CLDN5. These cells, which are widely used as an in vitro model of the human brain endothelium,20 were isolated from microvessel fragments of the temporal lobe of an adult with epilepsy and then used to coexpress human telomerase reverse transcriptase and simian vacuolating virus 40 (hTERT/SV40) large T antigen via a lentiviral vector transduction system.21 Cells with a passage number of 27‐29 were used as a representative model for the human BBB. This was done to mimic the clinical process of BM from lung cancer through the BBB. Because BM represents a great therapeutic challenge, it is indispensable to understand the mechanisms of CLDN5 in order to find targets to prevent brain metastasis formation. We also provided evidence that gene expression was changed as a result of CLDN5 overexpression. The correlated pathways and functions of CLDN5 in cell proliferation, migration, and hCMEC/D3 cell monolayer permeability were also investigated.
2. MATERIALS AND METHODS
2.1. Cell culture
hCMEC/D3 cells were cultured in EBM‐2 (Lonza, Allendale, NJ, USA) medium supplemented with the EGMTM‐2 BulletKit™ (Lonza, Allendale, NJ, USA). A549, a human lung adenocarcinoma cell line used in this study due to its prevalence in BM, was obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were cultured in 5α‐McCoy's medium (Sigma‐Aldrich Co., St Louis, MO, USA). The cultures were maintained in a humidified atmosphere (5% CO2/95% air) at 37°C, and the medium was refreshed every 2‐3 days. The medium was supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (15140‐122; Life Technologies, Carlsbad, CA, USA), and 10% fetal bovine serum (FBS; ThermoFisher Scientific, Waltham, MA, USA). The seeding density was 80 000 cells/cm2. Cells in the logarithmic growth phase were used to perform the experiments described in the following section or were subcultured. The ratio of the cell passage was 1:3‐4 after reaching confluence. The cells were washed with phosphate buffered saline (PBS; HyClone Thermo Fisher, Pittsburgh, PA, USA), detached using 0.5% trypsin (Gibco, Thermo Fisher, Pittsburgh, PA, USA), centrifuged at 130 g for 5 minutes, and seeded into new T25 flasks.
2.2. Gene knockdown and overexpression
Silencing of CLDN5 was achieved by applying small interfering RNA (siRNA); the siRNAs and their controls were synthesized by Gene Pharma (Suzhou, China). The target sequences are listed in Table 1. The hCMEC/D3 cells were seeded in six‐well culture plates. When the cells reached 50% confluency, the cells were transfected with 3.75 μL of siRNA, 7.5 μL of RNAiMAX (Invitrogen, Carlsbad, CA, USA), and 188.75 μL of serum‐free Opti‐MEM medium (Invitrogen) without antibiotics, according to the manufacturer's instructions. After incubation for 6 hours, the medium was replaced with the standard culture medium as previously described. After an additional 18‐hour incubation, the cells were used for the experiments described in the following section. The siRNA silencing experiment was repeated at least three times.
Table 1.
Primers used in this study
| Gene | Primer sequence |
|---|---|
| CLDN5 | |
| Sense | 5ʹ‐CTGGCGTTCGTTGCGCTCTT‐3ʹ |
| Antisense | 5ʹ‐GGGCACAGACGGGTCGTAAAA‐3ʹ |
| CLDN5(CDS) | |
| Sense | 5ʹ‐TAAGAATTCATGACCCGCGCACGGATTGG‐3ʹ |
| Antisense | 5ʹ‐GCCCTCGAGTCAGACGTAGTTCTTCTTGTCG‐3ʹ |
| GAPDH | |
| Sense | 5ʹ‐ATGATTCTACCCACGGCAAG‐3ʹ |
| Antisense | 5ʹ‐CTGGAAGATGGTGATGGGTT‐3ʹ |
| siCLDN5‐1 | |
| Sense | 5ʹ‐CCUUAACAGACGGAAUGAATT‐3ʹ |
| Antisense | 5ʹ‐UUCAUUCCGUCUGUUAAGGTT‐3ʹ |
| siCLDN5‐2 | |
| Sense | 5ʹ‐CUGCUGGUUCGCCAACAUUTT‐3ʹ |
| Antisense | 5ʹ‐UUCAUUCCGUCUGUUAAGGTT‐3ʹ |
| GAPDH(siRNA) | |
| Sense | 5ʹ‐UGACCUCAACUACAUGGUUTT‐3ʹ |
| Antisense | 5ʹ‐AACCAUGUAGUUGAGGUCATT‐3ʹ |
| Negative control | |
| Sense | 5ʹ‐UGACCUCAACUACAUGGUUTT‐3ʹ |
| Antisense | 5ʹ‐ACGUGACACGUUCGGAGAATT‐3ʹ |
For the overexpression of CLDN5, the full‐length cDNA of CLDN5 was designed (Origene Technologies, Montgomery, MD, USA) according to the coding sequence (CDS). The forward and reverse primers were annealed and cloned into the pLL3.7 vector through HpaI and XhoI sites. The plasmids were acquired after amplification and sequencing verification. Lentiviruses were generated by cotransfecting pLL3.7‐CLDN5 and the packaging vector (Plvx‐IRES‐GFP) into 293T cells using Lipofectamine 3000 Reagent (Invitrogen) according to the manufacturer's instructions. At 48 hours after transfection, the culture medium containing the viruses was filtered through a 0.45 mm filter and then used to transduce cells. The infection rate was evaluated through the expression of GFP after incubation with virus for 2 days. Green fluorescence could be observed under a fluorescence microscope in almost all of the cells transfected after cell sorting by a flow cytometer (FCM). Knockdown and overexpression of CLDN5 were confirmed by quantitative real‐time polymerase chain reaction (qRT‐PCR).
2.3. RT‐polymerase chain reaction and quantitative real‐time PCR
Total RNA of the cells was extracted using Trizol reagent (Invitrogen) following the manufacturer's instructions and dissolved in RNase‐Free H2O. The RNA concentrations were determined by an ultraviolet spectrophotometer (Biospec nano, SHIMADZU, Tokyo, JPN). Subsequently, reverse transcription to cDNA in a 20 μL reaction was performed with 1 μg total RNA using the PrimeScript RT Reagent Kit with gDNA Eraser from TAKARA (Shiga, JPN) according to the manufacturer's protocol. The mRNA expression was analyzed by qRT‐PCR, with the primer sequences shown in Table 1. A SYBR Premix Ex Taq II kit (TAKARA, Shiga, JPN) was used for gene amplification by a qRT‐PCR in the TAKARA system. The primers were designed by Primer5 software (Premier Biosoft, Palo Alto, CA, USA) on exon junctions to prevent co‐amplification of the genomic DNA. The qRT‐PCR assays were performed in triplicate, and the mean values were used to calculate mRNA expression. Gene expression was normalized to GAPDH mRNA. The results were calculated and are presented as 2−∆∆CT. The amplification efficiency of all primers in our study was tested with a rate of close to 100% (95%‐105%), so we use the delta delta Ct method of quantitation in qPCR experiments.
2.4. Western blot analysis of hCMEC/D3 lysates
For CLDN5 analyses, hCMEC/D3 cells were grown to confluence and then harvested with a Minute™ Plasma Membrane Protein Isolation Kit (Invent, Plymouth, MN, USA). For all other protein analyses, radio‐immunoprecipitation assay (RIPA) lysis buffer (ThermoFisher Scientific) with complete protease and phosphatase inhibitors (Roche, Basel, Switzerland) was used. The protein concentration of each sample was quantified using the bicinchoninic acid assay and then normalized; the samples were denatured at 95°C for 5 minutes, and then, the denatured protein (50 μg) was run in a 12% acrylamide gel for electrophoretic separation, followed by electrotransfer to a membrane. The nonspecific binding sites were blocked by incubating the membranes for 1 hour at 37°C with 5% nonfat dried milk in PBS containing 0.05% Tween‐20 (PBST). Following repeated washes (three times for 10 minutes each) with PBS containing 0.1% Tween‐20, the PVDF membranes were blocked and the blots were incubated overnight at 4°C with the primary antibodies (rabbit anti‐CLDN5 and anti‐β‐actin) (1:1000; Abcam, Cambridge, UK) in cold blocking buffer (1:1000). After three successive washes, the membranes were incubated with a HRP‐conjugated goat antirabbit secondary antibody (1:2000; Abcam). The bands were visualized using an enhanced chemiluminescence system (ECL; Pierce, Rockford, IL, USA). The band intensities were quantified by densitometry using the ImageJ (National Institutes of Health, Bethesda, MD, USA) software.
2.5. Cell proliferation assay
The hCMEC/D3 cell proliferation in different groups was analyzed using 5‐bromo‐2′‐deoxy‐uridine (BrdU) (BD Pharmingen, San Diego, CA, USA) incorporated into the newly synthesized DNA of replicating cells under growing conditions. In brief, the cells were cultured in a 6‐well plate with normal medium as indicated until reaching 60% confluency and were not treated with RNase or antibody until they were labeled with BrdU for 12 hours according to the manufacturer's instructions. The cells were then labeled with APC anti‐BrdU and 7‐AAD before the single cell suspension was taken to the FCM for cell proliferation and cell cycle analyses. Cell cycle distribution was determined by analyzing 10 000‐20 000 cells using a FACScan FCM and Flowjo software (FLOWJO, Ashland, OR, USA), in which positive BrdU‐labeled cells were detected. The percentage of proliferation cells was determined by the S phase and the G0/G1 phase in the DNA histogram. Each assay was repeated at least 3 times.
2.6. Cell migration assay
The 1600R model of the electric cell impedance sensing (ECIS) instrument (Applied Biophysics Inc, NJ, USA) was used for the migration assay. The 8W10E arrays (8 well format with 10 probes in each well) were used in this study. The array surface was treated with 200 μL of 10 mmol L−1 L‐cysteine solution for 20 minutes, which binds to the gold surface via its thiol group forming a monomolecular layer, followed by two washes in EBM2 medium. An electrode check was run to check the impedance value of the cell‐free wells containing only fresh medium and to assess the integrity of the arrays. The arrays were seeded at a density of 100 000 cells in 400 μL of EBM2 medium. The confluence of the cell monolayer was confirmed by a light microscope, and an electrode check was run to check the impedance value of the cells, as they were seeded on the chamber to document the adhesive capacity and contacts of the cells. When impedance reached a plateau, the cell monolayer was electrically wounded with a 5 V AC at 4000 Hz for 45 seconds. The impedance and resistance of the cell layer were immediately recorded every millisecond for a period of up to 24 hours. The data curve rises as the monolayer of cells reaches confluency, when the curve reaches a plateau, which indicates the formation of a cell monolayer and the healing of the wound, from which the data on the cells’ time‐of‐healing can be obtained.
2.7. Wound healing assay
For the wound healing assay, 5 types (PLL3.7, PLL3.7‐CLDN5, siCLDN5, NC, and WT) of hCMEC/D3 cells were seeded into 6‐well plates (500 000 cells in each well) and cultured to confluency. A P200 pipette tip was used to make three horizontal straight line simulation “wounds.” Subsequently, the wounded monolayers were washed several times with PBS to remove nonadherent cells, after which serum‐free medium was added to the wells. The extent of wound closure was monitored under the microscope. The cells were photographed 24 hours after wounding with a camera attached to a microscope at a 10× magnification. The distances migrated under the same conditions were analyzed. Each condition was evaluated in triplicate, and five different images were processed for each different assay; the mean distance migrated in both groups were measured in ImageJ software.
2.8. Cell invasion assay
The effect of the barrier function of the ECs treated in different ways was performed using Transwell tissue culture inserts (8 μmol L−1 pore size) (Corning Inc., Coring, NY, USA), with 1 × 105 hCMEC/D3 cells cultured on transwell membrane inserts to confluency at 37°C. Basal medium and FBS medium (20%) were added to the lower wells as a chemoattractant; then, the chamber was placed into a 24‐well plate and incubated at 37°C with 5% CO2. After reaching confluency, the cell suspension of the lung adenocarcinoma cell line A549, labeled with CM‐DIL (red; Invitrogen), was seeded onto the hCMEC/D3 monolayer; after incubation for 48 hours, the noninvading cells in the upper chamber were removed, and the invading cells were fixed with 4% paraformaldehyde for 20 minutes and stained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) (Sigma‐Aldrich, Steinheim, Germany) for 15 minutes. The samples were collected from both the upper (luminal) and the lower (abluminal) chambers for analysis. The number of stained cells on the undersurface of the polycarbonate membranes was then counted visually in five random image fields at a 20× magnification using a Zeiss Axio Observer Z1 confocal laser scanning microscopy (Oberkochen, Baden‐wurttemberg, Germany) equipped with Axiocam MRc5 (brightfield) and Hamamatsu Orca CCD (fluorescence, Hamamatsu, Shizuoka, JPN) cameras to measure the number of tumor cells invading in vitro: The cells stained with blue cores represented the hCMEC/D3, while the cells with blue cores and red membrane represented the A549 cells. The cells were counted and compared among the groups. The figures were generated using GraphPad Prism (GraphPad, La Jolla, CA, USA). The experiments were repeated three times.
2.9. Microarray data sequencing and verification of differential expression of genes
RNA extraction and microarray expression data generation used the Affymetrix GeneChip® Human Transcriptome Array 2.0 (Thermo Fisher, Pittsburgh, PA, USA) as part of the Decipher assay. The expression data were generated by the Affymetrix Expression Console software (Thermo Fisher, Pittsburgh, PA, USA) and normalized by the Robust Multichip Average method. An random variance model t test (BRB‐ArrayTools v4.5.1, National Institute of Health, Bethesda, MD, USA) was applied to filter the differentially expressed genes for the control and the experimental groups. The probes with a 100% match were collected for further data analysis. DEGs (Differentially Expressed Genes) were then identified based on fold change (FC) values of gene expression. The thresholds for DEGs were an FC >1.2 and a P value < 0.05. To estimate the accuracy of the microarray data, we evaluated the variation of up‐ and downregulated genes by RT‐PCR measurements to provide reliable validation measurements for comparison with the microarray data. In this study, GO and pathway enrichment analyses were conducted to determine the roles of these DEGs. The relationship between the DEGs’ functions in the pathways and the changes in the cell phenotype was also investigated.
2.10. Statistical analysis
All statistical analyses were performed using SPSS (version 16; SPSS Inc., Chicago, IL, USA). Mapping was performed using GraphPad Prism5.0 software (GraphPad, San Diego, CA, USA). The values were expressed as the means ± SEM, and the statistical significance was analyzed using a t test when the changes were compared between two groups. A P‐value of <0.05 was considered as significant and is indicated by asterisks in the figures.
3. RESULTS
3.1. Overexpression and knockdown of CLDN5 in an hCMEC/D3 cell line
The CLDN protein family is a key component of TJs. CLDNs are transmembrane proteins that span the cellular membrane 4 times, with the N‐terminal end and the C‐terminal end both located in the cytoplasm, and have two extracellular loops, which show high conservation. CLDN5 is an important CLDN protein and contains 4 transmembrane domains and 218 amino acids, with a molecular weight of 23 145 Da (Figure 1A). The CLDN5 protein is encoded by the CLDN5 gene, which is located on chromosome 22q11.21. The CLDN5 gene produces two variant transcripts that share the same CDS (911 bp) (Figure 1B).
Figure 1.
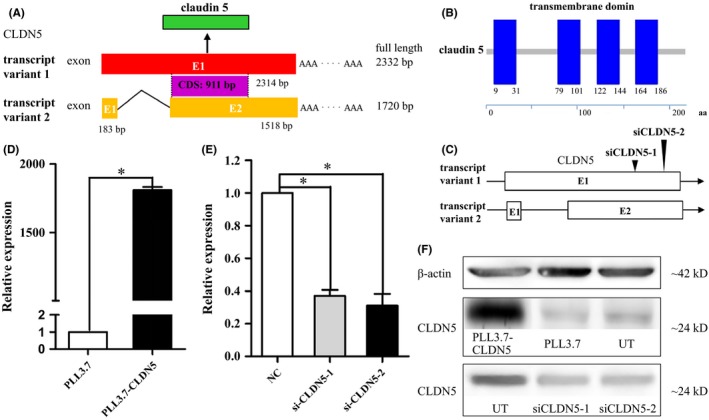
Structure analysis of human CLDN5 mRNA, protein, and expression of CLDN5 at mRNA and protein level. (A) Exon composition and the relationship between the two transcript variants within the CLDN5 gene. (B) Schematic representation of the CLDN5 protein, the blue boxes indicate protein domain of the transmembrane regions. (C) The position of the siCLDN5 oligonucleotides is indicated by black triangles. (D) CLDN5 expression was significantly high in the PLL3.7‐CLDN5 cells at mRNA level using qRT‐PCR. E, The expression was reduced in siCLDN5 cells. Data were normalized for GAPDH expression and expressed as fold change, values are mean ± SEM from three independent experiments. Statistical significance is indicated. F, Protein level of CLDN5 expressed in control cells was similar to untreated cells; however, it was significantly high in PLL3.7‐CLDN5 cells and low in siCLDN5 compared with control cells. β‐actin is used as a loading control in Western blot
To investigate the effect of CLDN5 on BBB permeability, we first established an hCMEC/D3 cell line stably overexpressing CLDN5. We then silenced CLDN5 using two transcript‐specific siRNAs (siCLDN5‐1 and siCLDN5‐2) that specifically targeted exon 1 of transcript variant 1 and exon 2 of transcript variant 2 in the hCMEC/D3 cells (Figure 1C).
The expression of CLDN5 mRNA and protein was measured using quantitative RT‐PCR (qPCR) and Western blots in both reinforced and knockdown hCMEC/D3 cells. The results indicated that the CLDN5 mRNA and protein were highly expressed in cells transfected with PLL3.7‐CLDN5 compared with PLL3.7 (P < 0.01) and that both siCLDN5‐1 and siCLDN5‐2 caused visible reductions in CLDN5 mRNA and protein levels (P < 0.05) (Figure 1D‐F).
3.2. Effects of CLDN5 on hCMEC/D3 cell proliferation and cell cycle progression
Brain EC proliferative capacity influences the permeability of the BBB. To explore the potential roles of CLDN5 in cell proliferation and the cell cycle in hCMEC/D3, the cell‐incorporated BrdU and total DNA content were measured by flow cytometry. There are four distinct stages in the cell cycle: G1, S, G2, and M, with DNA replication occurring in the S phase. A fifth stage, G0, is where the cell remains indefinitely until it begins the cell cycle again.
Our analysis confirmed that compared with the PLL3.7 hCMEC/D3 cells, the PLL3.7‐CLDN5 hCMEC/D3 cells showed an increased percentage of cells in the S phase and a decreased percentage of cells in the G0/G1 checkpoint, a difference that was statistically significant (P < 0.05) (Figure 2A‐B). Introduction of siCLDN5‐1 and siCLDN5‐2 in the hCMEC/D3 cells significantly resulted in a reduced percentage of cells in the S phase and an increased percentage of cells in the G0/G1 phase (P < 0.05) (Figure 2C‐D).
Figure 2.
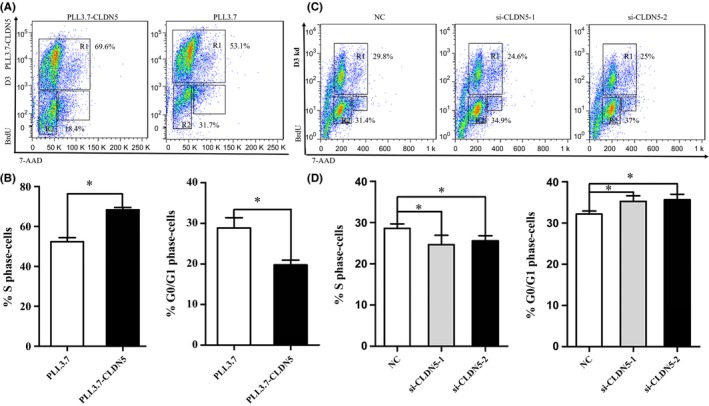
CLDN5 promotes hCMEC/D3 cell proliferation. Cells as indicated were incubated and subjected to BrdU assays. (A‐B) The proliferation of hCMEC/D3 cells was accelerated in PLL3.7‐CLDN5 cells compared with the PLL3.7 cells. The cells count in S phase increased, and cell ratio of G0/G1 phase decreased. (C‐D) The cell proliferation of siCLDN5 cells showed significant decline compared with the control cells. The number of S phase cells decreased, but G0/G1 phase cell ratio increased. Values are mean ± SEM from three independent experiments. Statistical significance is indicated
The results indicated that reinforced CLDN5 helped to promote the proliferation of the hCMEC/D3 cells and that the silencing of CLDN5 inhibited hCMEC/D3 cell proliferation by blocking the cell cycle at the G0/G1 phase.
3.3. CLDN5 induces an alteration in migration
The effect of CLDN5 on cellular migration was examined using an in vitro wound healing assay and an ECIS assay. A decreased distance in cell migration was seen in PLL3.7‐CLDN5 cells after 24 hours compared with PLL3.7 cells under a microscope at a 10× magnification. The data collected were processed using the ImageJ software (Figure 3A). Similar results were also found when ECIS was performed, which showed that the PLL3.7‐CLDN5 cells were significantly less motile compared with the PLL3.7 cells, as the resistance in the electrode increased as the cells reached confluency (P < 0.05), which took longer and demonstrated a low slope, with the time‐of‐healing in the PLL3.7‐CLDN5 cells being significantly shorter than that in the PLL3.7 cells (Figure 3B). The siCLDN5 cells showed a significantly increased cellular migration rate when compared with the control cells 24 hours after wounding. Data in all of these two groups were found to be significant (P < 0.05) (Figure 3C). These results indicate that CLDN5 might have a reverse promigratory effect on cells, whereas the siCLDN5 cells showed a markedly vigorous migration characteristic and took less time to come to confluency (P < 0.05) (Figure 3D).
Figure 3.
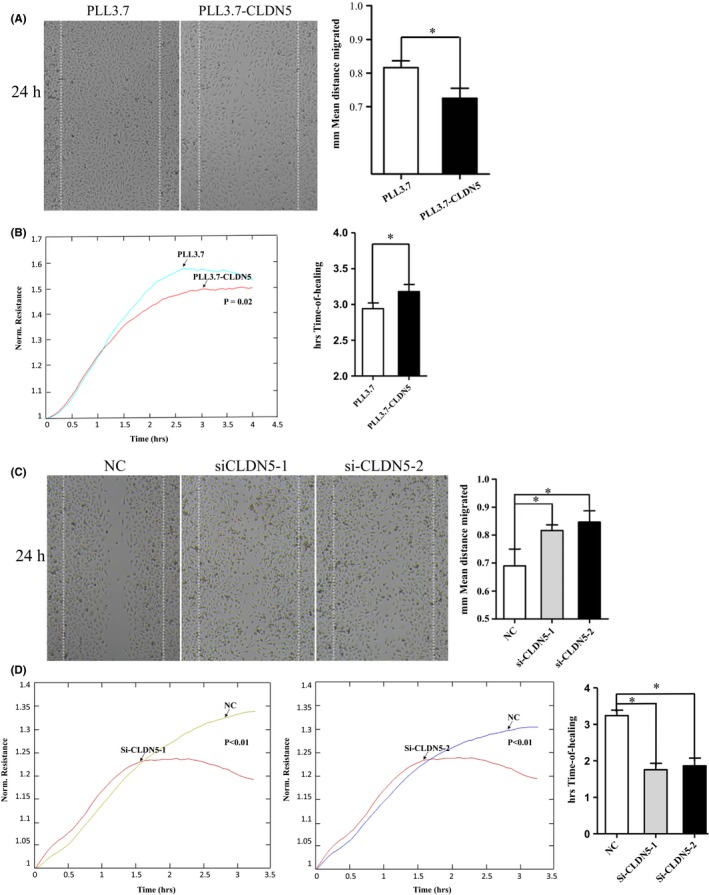
CLDN5 inhibited hCMEC/D3 cell migration. PLL3.7‐CLDN5 and PLL3.7 cells were subjected to wound healing assays. (A‐B) Larger gap was found in PLL3.7‐CLDN5 cells comparing to the control cells. The impaired motility of PLL3.7‐CLDN5 cells was also verified using electric cell impedance sensing (ECIS), more time was needed for PLL3.7‐CLDN5 cells to reach confluence after wounding created on the panel in ECIS assays. (C‐D) SiCLDN5 cells were more vigorous compared with the control ones. The siCLDN5 cells showed an increased migration rate comparing with the control group in that fewer time was needed for healing. Original magnification, 10×. Values are mean ± SEM from three independent experiments. Statistical significance is indicated
3.4. CLDN5 influences the permeability of the BBB and tumor cell invasion
A transwell assay was used to examine the possible role of the effect of CLDN5 on BBB permeability to the lung adenocarcinoma cell line A549 stained with CM‐DIL. It was observed that there were slightly fewer A549 cells stained in red in the PLL3.7‐CLDN5 group according to the core (in blue). However, there were significantly fewer A549 cells invading in the PLL3.7‐CLDN5 group compared with the PLL3.7 group (P < 0.05) (Figure 4A). More A549 cells could be seen under the microscope in the siCLDN5‐1 and siCLDN5‐2 groups compared with the control (P < 0.05) (Figure 4B). Our results show that overexpression of CLDN5 decreased the paracellular permeability of the hCMEC/D3 cells compared with the PLL3.7 cells, whereas the depletion of CLDN5 yielded the opposite results. The data represent the mean apparent permeability coefficients per group pooled from three independent experiments.
Figure 4.
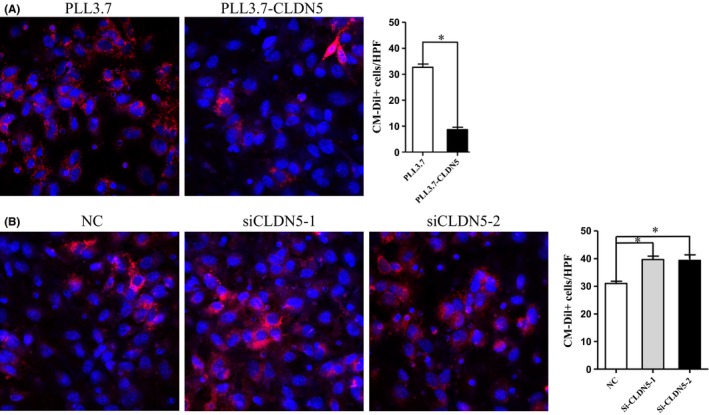
CLDN5 decreased the permeability of BBB to A549 cells. The effect of CLDN5 on barrier function of hCMEC/D3 cells to A549 cells was evaluated. Cells invaded were counted under confocal microscope after A549 cells were seeded on the monolayer of hCMEC/D3 cells for 48 hours. (A) Microscope images and quantitative analysis of cells invaded through the monolayer cell in PLL3.7‐CLDN5 and PLL3.7 group, and a significant decrease in cell stained in red(A549) was obtained. (B) The permeability increased in siCLDN5 group, which was manifested by more A549 cells with the membrane stained in red was observed in the lower panel. The A549 cells were stained with CM‐DIL (red), and nucleus was stain for DAPI (blue). Original magnification, 100×. Statistical significance is indicated
3.5. Investigation of the functional mechanism of CLDN5 in hCMEC/D3 cells
We compared the gene expression patterns between PLL3.7‐CLDN5 and PLL3.7 hCMEC/D3 cells. We identified 1685 DEGs in our microchip assay, of which 1378 were upregulated and 307 were downregulated. Hierarchical clustering revealed the nonrandom partitioning of samples into the PLL3.7 and PLL3.7‐CLDN5 groups (Figure 5A). The analysis showed systematic variations in the expression of the differential genes in the PLL3.7 and PLL3.7‐CLDN5 hCMEC/D3 cells. Thus, the gene expression signatures identified in this study were likely representative.
Figure 5.
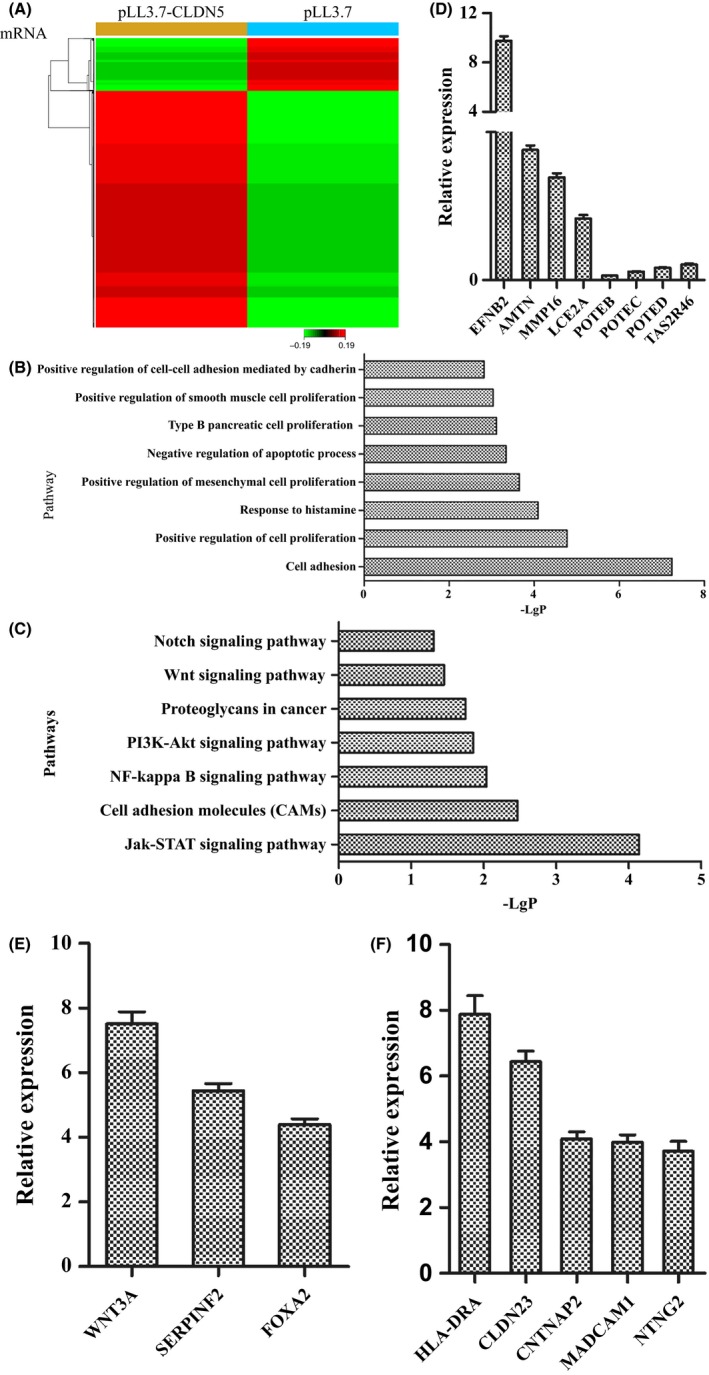
DEGs and pathways involved in the PLL3.7 hCMEC/D3 cells analyzed in microchip assay. (A) 1378 upregulated and 307 downregulated DEGs were presented in hierarchical clustering in the microchip assay. (B) The pathways that contributed to various cellular functions using GO analysis were listed according to the value of −LgP. (C) Seven typical significant pathways in all 48 ones which might play role in regulating the permeability of blood‐brain barrier were also listed. (D) The expression levels of DEGs with larger fold changes from differentially expressed datasets were analyzed by qRT‐PCR, with the first four genes upregulated and the last four‐ones downregulated. (E) Expression of 3 genes in gene function of the positive regulation of cell‐cell adhesion mediated by cadherin was revealed by qRT‐PCR. (F) There are 5 upregulated genes verified by qRT‐PCR in cell adhesion molecules pathway
The main functions of the 1685 DEGs in PLL3.7‐CLDN5 were analyzed using GO analysis. The results showed that DEGs were involved in 83 significant functions (P < 0.05; Table 2). The pathways of the 1685 DEGs involved were then analyzed according to KEGG, Biocarta, and Reatome. The DEGs were involved in the 48 enrichment‐related pathways (P < 0.05) (Table 3).
Table 2.
The pathways of DEGs involved using GO analysis
| No. | Pathway | −LgP | No. | Pathway | −LgP |
|---|---|---|---|---|---|
| 1 | G protein‐coupled receptor signaling pathway | 14.86 | 43 | positive regulation of G protein‐coupled receptor protein signaling pathway | 3.51 |
| 2 | Multicellular organismal development | 8.66 | 44 | Negative regulation of very‐low‐density lipoprotein particle clearance | 3.49 |
| 3 | Positive regulation of transcription from RNA polymerase II promoter | 8.02 | 45 | Negative regulation of lipid metabolic process | 3.49 |
| 4 | Neuropeptide signaling pathway | 7.50 | 46 | Regulation of dopamine uptake involved in synaptic transmission | 3.49 |
| 5 | Cell adhesion | 7.24 | 47 | Visual perception | 3.47 |
| 6 | Cell differentiation | 6.86 | 48 | Transmembrane transport | 3.43 |
| 7 | Synaptic transmission | 6.80 | 49 | Positive regulation of ERK1 and ERK2 cascade | 3.39 |
| 8 | Sensory perception of taste | 6.60 | 50 | Negative regulation of neuron differentiation | 3.36 |
| 9 | Brain development | 5.84 | 51 | Negative regulation of apoptotic process | 3.33 |
| 10 | G protein‐coupled receptor signaling pathway, coupled to cyclic nucleotide second messenger | 5.81 | 52 | Regulation of dopamine secretion | 3.30 |
| 11 | Cell‐cell signaling | 5.60 | 53 | Dopamine metabolic process | 3.30 |
| 12 | Keratinization | 5.40 | 54 | Glucose homeostasis | 3.30 |
| 13 | Proteolysis | 5.40 | 55 | Epidermis development | 3.25 |
| 14 | Transcription, DNA‐dependent | 5.37 | 56 | Negative regulation of phosphatase activity | 3.23 |
| 15 | Immune response | 5.36 | 57 | Response to amphetamine | 3.18 |
| 16 | Pattern specification process | 5.17 | 58 | Regulation of glucose metabolic process | 3.11 |
| 17 | Inflammatory response | 5.00 | 59 | Neuron fate specification | 3.11 |
| 18 | Positive regulation of cell proliferation | 4.77 | 60 | Negative regulation of protein secretion | 3.11 |
| 19 | Adenylate cyclase‐activating G protein‐coupled receptor signaling pathway | 4.75 | 61 | Negative regulation of inflammatory response | 3.11 |
| 20 | Regulation of immune response | 4.63 | 62 | Adenylate cyclase‐inhibiting dopamine receptor signaling pathway | 3.11 |
| 21 | Positive regulation of actin filament polymerization | 4.59 | 63 | Glycerol transport | 3.11 |
| 22 | Negative regulation of retinoic acid receptor signaling pathway | 4.59 | 64 | Negative regulation of interleukin‐5 production | 3.11 |
| 23 | Behavioral response to cocaine | 4.42 | 65 | Drinking behavior | 3.11 |
| 24 | T cell costimulation | 4.39 | 66 | Type B pancreatic cell proliferation | 3.11 |
| 25 | Signal transduction | 4.24 | 67 | Lateral mesoderm development | 3.11 |
| 26 | Synaptic transmission, dopaminergic | 4.20 | 68 | Feeding behavior | 3.09 |
| 27 | Cell surface receptor signaling pathway | 4.18 | 69 | Defense response | 3.07 |
| 28 | Response to histamine | 4.09 | 70 | Humoral immune response | 3.03 |
| 29 | Negative regulation of dopamine receptor signaling pathway | 4.09 | 71 | Positive regulation of smooth muscle cell proliferation | 3.03 |
| 30 | Biological process | 4.01 | 72 | Eating behavior | 3.01 |
| 31 | Potassium ion transport | 4.00 | 73 | Chondrocyte differentiation | 2.93 |
| 32 | Positive regulation of sequence‐specific DNA binding transcription factor activity | 3.98 | 74 | Negative regulation of adenylate cyclase activity | 2.90 |
| 33 | Nervous system development | 3.95 | 75 | Adult walking behavior | 2.85 |
| 34 | Positive regulation of canonical Wnt receptor signaling pathway | 3.95 | 76 | Forebrain development | 2.84 |
| 35 | Negative regulation of cell differentiation | 3.94 | 77 | Positive regulation of protein processing | 2.82 |
| 36 | Regulation of sequence‐specific DNA binding transcription factor activity | 3.89 | 78 | Chylomicron remnant clearance | 2.82 |
| 37 | Interferon‐gamma‐mediated signaling pathway | 3.83 | 79 | Negative regulation of receptor‐mediated endocytosis | 2.82 |
| 38 | Keratinocyte differentiation | 3.75 | 80 | Negative regulation of neuron death | 2.82 |
| 39 | Positive regulation of protein kinase B signaling cascade | 3.66 | 81 | Positive regulation of cell‐cell adhesion mediated by cadherin | 2.82 |
| 40 | Positive regulation of mesenchymal cell proliferation | 3.65 | 82 | Positive regulation of filopodium assembly | 2.80 |
| 41 | Learning or memory | 3.56 | 83 | Ion transmembrane transport | 2.79 |
| 42 | Oxygen transport | 3.51 |
Table 3.
The enrichment‐related pathways of DEGs involved
| No. | Pathway | −LgP | No. | Pathway | −LgP |
|---|---|---|---|---|---|
| 1 | Olfactory transduction | 26.8 | 25 | Glycine, serine, and threonine metabolism | 2.51 |
| 2 | Neuroactive ligand‐receptor interaction | 16.3 | 26 | Cell adhesion molecules (CAMs) | 2.46 |
| 3 | Hematopoietic cell lineage | 6.98 | 27 | Tuberculosis | 2.42 |
| 4 | Cytokine‐cytokine receptor interaction | 6.36 | 28 | Phototransduction | 2.24 |
| 5 | Taste transduction | 4.63 | 29 | Vascular smooth muscle contraction | 2.22 |
| 6 | Type I diabetes mellitus | 4.24 | 30 | Toxoplasmosis | 2.16 |
| 7 | Jak‐STAT signaling pathway | 4.14 | 31 | Phagosome | 2.16 |
| 8 | Dopaminergic synapse | 4.00 | 32 | Viral myocarditis | 2.10 |
| 9 | Intestinal immune network for IgA production | 3.95 | 33 | Staphylococcus aureus infection | 2.08 |
| 10 | Salivary secretion | 3.90 | 34 | NF‐kappa B signaling pathway | 2.04 |
| 11 | Rheumatoid arthritis | 3.71 | 35 | Leishmaniasis | 2.03 |
| 12 | Antigen processing and presentation | 3.62 | 36 | Morphine addiction | 2.01 |
| 13 | Asthma | 3.56 | 37 | African trypanosomiasis | 1.93 |
| 14 | Cholinergic synapse | 3.53 | 38 | Type II diabetes mellitus | 1.92 |
| 15 | Maturity‐onset diabetes of the young | 3.47 | 39 | Circadian entrainment | 1.89 |
| 16 | Malaria | 3.29 | 40 | PI3K‐Akt signaling pathway | 1.86 |
| 17 | Natural killer cell mediated cytotoxicity | 3.13 | 41 | Primary immunodeficiency | 1.82 |
| 18 | Allograft rejection | 3.03 | 42 | Hedgehog signaling pathway | 1.79 |
| 19 | HTLV‐I infection | 2.97 | 43 | Arachidonic acid metabolism | 1.78 |
| 20 | Transcriptional misregulation in cancer | 2.96 | 44 | Proteoglycans in cancer | 1.75 |
| 21 | Basal cell carcinoma | 2.94 | 45 | Serotonergic synapse | 1.46 |
| 22 | Melanogenesis | 2.83 | 46 | Wnt signaling pathway | 1.46 |
| 23 | Graft‐versus‐host disease | 2.78 | 47 | Vasopressin‐regulated water reabsorption | 1.42 |
| 24 | Hippo signaling pathway | 2.67 | 48 | Notch signaling pathway | 1.31 |
To explore the key gene functions and pathways that were regulated by CLDN‐5, we identified 8 significant gene functions that contributed to various cellular functions, including cell adhesion, proliferation, and apoptosis (P < 0.05, Figure 5B), and 7 significant pathways that were involved in the Jak‐STAT signaling pathway, the PI3K‐Akt signaling pathway, proteoglycans in cancer pathway, and cell adhesion molecules (CAMs), among others. These gene functions and pathways affected the function of the TJs to regulate the permeability of the BBB (P < 0.05, Figure 5C).
To validate the microarray analysis conclusions, we randomly selected 8 DEGs with larger FCs from the differentially expressed datasets and analyzed their expression levels by qRT‐PCR in the PLL3.7 and PLL3.7‐CLDN5 hCMEC/D3 cells. Our results confirmed the findings of the gene microarray data (Figure 5D).
First, we analyzed the expression of 3 genes with gene functions in the positive regulation of cell‐cell adhesion mediated by cadherin (GO: 2000049) in PLL3.7 and PLL3.7‐CLDN5 hCMEC/D3 cells. As shown in Figure 5E, the mRNA levels of WNT3A, SERPINF2, and FOXA2 were upregulated. Next, we analyzed the mRNA levels of 5 genes in the CAMs (KEGG: map04514) pathway in PLL3.7 and PLL3.7‐CLDN5 hCMEC/D3 cells. Analysis by qRT‐PCR showed that the expression of HLA‐DRA, CLDN23, CNTNAP2, MADCAM1, and NTNG2 were upregulated in PLL3.7‐CLDN5 hCMEC/D3 cells (Figure 5F).
4. DISCUSSION
The ability to invade into the BBB is a very important capacity for tumors in the formation of BM. The ECs not only account for a large proportion of composition of the BBB but also play a vital role in the barrier function of the BBB. CLDN5 is an important component of TJs, which exist in the paracellular space, and is highly expressed in ECs, especially in cerebral vascular ECs. To investigate the exact role CLDN5 plays in the formation of BM, a monolayer of hCMEC/D3 cells was used to mimic the BBB. The hCMEC/D3 cells show a spindle‐shaped elongated morphology similar to primary cultures of brain ECs and express a variety of brain endothelial markers typical of the brain epithelium.20 In the present study, hCMEC/D3 monolayer cells that were CLDN5‐reinforced and silenced were established as models. The influence of CLDN5 on paracellular permeability, cell proliferation, and cell migration was observed, and the underlying mechanisms and the interactions between genes after the overexpression of CLDN5 were investigated.
Previous studies22, 23, 24 have described that the downregulation of CLDN5 increased the permeability of both BBB and common ECs, although some studies have demonstrated that exposure of brain microvascular ECs to certain pathological states25, 26, 27 might increase BBB permeability in parallel with reduced expression level of CLDN5. In this study, we found that there was an increased number of A549 cells invading in the siCLDN5 group compared with the control group. However, this result was not consistent with a previous study28 that showed little change in either knockdown monolayer cells or knockout mice, which indicates that the CLDN5 knockdown might be compensated for functionally by other CLDNs.8 However, the increased permeability could be explained by the dysfunction of TJs caused by the lower expression of CLDN5 and the invasive characteristics of lung cancer, such as migration and the secretion of MMP, resulting in a synergistic effect created by both of these factors.
We can see that the barrier function was enhanced significantly in PLL3.7‐CLDN5 cells, in which the expression of CLDN5 was increased by nearly 2000‐fold, although results have been reported that the CLDN5 function is cell‐type‐dependent and is influenced by the context of expression.29 We presumed that because the monolayer of hCMEC/D3 cells had reached confluency, there were no differences in cell numbers and hardly any influence on proliferation, which indicates that the decrease in permeability was due to the comprehensive effects of genes caused by the overexpression of CLN5 in regulating the paracellular barrier. A total of 1685 DEGs and 48 pathways were detected in the microarray. One of these was the NF‐kappa B signaling pathway, which might be correlated with permeability; there is also a study20 consistent with this result suggesting that the underlying mechanism of increased paracellular permeability functioned via signaling pathways such as JNK, PKC, or NFkB. Among these pathways from the overexpression group, we found that the CAMs pathway was involved in the regulation of permeability, as genes in this pathway, such as SELL, were functioning to enhance the paracellular adhesiveness and junctions and barrier function, due to the improvement in cell‐cell adhesive functions that can be modulated by SELL in the upregulation of CD34, MADCAM1, and GlyCAM1. GO analysis also revealed a cell‐cell adhesion mediated by cadherin, so we can tentatively conclude that the decreased permeability was the result of the enhanced TJs mediated by the overexpression of CLDN5 via the CAMs pathway.
In addition, we found JAK‐STAT and PI3K pathways in CLDN5 overexpression that might be involved in modulating permeability by affecting cell proliferation. Previous studies30, 31 using other types of ECs have reported a positive role for CLDN5 in cell proliferation. Changes in CLDN5 expression have been previously connected to pathological angiogenesis.32 However, other research33 appeared to have a contradictory result, which suggests that CLDN5 plays a negative role in cell proliferation, although this trend did not reach a significant difference. In this study, our results showed that the knockdown of CLDN5 significantly suppressed cell proliferation in vitro and that the overexpression of CLDN5 had a positive role in the growth of the hCMEC/D3 cells. We observed more cells in the S phase in the CLDN5 overexpression group in our BrdU assay. Furthermore, in the microarray, we found that the overexpression of CLDN5 caused changes in IL7R and PI3KCG in the JAK‐STAT and PI3K pathways, respectively, which are directly involved in regulating cell proliferation. This is in agreement with a previous study, which showed that histamine and PI3K induce EC proliferation by upregulating CLDN5.34 However, the positive effects of CLDN5 on cell proliferation disappeared when the cells reached confluency because of contact inhibition,35, 36, 37 a phenomenon in which ECs stop proliferation as soon as they come into contact with another cell. The expression level of CLDN5 will then be stabilized and act as a functional barrier. Thus, the cell proliferation caused by the overexpression of CLDN5 might also contribute to the decreased permeability in PLL3.7 CLDN5 cells by forming a monolayer of hCMEC/D3 cells in a much shorter time. The actual mechanism of CLDN5‐induced proliferation still needs to be further investigated.
Cell motility is orchestrated by a variety of complicated signaling pathways, with most of these just now starting to be unraveled. Pathways that might influence cell motility were also revealed in our study. We found that cells showed a trend in reduction in mortality in PLL3.7‐CLDN5 cells compared with PLL3.7 cells and that the siCLDN5 cells showed an increase in motility, significantly different from the control cells. These results were verified by wound healing and ECIS assays. Previous studies have shown that CLDN5 has been connected to the regulation of EC motility and matrix adhesion and might interact with N‐WASP and ROCK 1 as well, but no direct effect on expression was found.33, 38 In addition, several genes required for EC migration were demonstrated by inducing the downregulation of CLDN5,39 which revealed a contradictory result between ECs and breast cancer cells. We presume that the impaired migration ability is good for maintaining stable circumstances, which is beneficial for cell proliferation and for formation of the cell‐cell interaction. To elucidate the underlying mechanisms, RNA microchip assays were performed in the CLDN5 overexpression group, in which we found the downregulation of ARHGEF12 located in the proteoglycans in cancer pathway and a decreased trend in cell migration due to the inhibition of the effect of ARHGEF12. Samarzija40 observed that Wnt3a induces proliferation and migration of ECs via canonical and noncanonical Wnt signaling pathways, and interestingly, the Wnt signaling pathway was also detected in our study. We might tentatively conclude from this that CLDN5 might be a regulator of migration, but the mechanism might be different between ECs and cancer cells, although this still needs to be investigated.
In this study, we provide a comprehensive analysis of the key gene functions and pathways that are regulated by CLDN‐5 in brain ECs, including cell proliferation, adhesion molecules, and the Jak‐STAT, PI3K‐Akt, Wnt, and Notch signaling pathways. In addition to affecting the function of the TJ that regulates the permeability of the BBB, these gene functions and pathways mediated interactions between metastatic tumor cells and brain ECs and regulated the motility and angiogenic potential of the tumor cells to initiate brain tumor metastasis.
In summary, we also showed that CLDN5 protected the permeability of the BBB by modulating certain genes in the CAMs pathway and demonstrated its role in enhancing cell proliferation and reducing cell migration, which might also reduce the permeability in different ways. We also confirmed the direct role of CLDN5 in the invasion of brain vascular ECs (hCMEC/D3) by lung cancer cells (A549), highlighting CLDN5 as an interesting target for treatment for BM.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This research was supported by National Natural Science Foundation of China (81471229). Shun‐Chang Ma expresses his sincere gratitude to Wang Jia for constant encouragement and guidance both in clinical work and in research project during the 3‐year study at Tiantan Hospital.
Ma S‐C, Li Q, Peng J‐Y, et al. Claudin‐5 regulates blood‐brain barrier permeability by modifying brain microvascular endothelial cell proliferation, migration, and adhesion to prevent lung cancer metastasis. CNS Neurosci Ther. 2017;23:947–960. 10.1111/cns.12764
REFERENCES
- 1. Crnic I, Christofori G. Novel technologies and recent advances in metastasis research. Int J Dev Biol. 2004;48:573‐581. [DOI] [PubMed] [Google Scholar]
- 2. Barnholtz‐Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22:2865‐2872. [DOI] [PubMed] [Google Scholar]
- 3. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94:2698‐2705. [DOI] [PubMed] [Google Scholar]
- 4. Wilhelm I, Molnar J, Fazakas C, Hasko J, Krizbai IA. Role of the blood‐brain barrier in the formation of brain metastases. Int J Mol Sci. 2013;14:1383‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005;75:5‐14. [DOI] [PubMed] [Google Scholar]
- 6. Sarkadi B, Homolya L, Szakacs G, Varadi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol Rev. 2006;86:1179‐1236. [DOI] [PubMed] [Google Scholar]
- 7. Wolburg H, Lippoldt A. Tight junctions of the blood‐brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323‐337. [DOI] [PubMed] [Google Scholar]
- 8. Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE. Transmembrane proteins of the tight junctions at the blood‐brain barrier: structural and functional aspects. Semin Cell Dev Biol. 2015;38:16‐25. [DOI] [PubMed] [Google Scholar]
- 9. Jia W, Lu R, Martin TA, Jiang WG. The role of claudin‐5 in blood‐brain barrier (BBB) and brain metastases (review). Mol Med Rep. 2014;9:779‐785. [DOI] [PubMed] [Google Scholar]
- 10. Tsukita S, Furuse M. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J Cell Biol. 2000;149:13‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohkubo T, Ozawa M. The transcription factor Snail downregulates the tight junction components independently of E‐cadherin downregulation. J Cell Sci. 2004;117:1675‐1685. [DOI] [PubMed] [Google Scholar]
- 12. Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin‐5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao F, Deng J, Yu X, Li D, Shi H, Zhao Y. Protective effects of vascular endothelial growth factor in cultured brain endothelial cells against hypoglycemia. Metab Brain Dis. 2015;30:999‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sandoval KE, Witt KA. Blood‐brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200‐219. [DOI] [PubMed] [Google Scholar]
- 15. Desai BS, Monahan AJ, Carvey PM, Hendey B. Blood‐brain barrier pathology in Alzheimer's and Parkinson's disease: implications for drug therapy. Cell Transplant. 2007;16:285‐299. [DOI] [PubMed] [Google Scholar]
- 16. Koto T, Takubo K, Ishida S, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin‐5 in endothelial cells. Am J Pathol. 2007;170:1389‐1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high‐energy diet on hippocampal function and blood‐brain barrier integrity in the rat. J Alzheimers Dis. 2010;21:207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell‐cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol. 2008;6:179‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luissint AC, Federici C, Guillonneau F, et al. Guanine nucleotide‐binding protein Galphai2: a new partner of claudin‐5 that regulates tight junction integrity in human brain endothelial cells. J Cereb Blood Flow Metab. 2012;32:860‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weksler BB, Subileau EA, Perriere N, et al. Blood‐brain barrier‐specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872‐1874. [DOI] [PubMed] [Google Scholar]
- 22. Liao Z, Yang Z, Piontek A, et al. Specific binding of a mutated fragment of Clostridium perfringens enterotoxin to endothelial claudin‐5 and its modulation of cerebral vascular permeability. Neuroscience. 2016;327:53‐63. [DOI] [PubMed] [Google Scholar]
- 23. Rosas‐Hernandez H, Ramirez M, Ramirez‐Lee MA, Ali SF, Gonzalez C. Inhibition of prolactin with bromocriptine for 28 days increases blood‐brain barrier permeability in the rat. Neuroscience. 2015;301:61‐70. [DOI] [PubMed] [Google Scholar]
- 24. Herr D, Fraser HM, Konrad R, Holzheu I, Kreienberg R, Wulff C. Human chorionic gonadotropin controls luteal vascular permeability via vascular endothelial growth factor by down‐regulation of a cascade of adhesion proteins. Fertil Steril. 2013;99:1749‐1758. [DOI] [PubMed] [Google Scholar]
- 25. Liu C, Wu J, Zou MH. Activation of AMP‐activated protein kinase alleviates high‐glucose‐induced dysfunction of brain microvascular endothelial cell tight‐junction dynamics. Free Radic Biol Med. 2012;53:1213‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae‐mediated internalization of occludin and claudin‐5 during CCL2‐induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053‐19066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stamatovic SM, Keep RF, Andjelkovic AV. Tracing the endocytosis of claudin‐5 in brain endothelial cells. Methods Mol Biol. 2011;762:303‐320. [DOI] [PubMed] [Google Scholar]
- 28. Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid‐beta‐induced occludin down‐regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14:1101‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schlingmann B, Overgaard CE, Molina SA, et al. Regulation of claudin/zonula occludens‐1 complexes by hetero‐claudin interactions. Nat Commun. 2016;7:12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen J, Stahl A, Krah NM, et al. Retinal expression of Wnt‐pathway mediated genes in low‐density lipoprotein receptor‐related protein 5 (Lrp5) knockout mice. PLoS ONE. 2012;7:e30203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krishnan S, Szabo E, Burghardt I, Frei K, Tabatabai G, Weller M. Modulation of cerebral endothelial cell function by TGF‐beta in glioblastoma: VEGF‐dependent angiogenesis versus endothelial mesenchymal transition. Oncotarget. 2015;6:22480‐22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herr D, Sallmann A, Bekes I, et al. VEGF induces ascites in ovarian cancer patients via increasing peritoneal permeability by downregulation of Claudin 5. Gynecol Oncol. 2012;127:210‐216. [DOI] [PubMed] [Google Scholar]
- 33. Escudero‐Esparza A, Jiang WG, Martin TA. Claudin‐5 participates in the regulation of endothelial cell motility. Mol Cell Biochem. 2012;362:71‐85. [DOI] [PubMed] [Google Scholar]
- 34. Laakkonen JP, Lappalainen JP, Theelen TL, et al. Differential regulation of angiogenic cellular processes and claudin‐5 by histamine and VEGF via PI3K‐signaling, transcription factor SNAI2 and interleukin‐8. Angiogenesis. 2017;20:109‐124. [DOI] [PubMed] [Google Scholar]
- 35. Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial‐cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15:2943‐2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR‐2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noseda M, Chang L, McLean G, et al. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol Cell Biol. 2004;24:8813‐8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Escudero‐Esparza A, Jiang WG, Martin TA. Claudin‐5 is involved in breast cancer cell motility through the N‐WASP and ROCK signalling pathways. J Exp Clin Cancer Res. 2012;31:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sobrado M, Ramirez BG, Neria F, et al. Regulator of calcineurin 1 (Rcan1) has a protective role in brain ischemia/reperfusion injury. J Neuroinflammation. 2012;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Samarzija I, Sini P, Schlange T, Macdonald G, Hynes NE. Wnt3a regulates proliferation and migration of HUVEC via canonical and non‐canonical Wnt signaling pathways. Biochem Biophys Res Comm. 2009;386:449‐454. [DOI] [PubMed] [Google Scholar]