

Summary
Aims
Sarsasapogenin has been reported to improve dementia symptoms somehow, probably through modulating the function of cholinergic system, suppressing neurofibrillary tangles, and inhibiting inflammation. However, the role of sarsasapogenin in response to beta‐amyloid (Aβ) remains to be delineated. This study aimed to determine the therapeutic effect of sarsasapogenin‐13 (AA13, a sarsasapogenin derivative) on learning and memory impairments in Aβ‐injected mice, as well as the role of AA13 in neuroglia‐mediated antiinflammation and Aβ clearance.
Methods
Focusing on the role of AA13 in regulating glial responses to Aβ, we conducted behavioral, morphological, and protein expression studies to explore the effects of AA13 on Aβ clearance and inflammatory regulation.
Results
The results indicated that oral administration of AA13 attenuated the memory deficits of intracerebroventricular (i.c.v.) Aβ‐injected mice; also, AA13 protected neuroglial cells against Aβ‐induced cytotoxicity. The further mechanical studies demonstrated that AA13 reversed the upregulation of proinflammatory M1 markers and increased the expression of antiinflammatory M2 markers in Aβ‐treated cells. Furthermore, AA13 facilitated Aβ clearance through promoting Aβ phagocytosis and degradation. AA13 modulated the expression of fatty acid translocase (CD36), insulin‐degrading enzyme (IDE), neprilysin (NEP), and endothelin‐converting enzyme (ECE) in neuroglia.
Conclusion
The present study indicated that the neuroprotective effect of AA13 might relate to its modulatory effects on microglia activation state, phagocytic ability, and expression of Aβ‐degrading enzymes, which makes it a promising therapeutic agent in the early stage of Alzheimer's disease (AD).
Keywords: antiinflammation, beta‐amyloid clearance, neuroglia, phagocytosis, sarsasapogenin‐AA13
1. Introduction
Alzheimer's disease (AD), an irreversible neurodegenerative disorder, is mainly characterized by the extracellular deposition of beta‐amyloid peptides (Aβ) and the intracellular accumulation of hyperphosphorylated tau protein. The pathological development of AD is accompanied with neuronal and synaptic loss, which finally leads to cognitive decline and memory loss.1, 2
The Aβ hypothesis proposes that an imbalance between Aβ production and clearance results in Aβ accumulation and deposition in the brain, which finally causes sporadic AD.3 With regard to inflammation hypothesis, a chronic neuroinflammatory response to Aβ deposits exacerbates the neurodegenerative pathology.4 Emerging evidences showed that activated microglia can be divided into “classic” or “alternative” activated populations, as also known as M1 and M2 microglia. M1 microglia secretes abundant proinflammatory cytokines, which can lead to tissue injury and contribute to pathogenesis, whereas M2 microglia tend to produce antiinflammatory factors and neurotrophins, and promote angiogenesis, tissue remodeling, and repair.5 Therefore, somehow enhancing the glial ability to eliminate Aβ and shifting glia from M1 to M2 phenotype are extremely interested.
Neuroglia, mainly referring to microglia and astrocytes, play an essential role in maintaining homeostasis and performing defense in the central nervous system (CNS). Previous studies have demonstrated that Aβ plaques are surrounded by abundant reactive glia.6, 7 Several mechanisms are involved in the initial defense against toxic Aβ accumulation, including enzymatic degradation, receptor‐mediated pathway and phagocytosis. Primary astrocytes were reported to bind and ingest Aβ in vitro.8 Thereinto, scavenger receptors (SRs), macrophage scavenger receptor 1 (MSR1), fatty acid translocase (CD36), and scavenger receptor class B member 1 (SCARB1) closely correlate with Aβ recognition and assimilation.9, 10 Besides, the high expression of Aβ‐degrading proteases, such as neprilysin (NEP), endothelin‐converting enzyme (ECE), and insulin‐degrading enzyme (IDE), effectively promotes the Aβ removal.11, 12 Preceding studies have found the deficient microglial elimination of toxic Aβ 13 and high level of proinflammatory factors in AD.14 The modest activated microglia produce inflammatory cytokines and chemokines and also raise phagocytic efficiency.15, 16 However, excessive activation of microglia conversely increases Aβ accumulation and injures neurons. The cytokines induced by Aβ in turn reduce the expression of Aβ‐degrading enzymes and Aβ‐binding receptors, which do harm to the brain function.17, 18 Taken together, the proper therapeutic method for AD is to alleviate the proinflammatory response and enhance elimination of Aβ.
Intracerebroventricular (i.c.v.) injection of Aβ 1‐42 in mice is a common model to mimic pathological and behavioral characterizations of AD. Although the model fails to induce amyloid plaque in the brain, the i.c.v. injection model enables estimation of drugs targeting Aβ 1‐42, inflammatory reactions, as well as learning and memory functions.19
Rhizoma anemarrhenae is isolated from the rhizome of Anemarrhena asphodeloides Bge. The saponins possess ample pharmacological activities, such as antiviral, antiinflammatory, antidiabetic, and antifungal.20, 21, 22, 23 Early investigation has discovered that timosaponin AIII ameliorates learning and memory deficits by inhibiting the expression of cholinesterase (AChE) and inflammatory cytokines.24, 25 Recent research showed that the “sarsasapogenin‐aglyconed” timosaponins played a role in lowering Aβ 1‐42 production and stimulating neurite outgrowth.26
Sarsasapogenin‐AA13 (AA13) was found to inhibit inflammation 27 and stimulate the secretion of neurotrophic factors (data not shown) in our previous study. This study aimed to determine the therapeutic effect of AA13 on learning and memory impairments in Aβ‐injected mice, as well as the role of AA13 in neuroglia‐mediated antiinflammation and Aβ clearance.
2. Materials and Methods
2.1. Materials
Dulbecco's modified Eagle's medium and F12 HAMS medium were purchased from Hyclone Laboratories (Logan, UT, USA). Fetal bovine serum (FBS) and GlutaMAX medium were from Gibco BRL (Grand Island, NY, USA). Dimethylsulfoxide (DMSO) and paraformaldehyde (PFA) were from Sigma Aldrich (St Louis, MO, USA). 3‐(4, 5‐Dimethythiazol‐2‐yl)‐2, 5‐diphenyl‐tetrazolium bromide (MTT), and trypsin were purchased from Sangon Biotech (Shanghai, China). EDTA‐2Na and glycerinum were from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Bovine serum albumin (BSA) was purchased from Aladdin Industrial Inc (Shanghai, China).
AA13, a derivative of natural compound sarsasapogenin, was synthesized by Lei Ma's laboratory from School of Pharmacy, East China University of Science and Technology (Figure 1A).28 A 5 mmol/L of stock solution of AA13 was dissolved in DMSO and stored at −80°C. The stock solution was diluted to the final concentrations before use, making sure that no detectable effect of DMSO was found in the experiments.
Figure 1.
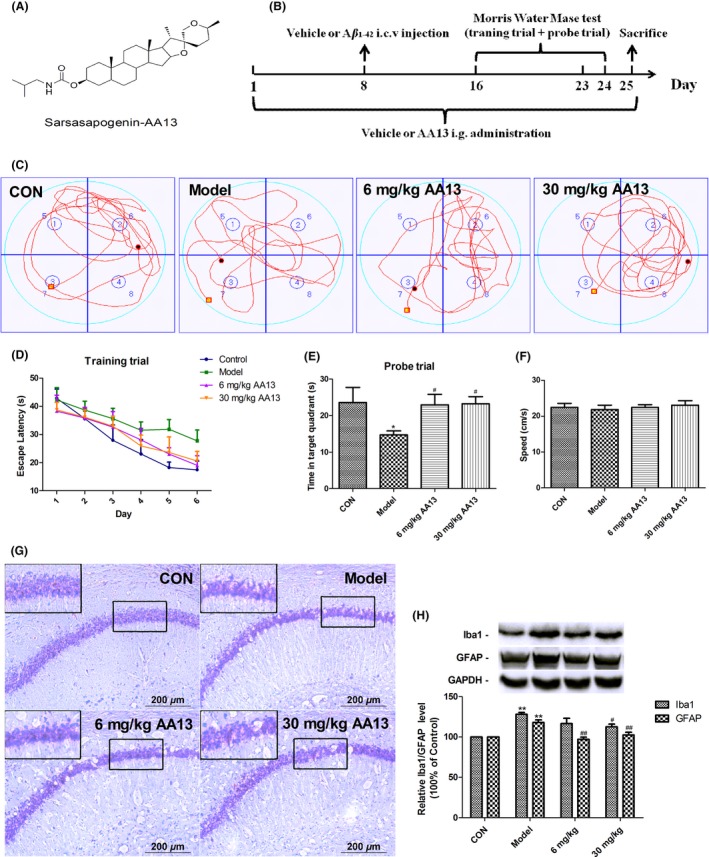
AA13 attenuated learning and memory impairments in Aβ 1‐42‐injected mice. (A) Chemical structure of sarsasapogenin‐AA13. (B) Experimental protocol diagram. Mice were randomly divided into four groups, namely Sham‐operated (vehicle+vehicle), Aβ 1‐42 model (vehicle+Aβ 1‐42), AA13 low‐dose group (6 mg/kg AA13+Aβ 1‐42), and AA13 high‐dose group (30 mg/kg AA13+Aβ 1‐42). The mice were orally administrated once a day for 7 d followed by Aβ 1‐42 i.c.v. injection surgery; then, morris water maze test was conducted. (C) The typical trajectory of each group in probe trial. (D) Time before reaching the hidden platform during 6 days of training trial. (E) Time spent in target quadrant in probe test. (F) The mean speed during the water maze test. (G) Nissl staining of hippocampal CA1 region of mice. (H) The alteration of Iba1 and GFAP protein expression in the brain. *P<.05 compared with control group. #P<.05 compared with model group . **P<.01 compared with control group. ##P<.01 compared with model group
2.2. Animals and drug treatments
Male ICR mice (30‐40 g, 8 weeks old) were purchased from Shanghai Sipper‐bk Laboratory Animal Co., LTD (Shanghai, China). Animals were raised at a temperature of 20‐25°C with relative humidity of 50%‐60% and a 12‐h light/dark cycle with free access to water and food. All experiments were performed during light phase on the day. The procedures of this study were conducted according to the guidelines of Care and Use of Laboratory Animals of China for animal experimentation.
Mice were randomly divided into four groups (n=8 animals per group): (1) sham‐operated (vehicle+vehicle); (2) Aβ 1‐42 model (vehicle+Aβ 1‐42): (3) AA13 low‐dose group (6 mg/kg AA13+Aβ 1‐42); and (4) AA13 high‐dose group (30 mg/kg AA13+Aβ 1‐42). The mice were orally administrated with sterile saline (groups 1, 2) and AA13 (groups 3, 4) once a day for 7 days. Then, mice received an i.c.v. injection of sterile saline (group 1) and Aβ 1‐42 (group 2, 3, 4). At the seventh day after the surgery, mice were subjected to Morris water maze test. Intragastric administration was continued until the mice were sacrificed.
2.3. Intracerebroventricular (i.c.v.) injection of Aβ 1‐42
The surgery of intracerebroventricular injection was performed as the procedure of Prakash et al. with little modification.29 Briefly, mice were anesthetized with isoflurane inhalation at an induced concentration of 2%‐2.5%. Then, the mice were placed in a stereotaxic apparatus (RWD Life Science, Shenzhen, China), and isoflurane in a maintained concentration of 1%‐1.5% was used to maintain anesthesia. After disinfecting with alcohol, the scalp was shaved to expose the skull. Cranial drill was inserted in the skull at the accurate location on the right over the lateral ventricle; 1 mg/mL of Aβ 1‐42 or 0.9% NaCl at a volume of 3 μL was injected gradually (1 μL/min) at the following coordinate from bregma: mediolateral (ML)=1 mm; anteroposterior (AP)=0.5 mm; and dorsoventral (DV)=2.4 mm. After injection, the skin was sutured followed by the application of antibiotics.
2.4. Morris water maze test
The apparatus, a circular tank (150 cm in diameter and 60 cm in height), was filled with water to a depth of 40 cm. The tank was conceptually divided into four quadrants. Black floating granules were tiled on the surface of water, and an escape platform invisible to the mice was placed 1 cm below the water level. The pool was placed in a soundproof and dim room with some brightly colored cues, by which the mice could fix the spatial orientation. The water maze test was started on the day 16 (Figure 1B), and the test consisted of two parts, seven consecutive days’ training trials and the following probe trial. Each mouse was trained to find the platform, with four trials a day starting at different entering quadrants. For training trial, mice were put into water to find the platform, which was visible on the first day and invisible over the following days. If the mouse located the escape platform in the given time period (60 seconds), it was allowed to stay on it for 10 seconds. If the mouse failed, it was guided to the platform and permitted to remain for 20 seconds. The time taken to find the platform (escape latency) was recorded using a video camera‐based Ethovision System (AniLab Software & Instruments Co., Ltd, Ningbo, Zhejiang, China) and averaged over four trials. For probe trial, the escape platform was removed. The mice were allowed to swim for 60 seconds in search of it. The time in the target quadrant was recorded.
2.5. Nissl staining
Mice were perfused with 4% paraformaldehyde (PFA), and then brains were cut into 20‐μm‐thick cerebral sections in a cryostat (Leica CM 1950, Germany). The frozen sections were mounted on gelatin‐coated slides, dried completely, and rehydrated in distilled water. Then, the sections were immersed in Nissl staining solution (Beyotime Biotechnology) for 10 minutes and rinsed in distilled water. After dehydrating in graded series of ethanol, the sections were submerged in xylene for 5 minutes, cover‐slipped with neutral balsam, and dried naturally.
2.6. Primary cell culture
Primary microglia and astrocytes were isolated from neonatal Sprague‐Dawley (SD) rats and cultured as previously described with some modification.30, 31 In brief, 8‐10 neonatal rats were decapitated after absolute disinfection. Cerebral cortex was separated from the brain and the outside blood vessels and meninges were removed. Then, the cortical tissue was kept in complete medium, minced with ophthalmic scissors, and transferred to a 50‐mL centrifugal tube (Corning, NY, USA). Then, the cells were dispersed through gently pipetting repeatedly. After filtration with a 70‐μm cell strainer (Biologix Group Ltd, Shandong, China), the cell suspension was then spun down at 200 g for 10 minutes. The cell pellets were resuspended with complete medium, plated in T75 flasks (Corning) at a concentration of 1.5×106 cells/mL, and maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The fresh medium was changed every 3 days.
After 9 days of culture, the microglia showed as small round cells on top of the monolayer of astrocytes, which could easily be isolated by shaking the culture flasks at 200 rpm for 2 hours. The percentage of Iba1‐positive cells was reached up to 95%. The flasks were shaken at 200 rpm on a rotary shaker for 24 hours to dislodge microglia and oligodendrocytes to obtain pure astrocytes. The medium was immediately discarded and replaced with a fresh medium. The percentage of glial fibrillary acidic protein (GFAP)‐positive cells was reached up to 98%.
2.7. Measurement of cell viability
Cell viability of microglia and astrocytes with different treatments was evaluated by MTT assay.32 Cells were seeded into 96‐well plates in an appropriate density. After various treatments for 24 hours, MTT (0.25 mg/mL) was added to each well. The plates were incubated for 4 hours at 37°C; then, the medium was discarded gently. The formazan particles were dissolved in DMSO. Finally, the plates were shaken mildly for 5 minutes to get the particles completely dissolved and the absorbance was read at 490 nm using a Synergy™ 2 multimode microplate readers (Bio Tek, Winooski, VT, USA).
2.8. Immunofluorescence staining
The cells seeded on the inserted coverslips (Nest Biotechnology, Wuxi, Jiangsu, China) were treated with AA13 (10 μmol/L) for 24 hours and fixed with 4% PFA for 15 minutes at room temperature (RT). Then, the cells on coverslips were rinsed with PBS to remove cellular debris, blocked with blocking buffer (5% BSA and 0.3% Triton‐× 100 in PBS) for 2 hours, and incubated with the rabbit anti‐Iba1 polyclonal antibody (Proteintech, Wuhan, China) overnight in a humidified chamber at 4°C. After washing three times in PBS, the cells were incubated with an Alexa Fluor 488‐conjugated secondary anti‐rabbit antibody (Invitrogen, MO, USA) for 2 hours at RT in dark. The fluorescent images were captured using a Nikon fluorescent microscope (Nikon, Tokyo, Japan).
2.9. Western blot
The various treated cells were harvested and lysed in cell lysis buffer (1 mol/L Tris‐HCl, PH 7.5, 0.5 mol/L EDTA, 100% NP‐40, NaCl, SDS, deoxycholate) containing protease inhibitor cocktail (Roche, Mannheim, Germany) for 30 minutes on ice. Thereafter, the lysate was collected and centrifuged at 10 000 g for 10 minutes. Protein concentration of supernatants was determined by Bradford assay. For Western blotting, 30‐40 μg protein was loaded for SDS‐PAGE electrophoresis and then transferred onto polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA). After blocking with 5% BSA in Tris‐buffered saline‐Tween 20 (TBST) for 2 hours at RT, the membranes were incubated with primary antibodies recognizing ECE, IDE (Santa‐Cruz Biotechnology, CA, USA), NEP, CD36, MSR1, iNOS, Arg‐1, Ym1/2 (Abcam, Cambridge, UK), TNF‐a (Cell signaling technology, USA), and Iba1 (Proteintech, Wuhan, China) at 4°C overnight. After washing three times in TBST, the membranes were incubated with horseradish peroxidase‐conjugated secondary antibodies (Invitrogen) for 2 hours. The bands were visualized using the enhanced chemiluminescence (ECL) (Millipore, USA) and semiquantified by densitometric analysis with ImageJ 1.46.
2.10. Clearance of Aβ 1‐42
The cells were plated into 24‐well plates overnight until the cells reached 80% confluence. Then, the medium was replaced by serum‐free DMEM/F12 supplemented with AA13 (1 μmol/L, 5 μmol/L, 10 μmol/L). After 24‐hours exposure, the cells were rinsed once with D‐Hanks followed by the addition of Aβ 1‐42 (0.2 μmol/L) per well for 4 hours. The supernatant was collected and the samples were preserved in the refrigerator at −80°C after liquid nitrogen freezing. The total cell protein was extracted as before. The Aβ 1‐42 levels were determined using a Aβ 1‐42 ELISA Kit (Invitrogen).
2.11. In vitro phagocytosis assay
The phagocytic ability of primary microglia was determined by the ingestion of fluorescent microspheres (Latex beads, carboxylate‐modified polystyrene, fluorescent red, aqueous suspension, L3030, Sigma).33 The pure microglia in complete medium were plated on coverslips in 12‐well plates. The medium was displaced by serum‐free DMEM/F12 with or without AA13 (10 μmol/L) after 24 hours. For phagocytosis measurement, 4 μL/mL of latex beads was added to each well and maintained at 37°C for 2 hours. The cells were washed three times in D‐Hanks to remove the free microspheres in the medium, and fixed with 4% PFA. Then, the nuclei were stained with DAPI (Roche Diagnostics) for 15 minutes. Images were visualized using a fluorescent microscope (Nikon), and three random fields per well were preserved for further analysis.
2.12. Statistical analysis
Results were presented as mean±standard error of the mean (SEM) of three independent experiments. One‐way analysis of variance (ANOVA) followed by post hoc (Student‐Newman‐Keuls) test was used for statistical analysis (SPSS 16.0 software, SPSS, USA). Two‐way ANOVA was performed for the statistical analysis of the escape latency in behavioral experiment. P<.05 was considered as significant.
3. Results
3.1. AA13 treatment ameliorates learning and memory impairment in Aβ 1‐42‐injected mice
The learning and memory function was evaluated by the Morris water maze test. As for escape latency, two‐way ANOVA indicated significant differences among different groups in the training trial [Fgroup (3168)=3.551, P=.016; Figure 1D]. Furthermore, the mean escape latency was prolonged along with the days progressed [Fday (5168)=14.567, P<.001]. But there was no significant interaction between group and day [Fgroup × day (15 168)=0.375, P=.984]. The Aβ 1‐42 i.c.v. injected mice showed significant impairment in escape latency to find the hidden platform as compared to mice in sham‐operated group (group 1 vs group 2: P=.002). And the treatment with AA13 significantly relieved the effect by decreasing the escape latency as compared to Aβ 1‐42 i.c.v. injection (group 2 vs group 3: P=.025; group 2 vs group 4: P=.031).
In the probe trial, the results demonstrated that the time spent in target quadrant significantly decreased in Aβ 1‐42 i.c.v. injected group compared with sham‐operated group (group 1 vs group 2: P=.037; Figure 1E). And mice treated with AA13 spent more time in target quadrant, suggesting poor performance induced by Aβ 1‐42 i.c.v. injection was significantly ameliorated by the chronic treatment with AA13 (group 2 vs group 3: P=.043; group 2 vs group 4: P=.037). Meanwhile, the mean speed did not differ between any of the groups (Figure 1F). And Figure 1C shows the typical trajectory of each group. The results indicated that AA13 treatment improved learning and memory functions in Aβ 1‐42‐injected mice.
The result of Nissl staining showed that the number of neural cells in the hippocampal CA1 region was significantly reduced in the model group, and cells were sparsely distributed compared with those in the sham‐operated group (Figure 1G). However, the hippocampal neuron loss was reduced by the treatment with AA13 (6 mg/kg and 30 mg/kg). Furthermore, Aβ 1‐42 significantly upregulated the expression of Iba1 and GFAP. Both 6 and 30 mg/kg AA13 treatment could effectively inhibit the upregulation of GFAP, and AA13 at high doses significantly decreased the Iba1 level (Figure 1H).
These results indicated that AA13 plays a neuroprotective effect in Aβ i.c.v. injected mice; also, it could effectively inhibit glial activation induced by Aβ.
3.2. AA13 alleviates Aβ 1‐42‐induced cytotoxicity in primary neuroglia
To evaluate the effect of AA13 on ameliorating Aβ 1‐42‐induced cytotoxicity, the glial cells were treated with Aβ 1‐42 (10 μmol/L), AA13 (10 μmol/L) and Aβ 1‐42 cotreated with AA13 for 24 hours, respectively. Results showed that there was a reduction in viability in Aβ 1‐42‐treated astrocytes (18.8%) and microglia (15.2%) (Figure 2A and B). AA13 apparently alleviated the Aβ 1‐42‐induced cytotoxicity (up to 16.3% in astrocytes and 23.0% in microglia).
Figure 2.
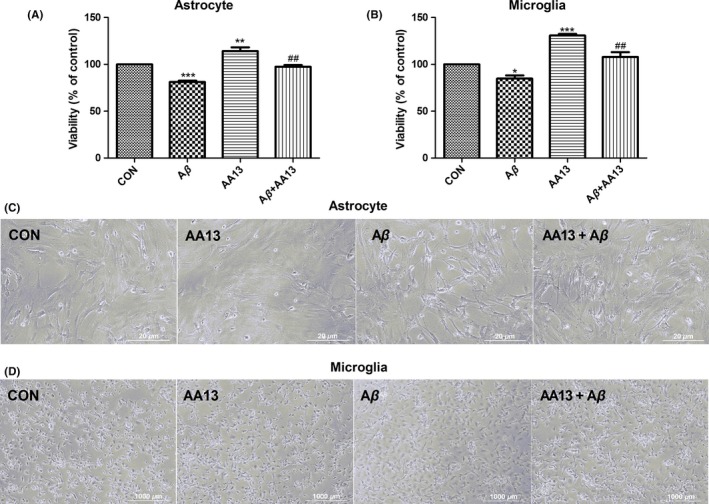
AA13 protected primary cultured neuroglia against Aβ‐induced cytotoxicity. (A, B) Both astrocytes and microglia were treated by 10 μmol/L Aβ 1‐42 with or without 10 μmol/L AA13 and AA13 alone, respectively, for 24 h. Cell viability was determined using MTT assay. (C, D) The morphological alterations of astrocytes and microglia were visualized by a phase‐contrast microscope. *P<.05, **P<.01, ***P<.001 compared with control group. ##P<.01 compared with Aβ‐treated group
The morphological alterations elicited by AA13 in Aβ 1‐42‐challenged cells were documented. As shown in Figure 2C, the Aβ 1‐42‐treated astrocytes showed obvious shrinkage, widened intercellular gaps, and increased ramified cells. In normal microglia, many long branches extended from cellular soma. Most of the Aβ 1‐42‐treated microglia displayed ameboid shape with enlarged soma and thickened processes (Figure 2D). However, the abovementioned morphological changes in astrocytes and microglia were partially reversed after AA13 treatment.
These results demonstrated that AA13 could protect neuroglia against Aβ 1‐42‐induced cellular damage.
3.3. AA13 promotes differentiation of glia from proinflammatory M1 to antiinflammatory M2 phenotype
As shown in results, Aβ alone did not affect Arg‐1 and Ym1/2 expressions obviously. However, AA13 pretreatment markedly increased Arg‐1 and Ym1/2 protein expression in astrocytes (Figure 3A and B) and microglia (Figure 3E and F). Besides, Aβ significantly increased the expression of TNF‐α and iNOS, which was then attenuated by incubation of AA13 in glia (Figure 3C, D, G, and H). The results indicated that AA13 exposure reversed the upregulation of M1 markers and increased the expression of M2 markers in Aβ‐treated glia.
Figure 3.
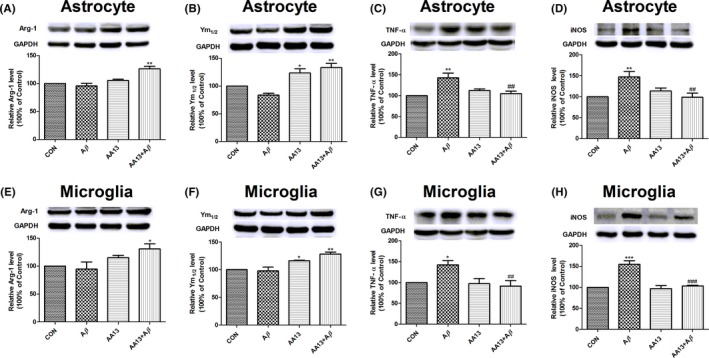
AA13 treatment inhibited the upregulation of M1 markers and increased expression of M2 markers in Aβ‐treated cells. Cells were pretreated with or without 10 μmol/L AA13, then exposed to 10 μmol/L Aβ 1‐42 for 24 h. Western blot was used. Error bar represented integrated optical density values normalized to the internal standard GAPDH. (A, B, E, and F) The M2 markers level, Arg‐1 and Ym1/2, increased in astrocytes and microglia. (C, D, G, and H) Meanwhile, AA13 restored the Aβ‐induced upregulation of TNF‐α and iNOS protein expression in astrocytes and microglia. *P<.05, **P<.01, ***P<.001 compared with control group. ##P<.01, ###P<.001 compared with Aβ‐treated group
3.4. AA13 increases Aβ 1‐42 clearance both in primary microglia and in astrocytes
To explore the effect of AA13 on Aβ clearance, unchallenged (control) and AA13‐challenged cells were incubated with Aβ 1‐42 (0.2 μmol/L) for 4 hours. The intracellular and extracellular Aβ 1‐42 levels were determined by ELISA kits. Results represented that the Aβ 1‐42 levels in the medium of AA13‐treated groups were greatly reduced in a dose‐dependent manner, both in astrocytes and in microglia (Figure 4A and B). However, slight and different changes in intracellular Aβ 1‐42 level were found in glia. The microglia seemed to promote the uptake of Aβ after AA13 treatment (Figure 4D), while the Aβ 1‐42 level decreased in the astrocytes (Figure 4C). It was worth noting that the total Aβ 1‐42 continued to fall as the drug concentration increased (Figure 4E and F). The finding indicated that AA13 exposure was conducive to eliminate Aβ around the glia.
Figure 4.
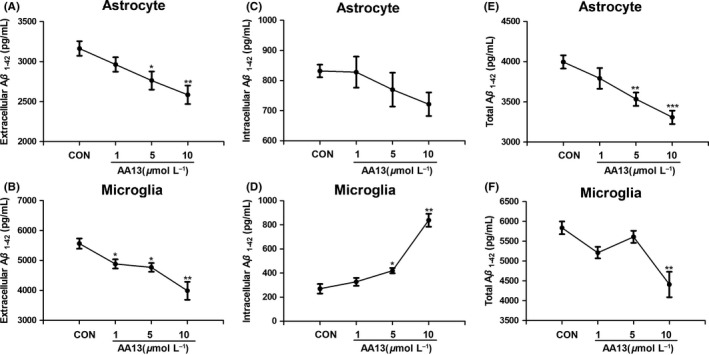
AA13 promoted Aβ 1‐42 clearance in primary cultured neuroglia. Astrocytes and microglia were pretreated with 0, 1, 5, 10 μmol/L AA13 for 24 h followed by the addition of 0.2 μmol/L Aβ 1‐42 for 4 h. (A, B) The extracellular Aβ 1‐42 level of astrocytes and microglia and (C, D) the intracellular Aβ 1‐42 level of astrocytes and microglia were determined using Aβ 1‐42 ELISA kit. (E, F) Combined the extracellular and cytosolic values together to gain the total decline in astrocytes and microglia. *P<.05, **P<.01, ***P<.001 compared with control group
3.5. AA13 promotes microglial activation and reinforces phagocytic efficiency
The activation of microglia results in transformation of cellular morphology and function.34 Results showed that neither 5 μmol/L nor 15 μmol/L AA13 treatment induced significant changes in Iba1 expression, while the Iba1 level increased in 10 μmol/L group (Figure 5A). AA13‐induced changes in cellular morphology and phagocytic efficiency were observed by fluorescence microscope. As shown in Figure 5B, AA13 exposure markedly increased the number of ameboid cells. With regard to phagocytic function, AA13‐treated cells swallowed much more fluorescent microspheres than the untreated cells (Figure 5C and D).
Figure 5.
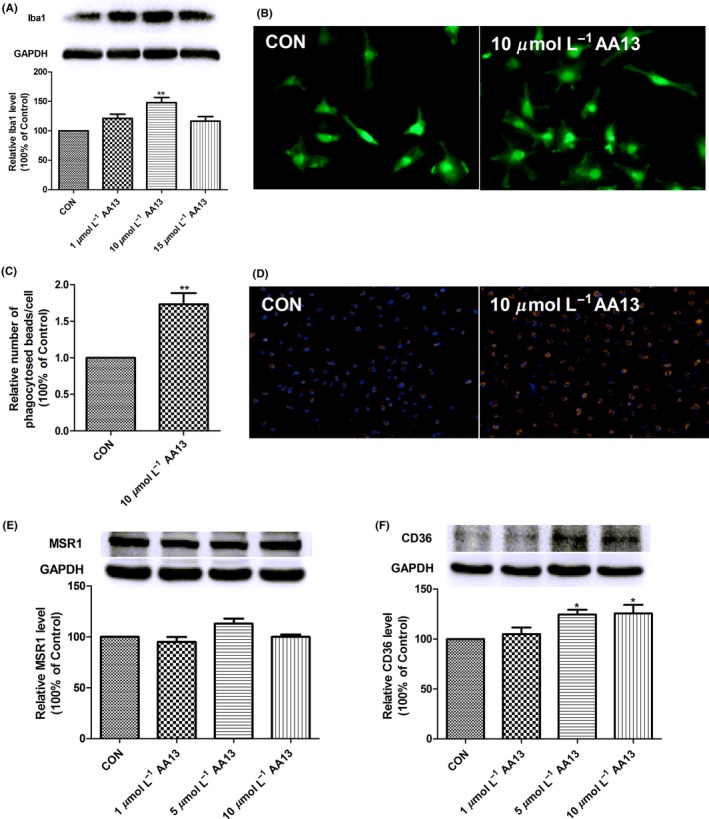
AA13 promoted microglia activation and phagocytosis. Primary microglia were treated with 0, 1, 5, and 10 μmol/L AA13 for 24 h. (A) Western blot was used to semiquantitatively determine the Iba1 immunoreactivity to illustrate the microglial activated state. Error bar represented integrated optical density values normalized to the internal standard GAPDH. (B) Compared with unchallenged cells, the effect of AA13 on microglia was visualized by typical cellular distribution of Iba1 immunoreactivity in green. (C, D) After incubation of 10 μmol/L AA13 for 24 h, fluorescent microbeads in red were added to challenged and unchallenged cells. DAPI‐labeled cell nuclei were shown in blue. (E) AA13 had no significant effect on the expression of MSR1 in microglia. (F) While CD36 level obviously enhanced after AA13 exposure. *P<.05, **P<.01 compared with control group
Scavenger receptors, CD36 and MSR1, are tightly correlated with Aβ recognition and clearance. The results showed that compared to control, the cells incubated with AA13 failed to change the protein expression of MSR1 in microglia (Figure 5E). Instead, CD36 level was significantly upregulated when the cells were treated with 5 μmol/L or 10 μΜ AA13 (Figure 5F). 1 μmol/L AA13 treatment had no effect. These results illustrated that AA13 could activate microglia and promote phagocytosis, which may be partially correlated with CD36.
3.6. AA13 treatment impacts on the expression of Aβ‐degrading enzymes
NEP, ECE, and IDE are potent Aβ‐degrading enzymes in the brain; their expression greatly influences Aβ clearance. We performed western blot to determine the effect of AA13 on the expression of Aβ‐degrading enzymes in cultured primary glia. As demonstrated in Figure 6A and B, the expression of IDE and NEP notably increased after AA13 treatment in a dose‐dependent manner in astrocytes. Inversely, AA13 exerted a negative effect on ECE; that is, 10 μmol/L AA13 decreased the expression of ECE (Figure 6C). Interestingly, microglial ECE protein was markedly augmented by AA13 (Figure 6F), but the expression of IDE and NEP was reduced by high‐dose AA13 (Figure 6D and E). These results suggested that AA13 could regulate the expression of Aβ‐degrading enzymes in astrocytes and microglia to promote elimination of Aβ protein.
Figure 6.
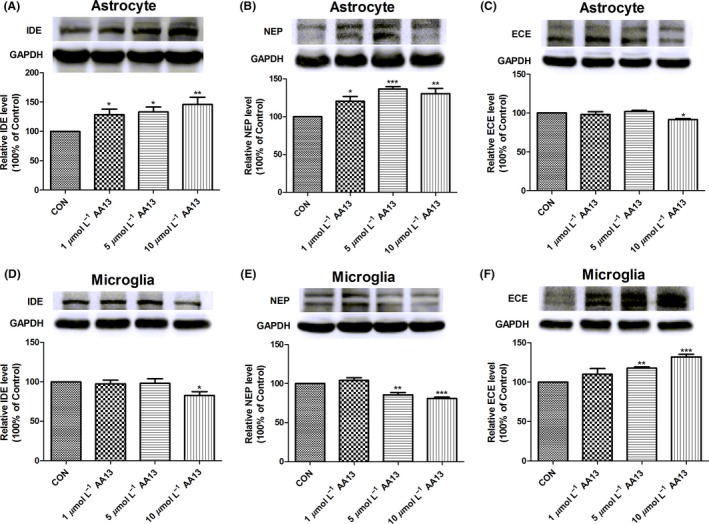
AA13 affected expression of Aβ‐degrading enzymes in primary cultured glia. Cells were treated with or without AA13 (1, 5, 10 μmol/L) for 36 h. Western blot was used. Error bar represented integrated optical density values normalized to the internal standard GAPDH. (A, B, and C) For astrocytes, AA13 significantly promoted the expression of IDE and neprilysin(NEP) in a dose‐dependent manner, but there was an opposite change in ECE expression. (D, E, and F) As for microglia, the expression of IDE and NEP was slightly inhibited by AA13 exposure, while ECE expression significantly increased in a dose‐dependent manner. *P<.05, **P<.01, ***P<.001 compared with control group
4. Discussion
Therapeutic strategies aimed to treat Alzheimer's disease (AD) may either produce an attenuation of symptoms or slow down deterioration by attenuating progression of the disease. The medication that benefits to eliminate Aβ relative toxicity or inflammatory responses would be a promising therapeutic candidate to slow down the progression of Alzheimer's disease. The present research suggested that AA13 exhibited a protective effect on impairment in Alzheimer‐like model and Aβ‐induced glial cytotoxicity. We observed that AA13 promoted exogenous Aβ clearance in primary glia, modulated microglial phagocytosis and the expression of IDE, NEP, and ECE in glia. Furthermore, AA13 alleviated glial proinflammatory responses on Aβ.
In the previous study, we have found that AA13 markedly ameliorated the cognitive deficit induced by scopolamine,28 mainly through upregulating the expression of BDNF and PSD95 in cortex (data not shown), which might prompt the neuroprotective potential of AA13. Neuroinflammation from aberrantly activated glia is an important mechanism that contributes to AD progression. To learn more about the potential mechanisms of AA13 on curing neurodegenerative diseases, in this study, we used Aβ 1‐42 i.c.v. injection‐induced Alzheimer‐like mice model, one of the common models, to characterize Aβ‐induced neuroinflammatory responses and enable preclinical evaluation of a drug targeting Aβ.19 Anand Kamal Sachdeva et al. proposed that lycopene alleviated the cognitive dysfunction through suppressing Aβ 1‐42‐induced neuroinflammatory cascade in Aβ 1‐42‐injected Alzheimer‐like model.35 Similarly, we found that a single i.c.v. injection of Aβ in mice impaired performance in Morris water maze, and AA13 treatment ameliorated the function impairments. The inflammation in the brain is predominantly mediated by astrocytes and microglia. Neuroglia activated by Aβ produce proinflammatory cytokines such as IL‐1β and TNF‐α, which in turn accelerate the accumulation of Aβ, activate glia, and cause neuronal cell damage.36, 37 Therefore, neuroglia have been considered to play a critical role in regulating Aβ‐induced toxicity.
In healthy CNS, microglia are always in a “quiescent” state and constantly survey their microenvironment. Under the trigger of disease or injury, microglia become activated and polarize to either M1 or M2 phenotype.38 In the early stage, microglial activation seems to be profitable through mechanisms including Aβ degradation, phagocytosis, and secretion of growth factors. However, persistent generation of proinflammatory factors is an initiator of neuroinflammation. And the chronic inflammatory responses would drive the progressive of AD.39 Previous finding has revealed that the use of NSAIDs decreases the secretion of proinflammatory factors and increases Aβ phagocytosis.40 Sebastian Jimenez et al. have reported that microglia surrounding the Aβ plaques for phagocytosis generally reveal M2 activation as labeled by Ym1. 41 Consistent with the previous findings, in our study, AA13 at 10 μmol/L activated microglia, which exhibited more ameboid‐shaped microglia with enlarged soma and thickened processes, and exaggerated intake of microbeads. In addition, we found that AA13 treatment inhibited the increase in M1 markers (TNF‐α and iNOS) in Aβ‐treated cells and promoted the expression of M2 markers (Arg‐1 and Ym1/2).
Scavenger receptors (SRs), a major class of pattern recognition receptors (PRRs), are equipped with high affinity to bind polyanionic ligands. SRs work together with other PRRs to defense against pathogens, and they facilitate ligand uptake via enhancing phagocytosis and endocytosis.42, 43 In AD, SRs primarily mediate Aβ clearance and activation of glia. Rodrigo Alarcon et al. proposed that MSR‐1 could be the scavenger receptor responsible for adhesion of astrocytes and microglia to Aβ.44 Compared to wild‐type microglia, CD36 knockout decreased Aβ clearance, followed by less activation of glia and reduction in inflammatory mediators.45 In this study, we found that expression of CD36 was increased after AA13 treatment in a concentration‐dependent manner in microglia. But MSR1 level was not altered. Taken together, it is speculated that AA13 promotes microglial phagocytosis probably through induction of M2 activation and upregulation of CD36. It is known that activated microglia accelerated the intake of toxic Aβ, but the excessive intracellular Aβ would damage the capacity of phagocytosis.16 Thus, further researches are required to explore the mechanisms underlying the effects of AA13 on glial activation.
A series of evidences have demonstrated that Aβ‐degrading enzymes play a critical role in removing toxic amyloid species to prevent the development of AD. Compared with wild‐type littermates, the Aβ level was sharply increased in IDE‐knockout animals.12 Previous studies have proposed that ECE‐1‐ or ECE‐2‐knockdown animals would increase Aβ level in the brain.46 And in the past few years, we have been focusing on the alteration of Aβ‐degrading enzymes in the progression of AD. The alteration of these enzymes was dynamic and variable. Zhou et al. found that the ratio of membrane to cytoplasmic NEP protein was significantly increased in AD. Moreover, treatment of human neuroblastoma SH‐SY5Y cells with Aβ produced upregulation of NEP beginning at 24 hours and persisting to 72 hours.47 Another study in our laboratory indicated that in APP/PS1 mice, NEP levels showed a significant decrease with age, while ECE protein significantly increased in the cerebral cortex and hippocampus.48 Similarly, Liu et al. found that astrocytes from AD mice expressed more NEP than the WT group, and NEP protein level gradually rose with time.49 In the current study, we found that AA13 regulated the Aβ‐degrading enzyme expression differently in microglia and astrocytes. AA13 treatment increased IDE and NEP expression, but decreased ECE expression in astrocytes. In contrast, expression of IDE and NEP was reduced in microglia, while the ECE level was significantly enhanced along with the increased drug concentration. These results indicated that AA13 treatment played an important role in Aβ degradation through regulating Aβ‐degrading enzyme expression. Interestingly, it seems that microglia tend to remove Aβ through phagocytosis, while astrocytes prefer to enzymatic degradation.
5. Conclusion
Our finding illustrated that oral administration of AA13 attenuated the cognitive impairment in Aβ‐injected mice; also, AA13 alleviated Aβ‐induced cytotoxicity in primary cultured glia. The mechanical studies found that AA13 promoted differentiation of neuroglial cells from proinflammatory M1 to antiinflammatory M2 phenotype. Moreover, AA13 facilitated Aβ clearance by means of promoting Aβ phagocytosis and degradation. AA13 modulated microglial activation and performed a beneficial effect on expression of CD36, NEP, IDE, and ECE in glial cells. In conclusion, the modulatory roles of AA13 in neuroglia make it a potential candidate for Alzheimer's disease.
Conflict of Interest
The authors have no competing interests.
Acknowledgments
This project was supported by grants from the National Natural Science Foundation of China (81072627) (81230090), the Shanghai Biomedical Technology Support Program (15401901100), and the Key Project from Shanghai Science and Technology Committee (12431900901). We thank the technical support of Professor Rui Wang. We also thank Professor Lei Ma for the gift of the Sarsasapogenin‐AA13.
Huang C, Dong D, Jiao Q, Pan H, Ma L, Wang R. Sarsasapogenin‐AA13 ameliorates Aβ‐induced cognitive deficits via improving neuroglial capacity on Aβ clearance and antiinflammation. CNS Neurosci Ther. 2017;23:498–509. 10.1111/cns.12697
The first two authors contributed equally to this work.
Contributor Information
Lei Ma, Email: malei@ecust.edu.cn.
Rui Wang, Email: ruiwang@ecust.edu.cn.
References
- 1. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741‐766. [DOI] [PubMed] [Google Scholar]
- 2. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329‐344. [DOI] [PubMed] [Google Scholar]
- 3. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science. 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heneka MT, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science. 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012;53:11‐24. [DOI] [PubMed] [Google Scholar]
- 6. Orre M, Kamphuis W, Dooves S, et al. Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain. 2013;136:1415‐1431. [DOI] [PubMed] [Google Scholar]
- 7. Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer's disease mouse model. Glia. 2012;60:615‐629. [DOI] [PubMed] [Google Scholar]
- 8. Nielsen HM, Mulder SD, Belien JA, Musters RJ, Eikelenboom P, Veerhuis R. Astrocytic A beta 1‐42 uptake is determined by A beta‐aggregation state and the presence of amyloid‐associated proteins. Glia. 2010;58:1235‐1246. [DOI] [PubMed] [Google Scholar]
- 9. Alarcon R, Fuenzalida C, Santibanez M, Von Bernhardi R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface‐bound beta‐amyloid. J Biol Chem. 2005;280:30406‐30415. [DOI] [PubMed] [Google Scholar]
- 10. Prior R, Wihl G, Urmoneit B. Apolipoprotein E, smooth muscle cells and the pathogenesis of cerebral amyloid angiopathy: the potential role of impaired cerebrovascular A beta clearance. Ann N Y Acad Sci. 2000;903:180‐186. [DOI] [PubMed] [Google Scholar]
- 11. Leissring MA. The AbetaCs of Abeta‐cleaving proteases. J Biol Chem. 2008;283:29645‐29649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farris W, Mansourian S, Chang Y, et al. Insulin‐degrading enzyme regulates the levels of insulin, amyloid beta‐protein, and the beta‐amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162‐4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lucin KM, O'Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron. 2013;79:873‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morales I, Guzman‐Martinez L, Cerda‐Troncoso C, Farias GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry. 2014;76:575‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96:29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354‐8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283‐4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Souza LC, Jesse CR, Antunes MS, et al. Indoleamine‐2,3‐dioxygenase mediates neurobehavioral alterations induced by an intracerebroventricular injection of amyloid‐beta1‐42 peptide in mice. Brain Behav Immun. 2016;56:363‐377. [DOI] [PubMed] [Google Scholar]
- 20. Hufford CD, Liu SC, Clark AM. Antifungal activity of Trillium grandiflorum constituents. J Nat Prod. 1988;51:94‐98. [DOI] [PubMed] [Google Scholar]
- 21. Ikeda T, Ando J, Miyazono A, et al. Anti‐herpes virus activity of Solanum steroidal glycosides. Biol Pharm Bull. 2000;23:363‐364. [DOI] [PubMed] [Google Scholar]
- 22. Kim SY, Son KH, Chang HW, Kang SS, Kim HP. Inhibition of mouse ear edema by steroidal and triterpenoid saponins. Arch Pharm Res. 1999;22:313‐316. [DOI] [PubMed] [Google Scholar]
- 23. Attele AS, Zhou YP, Xie JT, et al. Antidiabetic effects of Panax ginseng berry extract and the identification of an effective component. Diabetes. 2002;51:1851‐1858. [DOI] [PubMed] [Google Scholar]
- 24. Lee B, Jung K, Kim DH. Timosaponin AIII, a saponin isolated from Anemarrhena asphodeloides, ameliorates learning and memory deficits in mice. Pharmacol Biochem Behav. 2009;93:121‐127. [DOI] [PubMed] [Google Scholar]
- 25. Ouyang S, Sun LS, Guo SL, Liu X, Xu JP. Effects of timosaponins on learning and memory abilities of rats with dementia induced by lateral cerebral ventricular injection of amyloid beta‐ peptide. Chemical Science. 2016;7:3206‐3214. [PubMed] [Google Scholar]
- 26. Sy L‐K, Lok C‐N, Wang J‐Y, et al. Identification of “sarsasapogenin‐aglyconed” timosaponins as novel Aβ‐lowering modulators of amyloid precursor protein processing. Chemical Science. 2016;7:3206‐3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dong DONG, Nan‐nan ZHOU, Rui‐xuan LIU, et al. Sarsasapogenin‐AA13 reduces LPS‐induced inflammatory responses in vitro and relieves dimethylbenzene‐induced ear edema in mice. Acta Pharmacol Sin. 2016;160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan H, Van Khang P, Dong D, Wang R, Ma L. Synthesis and SAR study of novel sarsasapogenin derivatives as potent neuroprotective agents and NO production inhibitors. Bioorg Med Chem Lett. 2017;27:662‐665. [DOI] [PubMed] [Google Scholar]
- 29. Prakash A, Medhi B, Chopra K. Granulocyte colony stimulating factor (GCSF) improves memory and neurobehavior in an amyloid‐beta induced experimental model of Alzheimer's disease. Pharmacol Biochem Behav. 2013;110:46‐57. [DOI] [PubMed] [Google Scholar]
- 30. Keene CD, Chang R, Stephen C, et al. Protection of hippocampal neurogenesis from toll‐like receptor 4‐dependent innate immune activation by ablation of prostaglandin E2 receptor subtype EP1 or EP2. Am J Pathol. 2009;174:2300‐2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52:70‐77. [DOI] [PubMed] [Google Scholar]
- 32. Lee HJ, Maeng K, Dang HT, et al. Anti‐inflammatory effect of methyl dehydrojasmonate (J2) is mediated by the NF‐kappaB pathway. J Mol Med (Berl). 2011;89:83‐90. [DOI] [PubMed] [Google Scholar]
- 33. Szabo M, Gulya K. Development of the microglial phenotype in culture. Neuroscience. 2013;241:280‐295. [DOI] [PubMed] [Google Scholar]
- 34. Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sachdeva AK, Chopra K. Lycopene abrogates Abeta(1‐42)‐mediated neuroinflammatory cascade in an experimental model of Alzheimer's disease. J Nutr Biochem. 2015;26:736‐744. [DOI] [PubMed] [Google Scholar]
- 36. Bales KR, Du Y, Dodel RC, Yan GM, Hamilton‐Byrd E, Paul SM. The NF‐kappaB/Rel family of proteins mediates Abeta‐induced neurotoxicity and glial activation. Brain Res Mol Brain Res. 1998;57:63‐72. [DOI] [PubMed] [Google Scholar]
- 37. Hughes DT, Martel PM, Kinlaw WB, Eisenberg BL. The synthetic triterpenoid CDDO‐Im inhibits fatty acid synthase expression and has antiproliferative and proapoptotic effects in human liposarcoma cells. Cancer Invest. 2008;26:118‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752‐758. [DOI] [PubMed] [Google Scholar]
- 39. Sastre M, Gentleman SM. NSAIDs: how they Work and their Prospects as Therapeutics in Alzheimer's Disease. Front Aging Neurosci. 2010;2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lleo A, Galea E, Sastre M. Molecular targets of non‐steroidal anti‐inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci. 2007;64:1403‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jimenez S, Baglietto‐Vargas D, Caballero C, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age‐dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650‐11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stuart LM, Deng J, Silver JM, et al. Response to Staphylococcus aureus requires CD36‐mediated phagocytosis triggered by the COOH‐terminal cytoplasmic domain. J Cell Biol. 2005;170:477‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stuart LM. Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. 2005;22:539‐550. [DOI] [PubMed] [Google Scholar]
- 44. Zhang H, Su YJ, Zhou WW, et al. Activated scavenger receptor A promotes glial internalization of abeta. PLoS ONE. 2014;9:e94197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta‐amyloid. J Exp Med. 2003;197:1657‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease beta‐amyloid peptide is increased in mice deficient in endothelin‐converting enzyme. J Biol Chem. 2003;278:2081‐2084. [DOI] [PubMed] [Google Scholar]
- 47. Zhou L, Wei C, Huang W, et al. Distinct subcellular patterns of neprilysin protein and activity in the brains of Alzheimer's disease patients, transgenic mice and cultured human neuronal cells. Am J Transl Res. 2013;5:608‐621. [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou L, Liu J, Dong D, Wei C, Wang R. Dynamic alteration of neprilysin and endothelin‐converting enzyme in age‐dependent APPswe/PS1dE9 mouse model of Alzheimer's disease. Am J Transl Res. 2017;9:184‐196. [PMC free article] [PubMed] [Google Scholar]
- 49. Liu RX, Huang C, Bennett DA, Li H, Wang R. The characteristics of astrocyte on Abeta clearance altered in Alzheimer's disease were reversed by anti‐inflammatory agent (+)‐2‐(1‐hydroxyl‐4‐oxocyclohexyl) ethyl caffeate. Am J Transl Res. 2016;8:4082‐4094. [PMC free article] [PubMed] [Google Scholar]