

Summary
Aims
Traumatic brain injury (TBI) is induced by complex primary and secondary mechanisms that give rise to cell death, inflammation, and neurological dysfunction. Understanding the mechanisms that drive neurological damage as well as those that promote repair can guide the development of therapeutic drugs for TBI. Kruppel‐like factor 4 (KLF4) has been reported to negatively regulate axon regeneration of injured retinal ganglion cells (RGCs) through inhibition of JAK‐STAT3 signaling. However, the role of KLF4 in TBI remains unreported. Reactive oxygen species (ROS)‐induced neuronal death is a pathophysiological hallmark of TBI.
Methods
In this study, we used H2O2‐treated RGCs in vitro and the optic nerve crush model in vivo to simulate neuronal damage in TBI. The function of KLF4 in RGC survival and axon regeneration in these models was investigated. In addition, the effects of KLF4 knockdown on neuronal damage after a brain impact that mimics moderate TBI were studied.
Results
The results show that H2O2 induces p53‐dependent apoptosis of RGCs in vitro through upregulation of KLF4. Additionally, KLF4 knockdown in vivo significantly enhances CNTF‐induced axon regeneration of RGCs after optic nerve crush, and more importantly, prevents neuronal damage after a moderate brain impact in rats. Our Western blot analysis and immunoprecipitation assay results indicate that these effects of KLF4 knockdown are mediated by the p53 and JAK‐STAT3 pathways.
Conclusion
These findings provide evidence that KLF4 plays an important role in the pathophysiology of TBI. Blocking KLF4 may be a potential therapeutic strategy for the treatment of TBI, either alone or in combination with agents that target complementary mechanisms.
Keywords: Kruppel‐like factor 4, Nrf2, P53, STAT3, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) represents a major cause of mortality and morbidity in the world population, especially in children 1. The pathophysiology of TBI is complicated, and the brain damage is mainly divided into two phases: the primary injury and secondary injury. The primary injury occurs at the time of insult, and results from displacement of the physical structures of the brain such as laceration, contusion, and intracranial hemorrhage. This type of injury is sensitive to preventive measures but cannot be reversed by therapeutic treatment. The secondary injury is initiated at the moment of insult, but clinical symptoms gradually develop for a period of hours or days. The secondary injury mechanisms involve a complex array of cellular processes that include metabolic changes, oxidative damage, and neuroinflammation. These alterations might lead to massive neuronal cell death at the size of injury or diffuse brain region, exacerbating the initial damage 2. Evidence suggests that the secondary injury plays a key role in determining the ultimate extent of neurological injury and recovery after TBI 3. Understanding the molecular mechanisms of secondary injury may identify novel targets that could be harnessed to ameliorate TBI and promote functional recovery.
Mechanical stress to the brain causes direct tissue damage, disrupting the balance between cerebral blood flow and metabolism. This “ischemia‐like” stress activates a complex cascade of biochemical processes that result in excitotoxicity and excessive production of reactive oxygen species (ROS). A large body of evidence has demonstrated that ROS‐mediated damage plays a central role in secondary damage associated with TBI 4. P53, a master regulator of cell apoptosis, is dramatically upregulated by glutamate excitotoxicity and oxidative stress in neurons 5. P53 levels are elevated in experimental TBI 6. Additionally, blocking p53 protects against TBI‐associated neuronal loss 7. These findings suggest that p53‐dependent apoptosis plays a crucial role in the pathophysiology of TBI. The JAK/STAT pathway is also activated following experimental TBI 8, 9. The JAK2 inhibitor AG490 prevents post‐TBI behavioral recovery in rats 9. Therefore, this pathway may be at least partially responsible for the neurological recovery after TBI.
Kruppel‐like factor 4 (KLF4) is a zinc finger‐containing transcription factor that regulates cell proliferation, differentiation, apoptosis, and somatic cell reprogramming. Previous reports have shown that KLF4 exerts context‐dependent oncogenic or tumor‐suppressor functions in cancer, likely via regulation of p53 expression and its functional activation 10, 11, 12. In the nervous system, KLF4 is expressed in neural stem cells 13 and regulates neuronal differentiation and migration in the developing cerebral cortex through modulation of STAT3 14. Interestingly, a few recent studies have shown that KLF4 suppresses axon regeneration after injury through inhibition of JAK‐STAT3 signaling 15, 16. Restoration of damaged axons contributes to neurological recovery after TBI; however, the role of KLF4 in TBI remains unreported.
ROS generation is a hallmark of traumatic brain injury (TBI). In the present study, we investigated the function of KLF4 in H2O2‐induced apoptosis of retinal ganglion cells (RGCs) in vitro and axon regeneration of injured RGCs after optic nerves crush in vivo. In addition to these experiments that simulate neuronal injury associated with TBI, we studied the effects of KLF4 knockdown on brain neuronal damage and repair after a moderate brain impact in rats.
Materials and Methods
Animals
Sprague Dawley rats (7–8 weeks of age) were obtained from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (the Guide, NRC 2011). All animal study protocols were approved by the Ethics Committee of the Shanghai Tenth People's Hospital of Tongji University.
Cell Lines and Cultures
RGC‐5 cells, which are transformed rat RGCs expressing ganglion cell markers and exhibiting ganglion cell‐like behavior 17, were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 4500 mg/L glucose, 10% heat‐inactivated fetal bovine serum (FBS), and 1% penicillin/streptomycin at 5% CO2, 37°C in a humidified incubator. Cells were grown to 70% confluence and treated with H2O2 (Sigma‐Aldrich Co., Bellefonte, PA, USA) to induce apoptosis.
Cell Viability by the MTT Assay
Cell viability was determined using the MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyl‐tetrazolium bromide] assay. Briefly, cells seeded in 96‐well plates at 5 × 103 cells/well were grown to 70% confluence and incubated with H2O2 (0, 100, 250, or 500 μM) for 0, 6, 12, or 24 h. The medium was removed and replaced with fresh medium containing 10 μL MTT (5 mg/mL). After incubation for another 4 h, the medium was removed and the blue formazan crystal was dissolved in 100 μL dimethyl sulfoxide (DMSO). Absorbance at 570 nm was recorded on a fluorescence plate reader (Millipore Corp., Billerica, MA, USA). The relative cell viability (%) was calculated as a percentage relative to the untreated control.
Cell Apoptosis by Flow Cytometry
H2O2‐induced RGC‐5 apoptosis was determined by flow cytometry using the annexin V‐FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) following manufacturer's instructions. Briefly, cells seeded in 96‐well plates at 1.5 ×105/mL were grown to 70% confluence and incubated with H2O2 (0, 100, 250, or 500 μM) for 12 h or with 500 μM H2O2 for 0, 6, 12, or 24 h. After that, the cells were washed twice with ice‐cold PBS, stained in 5 μL of annexin V‐FITC and 1 μL of propidium iodide (PI, 1 mg/mL) for 15 min, and subjected to analysis on a flow cytometer (BD Biosciences).
Western Blot Analysis
Cell lysate samples were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to nitrocellulose membranes (Amersham Biosciences, Sunnyvale, CA, USA). After blocking in 5% nonfat milk in Tris‐buffered saline with Tween (TBST) for 2 h, the membranes were incubated with primary antibody (anti‐phospho‐STAT3 (p‐STAT3), 1:200; anti‐STAT3, 1:200; anti‐phospho‐p53 (p‐p53), 1:200; anti‐p53, 1:200; anti‐phospho‐p38MAPK (p‐p38MAPK), 1:500; anti‐p38MAPK, 1:500; anti‐Bcl‐2, 1:500; anti‐Bax, 1:500; anti‐Nrf2, 1:500; anti‐cleaved caspase‐3, 1:500; anti‐KLF4, 1:500; anti‐phospho‐KLF4 (p‐KLF4), 1:200; anti‐β‐APP, 1:200; anti‐GAPDH, 1:2000) at 4°C overnight. All primary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). After washing with PBS, the cells were incubated with horseradish peroxidase‐conjugated secondary antibody. Signals were detected using an ECL detection system (GE Healthcare, Boston, MA, USA) and analyzed using the ImageJ 1.42q software (National Institutes of Health, Rockville Pike Bethesda, MD, USA).
Immunoprecipitation
Cells or homogenized tissue samples were lysed in 20 mM Tris–HCl (pH 7.5) containing 1 mM EDTA, 1 M KCl, 5 mM MgCl2, 10% glycerol (v/v), 1% Triton X‐100 (v/v), 0.05% 2‐mercaptoethanol (v/v), and a cocktail of protease and phosphatase inhibitors. The lysates (1–4 mg) were precleared with protein G beads at 4°C for 30 min and subsequently incubated with protein G beads prebound with antibody at 4°C for 2–16 h. The beads were washed three times with 1% NP40, mixed with sample buffer, and subjected to SDS‐PAGE and immunoblotting. To detect the interaction between endogenous KLF4 and p‐STAT3, siKLF4‐transfected and untransfected RGCs were treated with ciliary neurotrophic factor (CNTF; 100 pM, Sigma) for 30 min. Cells were harvested and homogenized in lysis buffer. To detect the interaction between endogenous p53 and KLF4, sip53‐transfected and untransfected RGCs were treated with 500 μM H2O2. After 12 h, cells were harvested and homogenized in lysis buffer.
Lentiviral Plasmid and Small Interfering RNA (siRNA) Construction and Transfection
Rat KLF4 full‐length cDNA was generated by PCR using forward primer 5′‐CCAAGCTTATGAGGCAGCCACCTGGCGAGTCT‐3′ and reverse primer 5′‐CGGAATTCTTAAAAGTGCCTCTTCATGTGTAAG‐3′. The amplified fragment was subcloned into the lentiviral vector pGC‐FU‐EGFP‐3FLAG (GeneChem, Shanghai, China) after Hind III/EcoR I restriction enzyme digestion to produce the pGC‐FU‐KLF4‐1‐3FLAG plasmid. The small interfering RNAs (siRNA) specific for rat KLF4 (siKLF4, 5′‐CACCCACACTTGTGACTATTT‐3′), p53 (sip53, 5′‐GGACAGCCAAGTCTGTTATTT‐3′), and Nrf2 (siNrf2, 5′‐CCCTGTTGATGACTTCAATTT‐3′), and a scrambled siRNA (SCR, 5′‐CAAATACACTTCTGACTATTT‐3′) used as the negative control were synthesized by GenScript (China). RGC‐5 cells were transfected with plasmid or siRNA using oligofectamine (Invitrogen, Carlsbad, CA, USA) following manufacturer's instructions.
Reverse Transcription‐PCR
Total RNA was extracted using the RNA isolation kit (Ambion, Austin, TX, USA) following manufacturer's instructions. Reverse transcription polymerase chain reaction (RT‐PCR) was carried out on a GeneAmp® PCR System 9700 (Applied Biosystems, Alameda, CA, USA). PCR primers specific for KLF4 were 5′‐TCCAGCAGGTGCCCCGACTA‐3′ (forward) and 5′‐CTGCTAGCTGGGGAAGAGA‐3′ (reverse). PCR primers specific for p53 were 5′‐CCACCACAGCGACAGGGTCA‐3′ (forward) and 5′‐CCATAGTTGCCTTGGTAAGT‐3′ (reverse). Data were normalized to GAPDH.
TUNEL Assay
The in situ cell apoptosis in brain tissues was determined using the TUNEL assay kit from Boster (China). Brain tissue sections were deparaffinized in xylene, rehydrated through a graded alcohol series (95%, 90%, 80% (v/v)), digested in 20 μg/mL proteinase K solution for 20 min, and incubated with 0.3% (v/v) hydrogen peroxide in methanol for 30 min to inactivate endogenous peroxidase. After washing with sodium citrate buffer containing 0.1% (v/v) Triton X‐100 for 5 min, the sections were incubated with 50 μL TUNEL reaction mixture at 37°C for 1 h, washed with PBS, and incubated with 50 μL converter peroxidase at 37°C for 30 min. After washing again with PBS, the sections were incubated with 3,3′‐diaminobenzidine for 5–10 min and examined under a AxioVision 4AC microscope system (Carl Zeiss, Oberkochen, Germany).
Optic Nerve Crush
Two weeks after intravitreal injection of the lentiviral plasmids, the left intraorbital optic nerve was surgically exposed and crushed using forceps at 1 mm distal to the optic disk for 5 seconds with no damage to retinal blood vessels. After surgery, tobrex ophthalmic ointment was applied to prevent infection. CNTF (1 μL of 10 μg/mL in PBS; PeproTech, Rocky Hill, NJ, USA), AG490 (1 μL of 5 mM in 50% ethanol, Sigma), or vehicle control was intravitreally injected immediately following injury and 3 days later.
Traumatic Brain Injury (TBI) Model
Sprague Dawley rats were placed in a stereotaxic frame (Amscien Instruments, Richmond, VA, USA). A 3.5‐mm craniotomy was made in the right parietal bone midway between bregma and lambda with the medial edge 1 mm lateral to the midline, leaving the dura intact. Rats were impacted at 4.5 m/seconds with a 20‐ms dwell time and 1 mm depression using a 3‐mm‐diameter convex tip, mimicking a moderate TBI. Sham‐operated rats underwent the identical surgical procedures without the impact. The incision was closed with surgical staples and rats were allowed to recover. Throughout the entire procedure, body temperature was maintained at 37°C using a small animal temperature controller (Kopf Instruments, Tujunga, CA, USA). The STAT3 inhibitor AG490 (4 mg/kg, Sigma‐Aldrich) or saline was administered by intraperitoneal (IP) injections 20 min prior to the impact. Adenovirus with or without vector contain siRNA against Nrf2 or KLF4 were injected into the subarachnoid space 20 min prior to the impact.
In situ Detection of Superoxide Production
The production of superoxide (O2−) radicals was determined using hydroethidine (HEt; Molecular Probes, USA) staining as previously described 18. Briefly, HEt (1 mg/mL in 200 μL PBS) was administered intravenously 30 min before brain impact. Fluorescence emission from the oxidized HEt (excitation: 543 nm; emission: 560–590 nm) was recorded on a confocal laser fluorescence microscope (Leica, Solms, Germany).
Assessment of Surviving Cells
Cerebral cortex sections (25 μm thick) were collected from animals sacrificed 3 d after TBI. Cell nuclei were stained with DAPI (blue), and neuron density (I) of cerebral cortex was determined by confocal fluorescence imaging. The counting parameters utilized were as follows: the distance between counting frames, 75 μm; the counting frame size, 200 × 200 μm; the disector height, 10 μm; and the guard zone thickness, 2 μm. Cells with positively stained nuclei were counted as surviving cells. The number of surviving cells was calculated as the percentage of sham control.
Statistical Analysis
Data are presented as means ± SD (standard deviation). One‐way ANOVA followed by Student–Newman–Keuls post hoc tests was used to determine the significance of difference between results. Differences with a P value of <0.05 were considered statistically significant.
Results
H2O2 Induces p53‐Dependent Apoptosis in RGCs by Upregulating KLF4
The effects of H2O2 on the viability and apoptosis of RGCs in vitro were examined using the MTT assay and the annexin V‐FITC flow cytometric apoptosis assay, respectively. Treatment of RGCs with H2O2 significantly decreased the viability (Figure 1A,B) and increased the apoptosis of RGCs (Figure 1C,D) in a dose‐ and time‐dependent manner. Western blot analysis revealed that H2O2‐induced RGC apoptosis was accompanied by increased p53 phosphorylation, increased expression of Bax, a p53‐regulated pro‐apoptotic Bcl‐2 family protein, and increased caspase‐3 cleavage, as well as decreased expression of the antiapoptotic Bcl‐2 protein (Figure 2C–H). Transfection of RGCs with sip53 significantly reduced p53 expression (Figure 2J,K) and reversed H2O2‐induced changes in the levels of apoptosis‐related proteins (Bax, Bcl‐2, and cleaved caspase‐3) (Figure 2L–O). These data indicate that H2O2‐induced RGC apoptosis is p53 dependent.
Figure 1.
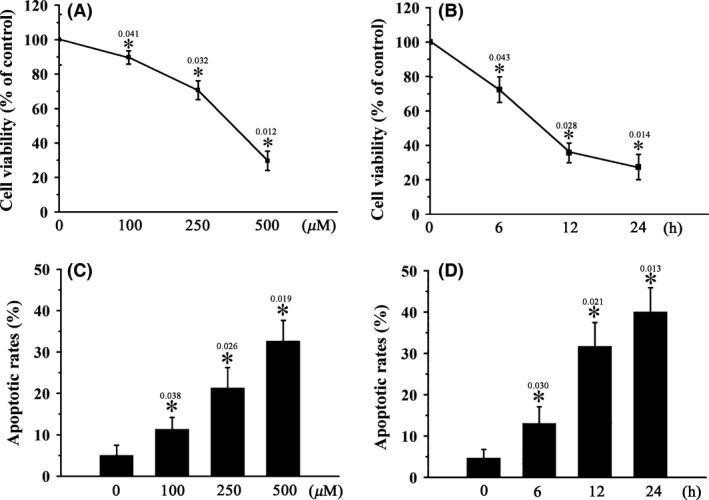
H2O2 induces RGC apoptosis in vitro. RGCs were treated with 0–500 μM H2O2 for 12 h (A, C) or 500 μM H2O2 for 0–24 h (B, D). Cell viability (A, B) and cell apoptosis (C, D) were determined using the MTT assay and the annexin V‐FITC flow cytometric apoptosis assay, respectively. n = 3, *P < 0.05 vs. control.
Figure 2.
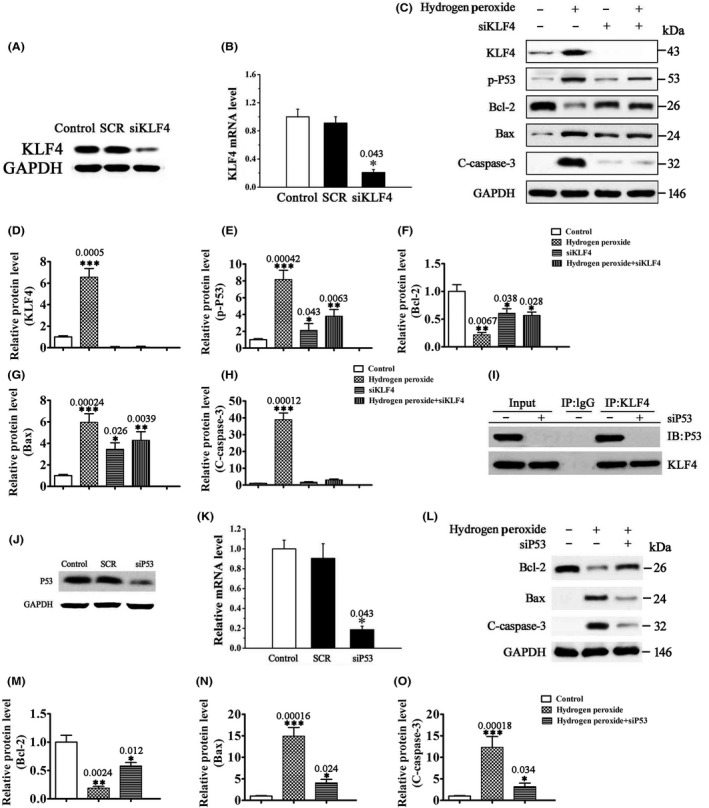
H2O2 induces p53‐dependent RGC apoptosis through upregulation of KLF4. (A, B) The mRNA (B) and protein (A) expression of KLF4 in SCR or siKLF4‐transfected RGCs by RT‐PCR and Western blot analysis, respectively. Untransfected cells were included as control. Data were normalized to GAPDH. n = 3, *P < 0.05 vs. control. (C–H) The protein levels of KLF4, p‐p53, and apoptosis‐related proteins (Bcl‐2, Bax, and cleaved caspase‐3 (C‐caspase‐3)) by Western blot analysis. RGCs transfected with siKLF4 or SCR were treated with 500 μM H2O2 or vehicle for 12 h. Data were normalized to GAPDH. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. (I) Coimmunoprecipitation analysis of the interaction between KLF4 and p53 in RGCs. IgG was used as a control. (J, K) The mRNA (K) and protein (J) expression of p53 in SCR or sip53‐transfected RGCs by RT‐PCR and Western blot analysis, respectively. Untransfected cells were included as control. Data were normalized to GAPDH. n = 3, *P < 0.05 vs. control. (L–O) The protein levels of apoptosis‐related proteins Bcl‐2, Bax, and C‐caspase‐3 by Western blot analysis. RGCs transfected with sip53 or SCR were treated with 500 μM H2O2 or vehicle for 12 h. Data were normalized to GAPDH. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Interestingly, our results showed that H2O2 also significantly induced KLF4 expression in RGCs (Figure 2C–H). KLF4 knockdown by siKLF4 prevented H2O2‐induced p53 phosphorylation, as well as changes in the levels of apoptosis‐related proteins (Figure 2A–H), indicating that KLF4 is a positive regulator of p53‐dependent apoptosis induced by H2O2. KLF4 has been reported to increase the DNA‐binding affinity of p53 through the formation of a ternary complex on DNA 12. Consistent with these previous findings, our coimmunoprecipitation assay revealed direct interaction between KLF4 and p53 in H2O2‐treated RGCs (Figure 2I), suggesting that KLF4 activates p53 by directly binding to p53.
The ROS‐p38MAPK‐p53/Nrf2 Signaling Contributes to H2O2‐Induced RGC Apoptosis
Nrf2 is a transcription factor that regulates the expression of many antioxidant proteins. It is activated by oxidative stress and protects against oxidative damage triggered by injury or inflammation 19. Previous studies have shown that p38 mitogen‐activated protein kinase (p38MAPK) is activated by oxidative stress, which in turn activates p53 and inhibits Nrf2, and this ROS‐p38‐p53/Nrf2 signaling pathway contributes to ROS‐induced cell apoptosis 20, 21. In this study, H2O2 treatment significantly increased p38MAPK expression and phosphorylation, as well as p53 and Nrf2 phosphorylation (Figure 3A–G), signaling activation of these three pathways. Blocking p38MAPK with the p38MAPK inhibitor SB203580 attenuated H2O2‐induced p53 phosphorylation and changes in the levels of apoptosis‐related proteins (Bcl‐2, Bax, and cleaved caspase‐3) (Figure 3A–L). Additionally, SB203580 further increased Nrf2 phosphorylation in H2O2‐treated RGCs (Figure 3A,F,G). As SB203580 also inhibits Raf, we repeated the experiments with a more specific p38MAPK inhibitor SB239063. SB239063 showed effects that were almost identical to SB203580 (Figure 3T). These data suggest that the Nrf2 phosphorylation is regulated by p38MAPK, and H2O2‐induced RGC apoptosis is controlled by the ROS‐p38MAPK‐p53/Nrf2 signaling pathway.
Figure 3.
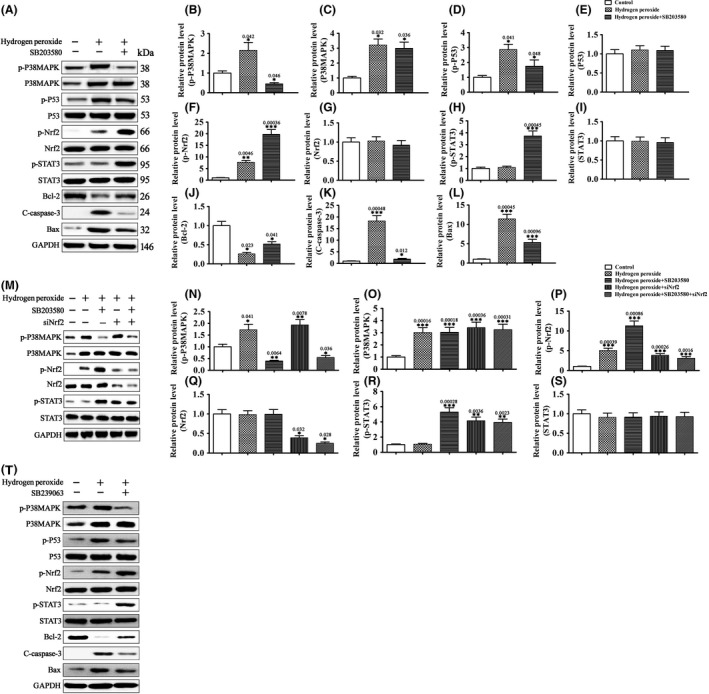
H2O2‐activated p38MAPK promotes RGC apoptosis by activating p53 and inhibiting Nrf2. (A–L and T) RGCs were pretreated with the p38MAPK inhibitor SB203580 (100 μM), SB239063 (100 μM), or vehicle for 10 min and then treated with H2O2 (500 μM) or vehicle for 30 min. The levels of p38MAPK, p‐p38MAPK, p53, p‐p53, Nrf2, p‐Nrf2, STAT3, p‐STAT3, and apoptosis‐related proteins Bcl‐2, C‐caspase‐3, and Bax were determined by Western blot analysis. Data were normalized to GAPDH. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001. NS means no significant difference (P > 0.05). (M–P) RGCs transfected with siNrf2 or SCR were pretreated with the p38MAPK inhibitor SB203580 (100 μM) or vehicle for 10 min and then treated with H2O2 (500 μM) or vehicle for 30 min. The levels of p38MAPK, p‐p38MAPK, Nrf2, p‐Nrf2, STAT3, and p‐STAT3 were determined by Western blot analysis. Data were normalized to GAPDH. n = 3, *P < 0.05. NS means no significant difference (P > 0.05).
Cytokine‐activated JAK‐STAT3 signaling promotes neuronal survival after injury 21, 22. In this study, H2O2 treatment did not change STAT3 expression or phosphorylation in RGCs (Figure 3A–L). However, blocking p38MAPK with SB203580 or silencing of Nrf2 by siRNA transfection greatly increased the level of p‐STAT3 in H2O2‐treated RGCs (Figure 3M–S). These findings suggest that H2O2‐activated p38MAPK and Nrf2 signalings negatively regulate the pro‐survival JAK‐STAT3 pathway in these cells.
H2O2‐Induced KLF4 Expression Inhibits CNTF‐Induced STAT3 Activation in RGCs
KLF4 has been reported to suppress CNTF‐induced axon regeneration of RGCs in vivo by directly binding to p‐STAT3 and inhibiting STAT3‐dependent gene expression 15, 16. In this study, we investigated the regulatory relationship between KLF4 and STAT3 in CNTF‐stimulated RGCs in vitro. Stimulation of RGCs with 100 pM CNTF for 30 min led to STAT3 activation as indicated in increased STAT3 phosphorylation and p‐STAT3 nuclear translocation (Figure 4A,C). Pretreatment with H2O2 significantly attenuated CNTF‐induced p‐STAT3 nuclear translocation (Figure 4B,C). As H2O2 induces KLF4 expression in RGCs, we thought that these inhibitory effects of H2O2 on CNTF‐induced p‐STAT3 nuclear translocation might be mediated by upregulation of KLF4. Indeed, KLF4 knockdown by siKLF4 restored CNTF‐induced p‐STAT3 nuclear translocation downregulated by H2O2 (Figure 4B,C). Similar to results from previous in vivo studies 16, our coimmunoprecipitation assay revealed direct interaction between KLF4 and p‐STAT3 in CNTF‐stimulated RGCs (Figure 4D). Interaction between KLF4 and p‐STAT3 was not detected in unstimulated cells, presumably due to the low p‐STAT3 level in these cells. Thus, similar to H2O2‐activated p38MAPK, H2O2‐induced KLF4 expression negatively regulates the pro‐survival JAK‐STAT3 pathway in H2O2‐treated RGCs. This inhibition is likely mediated by direct interaction between KLF4 and p‐STAT3, which results in reduced p‐STAT3 nuclear translocation.
Figure 4.
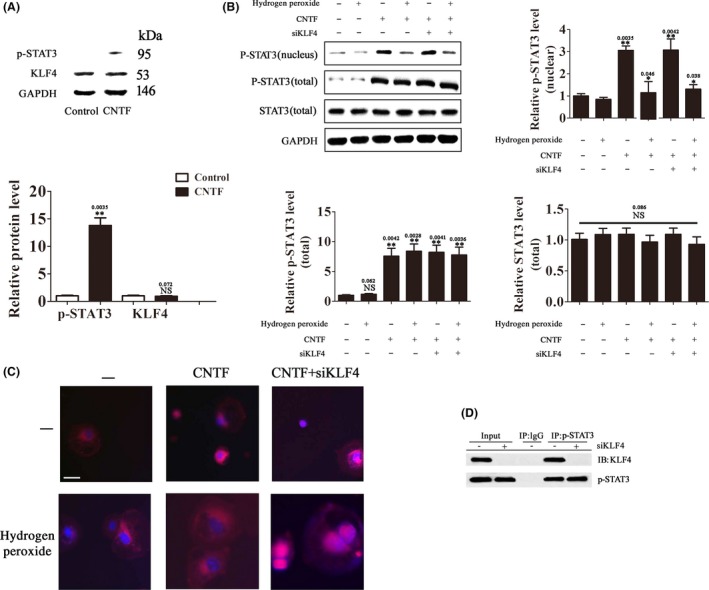
H2O2‐induced KLF4 expression inhibits CNTF‐induced STAT3 activation. (A) RGCs were treated with CNTF (100 pM) or vehicle for 30 min. The protein levels of p‐STAT3 and KLF4 were determined by Western blot analysis. Data were normalized to GAPDH. n = 3, *P < 0.05. NS means no significant difference (P > 0.05). (B, C) RGCs transfected with siKLF4 or SCR were treated with H2O2 (500 μM) or vehicle for 30 min and then with CNTF (100 pM) or vehicle for 30 min. (B) The protein levels of total p‐STAT3, nuclear p‐STAT3, and total STAT3 were determined by Western blot analysis. Data were normalized to GAPDH. n = 3, *P < 0.05, **P < 0.01 vs. control. NS means no significant difference (P > 0.05). (C) Nuclear translocation of p‐STAT3 was examined by confocal fluorescence microscopy. The images are representatives of more than 50 cells examined. Scale bar = 20 μm. (D) Coimmunoprecipitation analysis of the interaction between KLF4 and p‐STAT3 in CNTF‐treated RGCs (100 pM for 30 min).
KLF4 Overexpression Induces RGC Apoptosis
Our results above have shown that H2O2‐induced KLF4 expression activates the pro‐apoptotic p53 signaling and inhibits CNTF‐induced pro‐survival STAT3 signaling in RGCs. To investigate the role of KLF4 in CNTF‐stimulated cells, we generated RGCs stably expressing KLF4 by lentiviral transfection (Figure 5A,B). CNTF stimulation (10 μg/mL, 24 h) alone had no significant effects on p53 phosphorylation or cell apoptosis; however, KLF4 overexpression significantly increased p53 phosphorylation and apoptosis in CNTF‐stimulated cells (Figure 5C–I). Compared with KLF4 overexpression, the JAK2 inhibitor AG490 induced a higher rate of cell apoptosis with no significant effect on p53‐dependent apoptotic pathway (Figure 5C–F). The pro‐apoptotic effects of AG490 were presumably mediated by inhibition of the pro‐survival JAK‐STAT signaling. Indeed, KLF4 overexpression and AG490 worked additively to enhance apoptosis in CNTF‐treated RGCs (Figure 5C–I), presumably through simultaneous p53 activation and JAK‐STAT3 inhibition.
Figure 5.
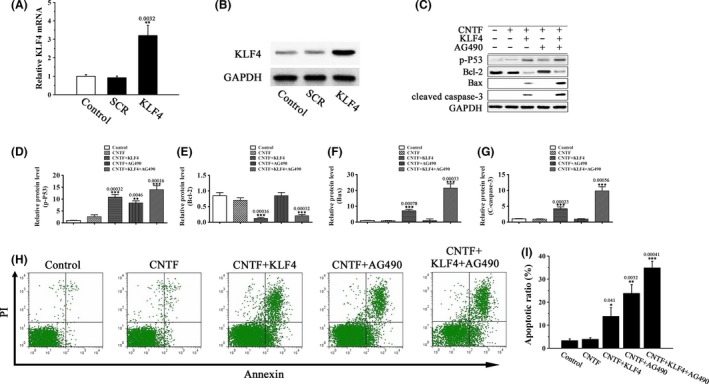
KLF4 overexpression induces RGC apoptosis. (A and B) The mRNA (A) and protein (B) levels of KLF4 in KLF4 or vector‐transfected RGCs by RT‐PCR and Western blot analysis, respectively. Untransfected cells were included as control. Data were normalized to GAPDH. n = 3, **P < 0.01 vs. control. (C–I) KLF4 or vector‐transfected RGCs were pretreated with the JAK2 inhibitor AG490 (0.1 μM) or vehicle for 5 h and then treated with 10 μg/mL CNTF or vehicle for 24 h. (C–G) The levels of p‐p53 and the apoptosis‐related proteins Bcl‐2, Bax, and C‐caspase‐3 were determined by Western blot analysis. Data were normalized to GAPDH. n = 3, **P < 0.01, ***P < 0.001 vs. control. (H, I) Cell apoptosis was detected by flow cytometry using annexin V‐FITC/PI double staining. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
KLF4 Knockdown Promotes Axon Regeneration in vivo
Previous studies have shown that KLF4 deletion in vivo enhances CNTF‐induced axon repair of RGCs via JAK‐STAT3 signaling 16. In this study, we investigated the effects of KLF4 knockdown on axon regeneration of RGCs after optic nerve crush. Superoxide levels in RGCs increased with time after injury, reaching a peak value that was about fourfold of the uninjured cells 3 days later (Figure 6A). Intravitreal injection of CNTF (1 μL of 10 μg/mL) immediately after the injury and 3 days later activated STAT3 phosphorylation and significantly increased the number of regenerating fibers (Figure 6B–D). The JAK2 inhibitor AG490 completely blocked CNTF‐induced axon regrowth (Figure 6C,D), underscoring the key role of JAK‐STAT signaling in this axon repair process. Similar to KLF4 deletion 16, intravitreal injection of siKLF4 lentiviral plasmids 2 weeks prior to optic nerve crush dramatically enhanced axon regeneration in CNTF‐treated retina (Figure 6C, 6D). Compared with CNTF‐treated control (vector‐injected) RGCs, KLF4 knockdown led to more than twofold increase in the number of regenerating fibers at 1.5 mm distal to the crush site (Figure 6D). Our results provided further evidence that KLF4 functions as a negative regulator of axon regeneration in vivo.
Figure 6.
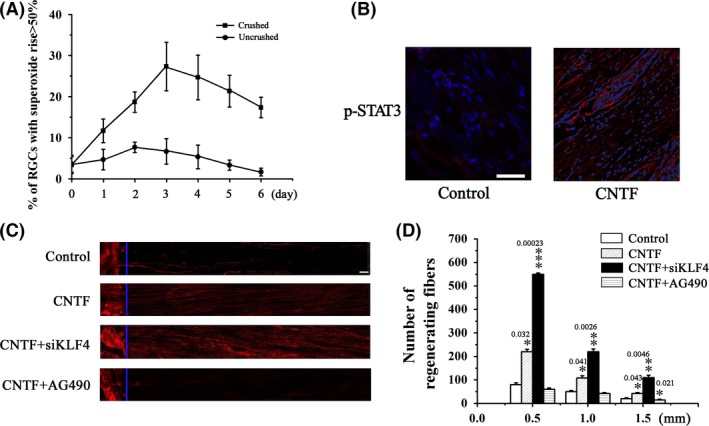
Axotomy of RGCs by optic nerve crush induces oxidative stress. (A) RGCs were dissociated from the crushed retina at the indicated time postinjury. Superoxide levels were determined using HEt staining 1 h after dissociation. RGCs dissociated from the uninjured retina were used as control. n = 5. (B) CNTF (1 μL of 10 μg/mL) or vehicle was intravitreally injected immediately after injury and 3 days later. Retina was dissected 24 h after the second injection and p‐STAT3 was detected by immunohistochemical analysis. (C, D) Immediately following the injury, CNTF (1 μL of 10 μg/mL), CNTF (1 μL of 10 μg/mL) in combination with siKLF4 (100 μL of 108 TU/mL), CNTF (1 μL of 10 μg/mL) in combination with AG490 (30 μg), or vehicle alone was intravitreally injected. Animals were sacrificed on day 21. Optic nerves were dissected from the injured eye. Axon regeneration was assessed by immunohistochemical analysis based on GAP43 staining. (C) Representative fluorescence images of optic fibers. Scale = 100 μm. (D) Numbers of regenerating axons at the indicated distance distal to the crush sites. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
KLF4 Knockdown Prevents Neuronal Damage after TBI
Our results have shown that KLF4 positively regulates p53‐dependent apoptosis of RGCs in vitro while negatively regulates JAK/STAT3‐mediated axon regeneration of RGCs in vivo. In these in vitro and in vivo studies, RGCs were exposed to elevated ROS levels. Considering that ROS production, p53‐dependent neuronal apoptosis, and JAK/STAT‐mediated neurological recovery are key components of secondary injury mechanisms of TBI, we speculated that KLF4 might play a role in the pathophysiology of TBI. To test this hypothesis, we established a brain impact model that mimics moderate TBI. In this model, brain impact led to a rapid increase in superoxide levels in cerebral cortex and hippocampus (Figure 7A–D), along with significantly increased levels of p‐p53, Bax, and cleaved caspase‐3, decreased levels of Bcl‐2, and increased levels of the axonal injury marker β‐APP (Figure 7E). Consistent with these changes in protein levels, increased neuron apoptosis and axon injury, as well as decreased neuron density, were detected in cerebral cortex and hippocampus after the impact (Figure 8R–V). These impact‐induced neuronal damages were accompanied by increased KLF4 expression, as well as increased p38MAPK phosphorylation in the same brain regions (Figure 8A,B, and 8K). Injection of siKLF4 prior to brain impact significantly attenuated impact‐induced neuron apoptosis and axon injury and restored neuron density in cerebral cortex and hippocampus (Figure 8R–V). These neuroprotective effects of siKLF4 were accompanied by decreased p‐p53 and increased p‐STAT3 levels (Figure 8A,C,G), signaling the involvement of these two pathways. Injection of the JAK2 inhibitor AG490 completely blocked siKLF4‐induced axon regeneration and neuron repair (Figure 8R,S,U,V), and these effects were accompanied by decreased STAT3 phosphorylation (Figure 8G). Furthermore, AG490 partially reversed siKLF4‐induced inhibition of neuron apoptosis after the impact (Figure 8T), implying that the antiapoptotic effects of siKLF4 were partially mediated by activation of JAK‐STAT3. Collectively, these results suggest that impact‐induced KLF4 expression positively regulates neuron apoptosis through both p53 and JAK‐STAT3 and negatively regulates axon repair through JAK‐STAT3.
Figure 7.
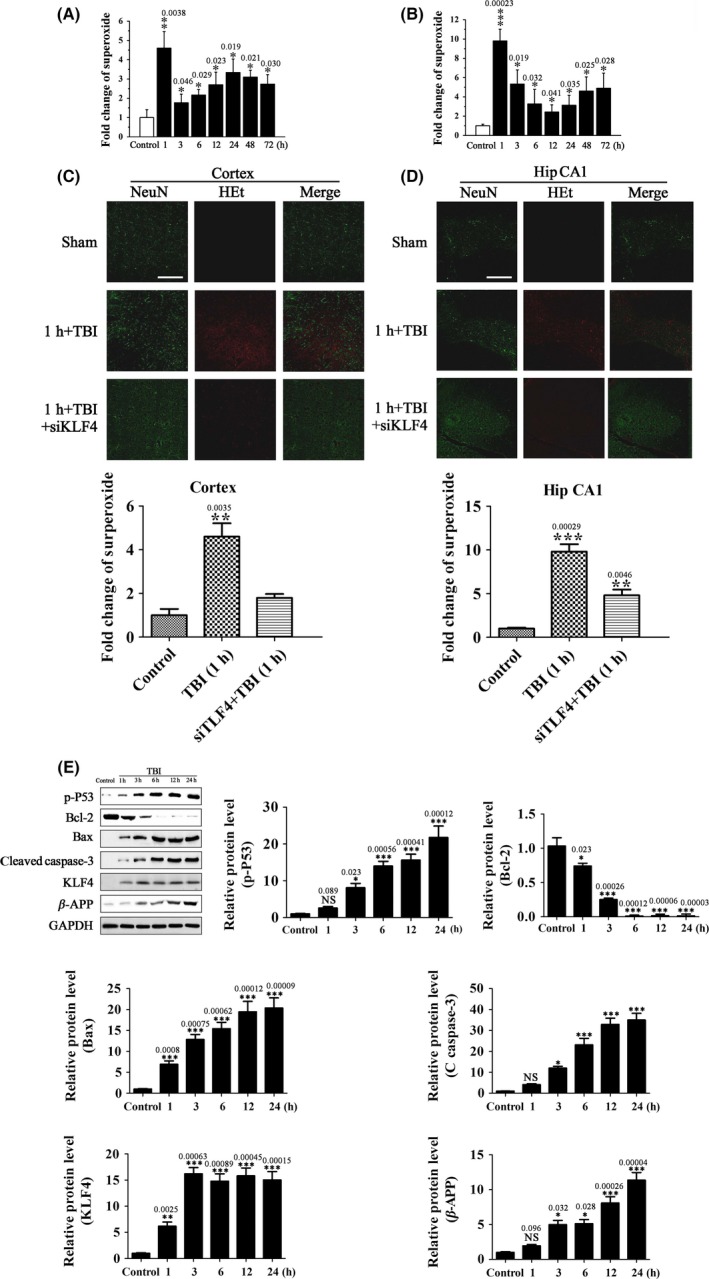
KLF4 knockdown attenuates TBI‐induced oxidative stress and protein expression associated with apoptosis and axonal injury. (A, B) Superoxide levels in cerebral cortex (A) and hippocampus (B) determined by immunohistochemical analysis using HEt staining at the indicated time after brain impact. Sham‐operated animals were used as control. Data are presented as fold change over control. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. (C, D) Representative confocal fluorescence images of cortex (C) and the medial hippocampal CA1 region (Hip CA1) (D) 1 h after brain impact. SiKLF4 was injected into the subarachnoid space 20 min prior to the impact. Tissue samples were stained with HEt (oxidized HEt, red) and NeuN (green). Scale = 100 μm. The relative superoxide levels in cerebral cortex (C) and hippocampus (D) were determined. n = 5, **P < 0.01, ***P < 0.001 vs. control. (E) The protein levels of KLF4, p‐p53, the apoptosis‐related proteins, Bcl‐2, Bax, and C‐caspase‐3, and the axonal injury marker β‐APP in cerebral cortex by Western blot analysis at the indicated time after brain impact. Sham‐operated animals were used as control. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Figure 8.
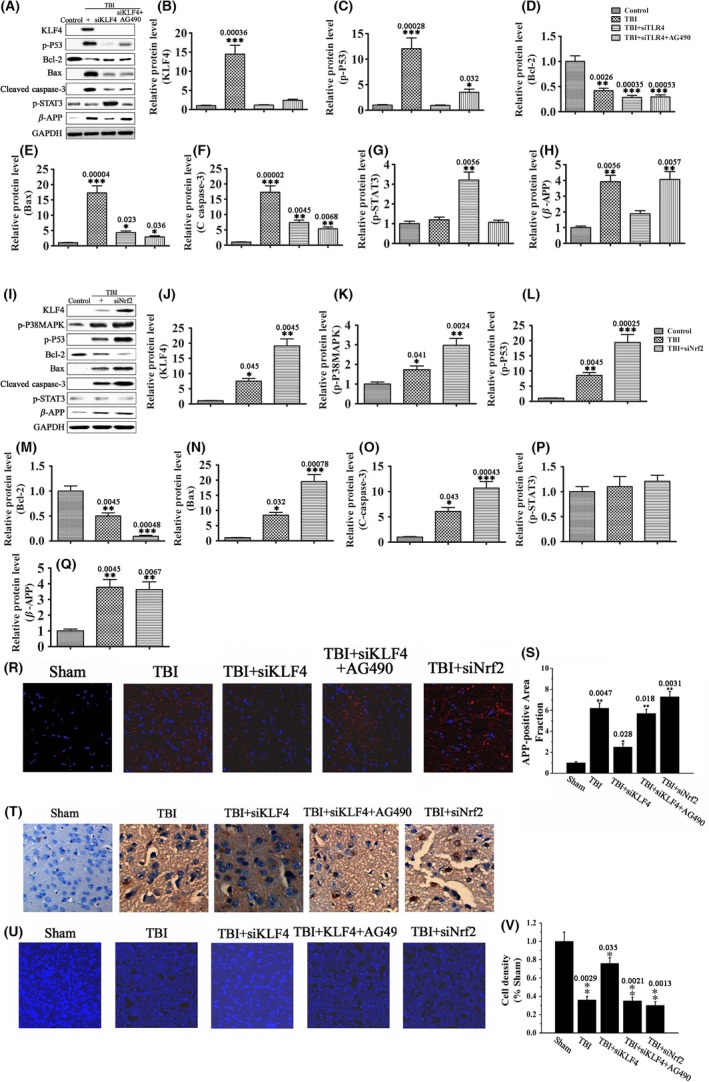
KLF4 knockdown attenuates TBI‐induced neuronal damage through p53 and JAK‐STAT3. SiKLF4, siNrf2, SCR, and AG490, either alone or in combination as indicated, were injected into the subarachnoid space 20 min prior to brain impact. (A–Q) The protein levels of KLF4, p‐p38MAPK, p‐p53, and the apoptosis‐related proteins Bcl‐2, Bax, and C‐caspase‐3, the axonal injury marker β‐APP, and p‐STAT3 in cerebral cortex and hippocampus 72 h after brain impact by Western blot analysis. Data were normalized to GAPDH. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. (R, S) Immunohistochemical analysis of β‐APP levels in cerebral cortex and hippocampus 3 d after brain impact. Nuclei were stained with DAPI (blue). n = 6, *P < 0.05, **P < 0.01 vs. sham. Scale = 50 μm. (T) In situ neuron apoptosis in cerebral cortex 3 d after brain impact by TUNEL staining. Scale = 20 μm. (U, V) Confocal images (U) and neuron density (V) of cerebral cortex 3 d after brain impact. Nuclei were stained with DAPI (blue). n = 5–7, *P < 0.05, **P < 0.01 vs. sham. Scale = 20 μm.
In contrast to siKLF4, injection of siNrf2 exacerbated impact‐induced neuronal damages (Figure 8R–V). Moreover, siNrf2 enhanced impact‐induced changes in KLF4, p‐p38MAPK, p‐p53, and the apoptosis‐related proteins Bcl‐2, Bax, and cleaved caspase‐3, while showing no significant effect on p‐STAT3 and β‐APP (Figure 8I–Q). KLF4 induction by siNrf2 was likely mediated by exacerbated oxidative stress due to downregulation of antioxidant gene expression. These data suggest that the neurotoxic effects of siNrf2 were at least partially mediated by upregulation of KLF4.
Discussion
TBI is induced by complex primary and secondary mechanisms that give rise to cell death, inflammation, and neurological dysfunction. The p53 and JAK‐STAT signaling, which are activated after TBI, contribute to neuronal damage and cognitive function recovery, respectively 7, 9. Understanding the molecular mechanisms that regulate the activation of these pathways can guide future research to develop therapeutic agents for TBI.
The transcription factor KLF4 is associated with both tumor suppression and oncogenesis. In normal cells, KLF4 inhibits proliferation by increasing the DNA‐binding affinity of p53 through the formation of a ternary complex on DNA 12. Conversely, KLF4 directly binds to the p53 promoter and suppresses p53 expression, thereby causing resistance to DNA‐damage‐induced apoptosis and allowing for oncogenic transformation 11. KLF4 also contributes to the development of vascular diseases by activating or repressing gene transcription via interacting with a variety of protein partners. KLF4 interacts with serum response factor, ELK1, and histone deacetylases to inhibit smooth muscle cell (SMC) differentiation 23, 24. KLF4 also inhibits SMC proliferation by associating with p53 and enhancing its transcriptional activity 25, 26. In addition, KLF4 regulates SMC calcification by interacting with RUNX2 27, 28. Furthermore, KLF4 plays an antiinflammatory role in endothelial cells (ECs). KLF4 attenuates inflammation‐related induction of the VCAM1 gene in cultured ECs by binding to p65 29, 30. EC‐specific KLF4 deletion increases while its overexpression decreases atherosclerotic lesion formation in Apoe background mice fed a high‐fat diet 29. These findings on KLF4‐mediated molecular mechanisms may contribute to the advancement of therapeutic strategies for treating cancer and vascular diseases.
Recently, KLF4 has been reported to negatively regulate axon regeneration of injured RGCs through inhibition of JAK/STAT signaling 16. In this study, we investigated the function of KLF4 in in vitro and in vivo models that mimics TBI. ROS generation in injured neural tissues is a hallmark feature of TBI; thus, we used H2O2‐treated RGCs for our in vitro studies. P53‐dependent apoptosis is a leading cause of neuronal loss after TBI 7. In this study, we found that H2O2 induces p53‐dependent apoptosis in RGCs through upregulation of KLF4. Previous studies have shown that KLF4 can either suppress the expression of p53 by directly acting on its promoter or activate p53‐dependent gene expression by directly binding to p53 and increasing its DNA‐binding affinity 11, 12. We detected direct interaction between KLF4 and p53 in H2O2‐treated RGCs; thus, KLF4 likely activates p53 signaling by directly binding to p53 in these cells. Besides, we found that the ROS‐p38MAPK‐p53/Nrf2 signaling pathway, which has been reported to control ROS‐induced cell apoptosis 20, 21, is involved in H2O2‐induced RGC apoptosis. Additionally, p38MAPK and Nrf2 are activated by H2O2 and negatively regulate the pro‐survival STAT3 signaling in these cells.
Cytokines such as CNTF promote survival and axon regeneration of injured RGCs via activation of JAK‐STAT3 signaling 31. KLF4 has been shown to directly bind to p‐STAT3 and inhibit STAT3‐dependent gene expression in CNTF‐stimulated retinal cells in vivo 16. KLF4 deletion drastically enhances CNTF‐induced axon repair in retinas after optic nerve crush 16. We found that KLF4 also directly binds to p‐STAT3 in RGCs in vitro upon CNTF stimulation. Additionally, KLF4 induced by H2O2 inhibits CNTF‐induced STAT3 activation in RGCs. In our in vivo optic nerve crush study, we detected elevated superoxide levels in crushed retinas. Importantly, KLF4 knockdown significantly enhances CNTF‐induced axon regeneration of RGCs in injured retinas, although to a lesser extent compared with KLF4 deletion 16.
Collectively, these findings demonstrate that KLF4 promotes ROS‐induced RGC apoptosis in vitro by activating p53 signaling and suppresses cytokine‐induced axon repair in vivo by inhibiting JAK‐STAT3 signaling. The complex signaling networks involved are depicted in Figure 9. Finally, we found that KLF4 knockdown prevents TBI‐induced neuronal damage via p53 and JAK‐STAT pathways in a murine TBI model. These findings indicate that KLF4 is involved in the pathophysiology of TBI. Blocking KLF4 with transgenic knockdown or agents that disrupt its binding with p53 and/or p‐STAT3 may prevent neuron loss and promote functional recovery after TBI.
Figure 9.
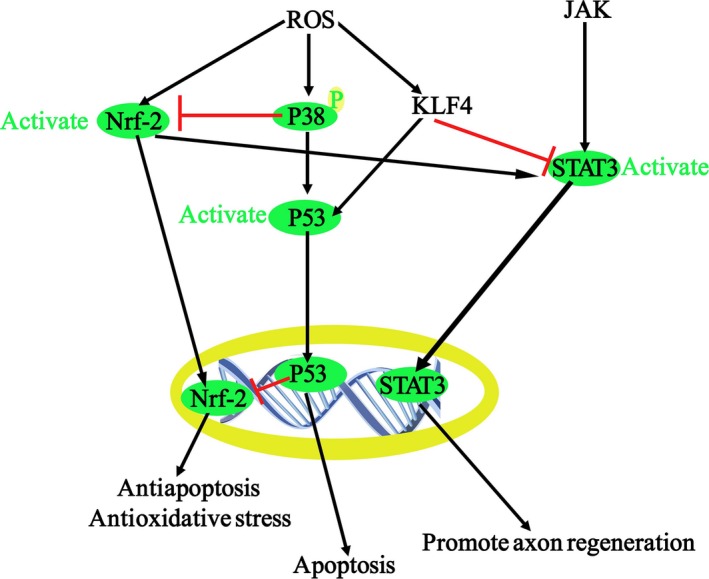
Molecular mechanisms of apoptosis and axon regeneration of RGCs under oxidative stress.
There is currently no therapeutic drug treatment for TBI despite decades of clinical development. Mechanisms controlling pathophysiological processes of secondary brain injury are extremely complex. Combinatorial treatment that target complementary mechanisms might be a potential therapeutic strategy. Combining KLF4 modulators with antioxidants or antiinflammatory agents might be a practical approach to improve clinical outcome of TBI.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the Youth Fund of the National Natural Science Foundation of China (81201979).
The first two authors contributed equally to this work.
References
- 1. CDC . Injury Prevention & Control: Traumatic Brain Injury. http://wwwcdcgov/traumaticbraininjury/. 2014.
- 2. Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol 2004;14:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee S, Park S, Won J, Lee SR, Chang KT, Hong Y. The incremental induction of neuroprotective properties by multiple therapeutic strategies for primary and secondary neural injury. Int J Mol Sci 2015;16:19657–19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cornelius C, Crupi R, Calabrese V, et al. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid Redox Signal 2013;19:836–853. [DOI] [PubMed] [Google Scholar]
- 5. Culmsee C, Zhu X, Yu QS, et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta‐peptide. J Neurochem 2001;77:220–228. [DOI] [PubMed] [Google Scholar]
- 6. Napieralski JA, Raghupathi R, McIntosh TK. The tumor‐suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain Res Mol Brain Res 1999;71:78–86. [DOI] [PubMed] [Google Scholar]
- 7. Rachmany L, Tweedie D, Rubovitch V, et al. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53‐dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT‐alpha. PLoS One 2013;8:e79837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oliva AA Jr, Kang Y, Sanchez‐Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem 2012;120:710–720. [DOI] [PubMed] [Google Scholar]
- 9. Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett 2011;498:147–152. [DOI] [PubMed] [Google Scholar]
- 10. Rowland BD, Peeper DS. KLF4, p21 and context‐dependent opposing forces in cancer. Nat Rev Cancer 2006;6:11–23. [DOI] [PubMed] [Google Scholar]
- 11. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context‐dependent oncogene. Nat Cell Biol 2005;7:1074–1082. [DOI] [PubMed] [Google Scholar]
- 12. Brandt T, Townsley FM, Teufel DP, Freund SM, Veprintsev DB. Molecular basis for modulation of the p53 target selectivity by KLF4. PLoS One 2012;7:e48252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qin S, Liu M, Niu W, Zhang CL. Dysregulation of Kruppel‐like factor 4 during brain development leads to hydrocephalus in mice. Proc Natl Acad Sci U S A 2011;108:21117–21121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qin S, Zhang CL. Role of Kruppel‐like factor 4 in neurogenesis and radial neuronal migration in the developing cerebral cortex. Mol Cell Biol 2012;32:4297–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore DL, Blackmore MG, Hu Y, et al. KLF family members regulate intrinsic axon regeneration ability. Science 2009;326:298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin S, Zou Y, Zhang CL. Cross‐talk between KLF4 and STAT3 regulates axon regeneration. Nat Commun 2013;4:2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim SJ, Ko JH, Yun JH, et al. Stanniocalcin‐1 protects retinal ganglion cells by inhibiting apoptosis and oxidative damage. PLoS One 2013;8:e63749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang QG, Laird MD, Han D, et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One 2012;7:e34504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol Aspects Med 2011;32:234–246. [DOI] [PubMed] [Google Scholar]
- 20. Song Y, Li X, Li Y, et al. Non‐esterified fatty acids activate the ROS‐p38‐p53/Nrf2 signaling pathway to induce bovine hepatocyte apoptosis in vitro. Apoptosis 2014;19:984–997. [DOI] [PubMed] [Google Scholar]
- 21. Cardaci S, Filomeni G, Rotilio G, Ciriolo MR. p38(MAPK)/p53 signalling axis mediates neuronal apoptosis in response to tetrahydrobiopterin‐induced oxidative stress and glucose uptake inhibition: Implication for neurodegeneration. Biochem J 2010;430:439–451. [DOI] [PubMed] [Google Scholar]
- 22. Smith PD, Sun F, Park KK, et al. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 2009;64:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem 2005;280:9719–9727. [DOI] [PubMed] [Google Scholar]
- 24. Yoshida T, Gan Q, Owens GK. Kruppel‐like factor 4, Elk‐1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol 2008;295:C1175–C1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang W, Geiman DE, Shields JM, et al. The gut‐enriched Kruppel‐like factor (Kruppel‐like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem 2000;275:18391–18398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wassmann S, Wassmann K, Jung A, et al. Induction of p53 by GKLF is essential for inhibition of proliferation of vascular smooth muscle cells. J Mol Cell Cardiol 2007;43:301–307. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida T, Yamashita M, Hayashi M. Kruppel‐like factor 4 contributes to high phosphate‐induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem 2012;287:25706–25714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michikami I, Fukushi T, Tanaka M, et al. Kruppel‐like factor 4 regulates membranous and endochondral ossification. Exp Cell Res 2012;318:311–325. [DOI] [PubMed] [Google Scholar]
- 29. Zhou G, Hamik A, Nayak L, et al. Endothelial Kruppel‐like factor 4 protects against atherothrombosis in mice. J Clin Invest 2012;122:4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshida T, Yamashita M, Horimai C, Hayashi M. Deletion of Kruppel‐like factor 4 in endothelial and hematopoietic cells enhances neointimal formation following vascular injury. J Am Heart Assoc 2014;3:e000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leibinger M, Muller A, Andreadaki A, Hauk TG, Kirsch M, Fischer D. Neuroprotective and axon growth‐promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. J Neurosci 2009;29:14334–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]