

Summary
Aims
To evaluate whether activating α7 nicotinic acetylcholine receptor (α7nAChR) could inhibit the NOD‐like receptor family, pyrin domain containing 3 (NLRP3) inflammasome through regulation of β‐arrestin‐1 in monocyte/macrophage system, thus contributing to the control of neuroinflammation.
Methods
The protein levels of NLRP3, caspase‐1 (Casp‐1) p20 and proCasp‐1, interleukin‐1β (IL‐1β) p17 and proIL‐1β, IL‐18 and proIL‐18 were measured using Western blotting. The mRNA levels of Casp‐1 and IL‐1β were detected by real‐time PCR (RT‐PCR). The colocalization and interaction of NLRP3 protein and β‐arrestin‐1 were measured by immunofluorescence staining and immunoprecipitation.
Results
The expression of β‐arrestin‐1 was significantly increased and colocalized with CD45‐positive cells in spinal cord of experimental auto‐immune encephalomyelitis (EAE) mice when compared with the sham mice, which was attenuated by pretreatment with PNU282987, a specific α7nAChR agonist. PNU282987 also significantly inhibited the activation of NLRP3 inflammasome and thus decreased the production of IL‐1β and IL‐18 both in lipopolysaccharide (LPS)/ATP‐stimulated BV2 microglia in vitro and spinal cord from EAE mice in vivo, while inverse effects were observed in α7nAChR knockout mice. Furthermore, overexpression of β‐arrestin‐1 attenuated the inhibitory effect of PNU282987 on NLRP3 inflammasome activation in LPS/ATP‐stimulated BV2 microglia. PNU282987 inhibited the interaction between β‐arrestin‐1 and NLRP3 protein in vitro.
Conclusions
The present study demonstrates that activating α7nAChR can lead to NLRP3 inflammasome inhibition via regulation of β‐arrestin‐1 in monocyte/microglia system.
Keywords: α7nAChR, β‐arrestin‐1, neuroinflammation, NLRP3 inflammasome
1. INTRODUCTION
Multiple sclerosis (MS) is one of the world's most serious neurological disorders among young people, commonly leading to progressive disabilities.1 It is mainly characterized by the inflammation‐ and immune‐related demyelination and neurodegeneration in the central nervous system (CNS).2, 3 For the study of MS, experimental auto‐immune encephalomyelitis (EAE) is most widely used animal model in fundamental researches. Although a large number of studies have been conducted on this disease, so far the specific mechanisms remain unclear and there is still no effective therapeutic strategy for the treatment of MS.
α7 nicotinic acetylcholine receptor (α7nAChR) is a subunit of nAChRs.4 It has been previously reported that activating α7nAChR can control the production of inflammatory cytokines in lipopolysaccharide (LPS)‐stimulated macrophages.5 Studies from our laboratory previously showed that α7nAChR was associated with various cardiovascular diseases.6, 7, 8 As α7nAChR has been found to be expressed in neuronal cells including microglia and astrocytes, it is considered to be highly related to CNS diseases including MS. Recently, Nizri et al9 reported that activating α7nAChR contributes to the alleviation of neuroinflammation in EAE model, but the underlying mechanism has not been fully clarified.
Inflammasomes are newly recognized, vital players in innate immunity. Among all these forms of inflammatory reaction, the NOD‐like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is one of the most popular forms nowadays, for it is considered as the connection between inflammation and innate immunity, which is involved in the initiation and progression of auto‐immune and auto‐inflammatory diseases including multiple sclerosis, inflammatory bowel disease, cryopyrin‐associated periodic fever syndrome and so on.10, 11, 12 Recently, Lu et al13 reported that activating α7nAChR can inhibit the NLRP3 inflammasome by preventing mitochondrial DNA release in peripheral macrophage. However, whether activating α7nAChR could inhibit NLRP3 inflammasome in CNS and the underlying mechanism remain unclear.
β‐arrestin‐1 is a member of multifunctional small molecular protein, mediating various signaling pathways.14 It is reported that β‐arrestin‐1 positively regulated activated CD4+ T‐cell survival and mice deficient in β‐arrestin‐1 were much more resistant to EAE.15 Recently, Mao et al16 reported that β‐arrestin‐1 played a vital role in the assembly and activation of NLRP3 inflammasome in the condition of inflammatory stimulation. However, whether β‐arrestin‐1 is involved in α7nAChR‐mediated NLRP3 inflammasome inhibition in CNS inflammation remains unclarified.
In this study, we raised the hypothesis that activating α7nAChR can inhibit NLRP3 inflammasome via the regulation of β‐arrestin‐1 in microglia, thus contributing to the suppression of neuroinflammation and attenuation of EAE severity. Our study may provide a novel therapeutic strategy for the treatment of MS.
2. MATERIALS AND METHODS
2.1. Animals
C57BL/6 mice were purchased from Shanghai Super‐B&K Laboratory Animal Corp., Ltd. (Shanghai, China). Mice deficient in the α7 nicotinic acetylcholine receptors were purchased from Jackson laboratory (Bar Harbor, MA, USA) (B6.129P2‐Cnr2tm1Dgen/J, Stock Number: 005786). All animals were kept at 22°C under a 12‐hours light/dark cycle with unlimited access to water and standard rodent diet. All experiments were approved and conducted in accordance with the guidelines for the Animal Care Committee of Second Military Medical University and guidelines for Care and Use of Laboratory Animals published by the National Institutes of Health, USA.
2.2. Experimental auto‐immune encephalomyelitis induction and assessment
Experimental auto‐immune encephalomyelitis (EAE) was induced in C57BL/6, α7nAChR knockout (KO), and wild‐type (WT) mice at 8‐9 weeks as previously reported.17, 18, 19 In brief, mice were subcutaneously immunized with 200 μg MOG35‐55 in complete Freund's adjuvant (Sigma‐Aldrich, St. Louis, MO, USA) containing heat‐killed mycobacterium tuberculosis (H37RA strain; 5 mg/mL; BD Diagnostics, Franklin Lakes, NJ, USA). Pertussis toxin (200 ng/mouse; Calbiochem, Billerica, MA, USA) was injected via intraperitoneal injection (i.p.) on days 0 and 2. For drug treatment, PNU282987 (Sigma‐Aldrich), which was predissolved in DMSO and diluted in saline, was injected via i.p. daily from day 3 till the end of the study in the dose of 0.1 mg/kg body weight. Saline was given as vehicle.
2.3. Culture and treatment of BV2 microglia
Murine BV2 microglia were cultured with Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% (vol/vol) fetal bovine serum (Gibco) at 37°C in a humidified incubator with 5% CO2. Cells were pre‐incubated with vehicle or PNU282987 (1, 3, or 10 μmol/L) for 10 minutes before challenged by lipopolysaccharides (100 ng/mL, LPS, Sigma, Louis, MO, USA) and adenosine triphosphate (1 mmol/L, ATP) for 12 hours.20
2.4. Transient transfection
We used the pLenti‐CMV‐EGFP‐3FLAG vector to encode β‐arrestin‐1 and in vitro transfection was carried out as described previously.20, 21
2.5. RT‐PCR
Total RNA from BV2 microglia or spinal cord was isolated by TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The yield and purity of the RNA were determined by spectroscopic analysis, and the concentration of total RNA was equilibrated. The first‐strand cDNA was synthesized using PrimeScript RT Master Mix (Takara, Dalian, China). 7500 Real Time PCR System and the Fast Start Universal SYBR Green Master (Roche, Basel, Switzerland) were used for RT‐PCR according to the manufacturer recommendations. Primers for murine IL‐1β, Caspase‐1, β‐arrestin‐1 and Gapdh were listed in Table 1.
Table 1.
The sequences of the primers used in real‐time PCR (RT‐PCR) were listed as below
| Sense (5′‐3′) | Anti‐sense (5′‐3′) | |
|---|---|---|
| IL‐1β | CTCGTGCTGTCGGACCCCAT | AGTGTTCGTCTCGTGTTCGGAC |
| Caspase‐1 | CCCCAGGCAAGCCAAATC | CCTGACTGACCCTGGGAGT |
| β‐arrestin‐1 | ATACGCTGACTCCCTTCCTG | GGACACCACCAGTTTCACCT |
| Gapdh | GTATGACTCCACTCACGGCAAA | GGTCTCGCTCCTGGAAGATG |
2.6. Immunofluorescence staining
Spinal cords were fixed in 4% (w/v) paraformaldehyde overnight and embedded in paraffin, and then 15‐μm sections were sent to dewaxing and rehydration. After blocked with 5% bovine serum albumin in PBST for 2 hours, the sections were incubated with rabbit anti‐β‐arrestin‐1 (1:200, Abcam, Cambridge, MA, USA) and mouse anti‐CD45 (1:200, Abcam) antibody overnight at 4°C. For BV2 microglia, cells were fixed in 4% paraformaldehyde for 10 minutes and then washed with PBS. After permeabilized with 0.1% Triton X‐100 for 5 minutes, cells were blocked with 5% bovine serum albumin in PBST for 30 minutes and then incubated with rabbit anti‐NLRP3 (1:200, CST, Danvers, MA, USA) and mouse anti‐β‐arrestin‐1 (1:100, Santa Cruz Biotechnology, Dallas, TX, USA) antibody for 2 hours at 37°C. Double‐immunofluorescent staining was completed by Alexa‐488 or Alexa‐Cy3‐labeled secondary antibody (1:500, Jackson ImmunoResearch Inc, West Grove, PA, USA) incubation for 30 minutes at 37°C. After being washed, slides were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA), and colocalization was observed using a confocal laser scanning microscope (Fluoview FV1000, Olympus, Tokyo, Japan). Pearson's correlation coefficient was analyzed with ImageJ. In this case, the experiments were performed in a double‐blind manner.
2.7. Immunoprecipitation (IP)
After treatment as described above, the cell lysates were prepared in protein and IP lysis buffer (Biyuntian, Shanghai, China) and incubated with anti‐NLRP3 or anti‐β‐arrestin‐1 antibody together with protein A/G Plus‐agarose IP reagent (Invitrogen) at 4°C overnight. After three‐time washes, the immunoprecipitates were boiled in SDS sample buffer for 5 minutes and analyzed by immunoblot.
2.8. Immunoblot analysis
Spinal cord and BV2 microglia were lysated and used for immunoblotting. Blots were incubated with the primary antibodies overnight at 4°C. The primary antibodies were used as follows: NLRP3 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), Casp‐1 (1:500, Abcam), IL‐1β (1:500, R&D, Minneapolis, MN, USA), IL‐18 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), and β‐arrestin‐1 (1:500, Abcam). Then, the membranes were incubated with IRDye800CW‐conjugated secondary antibody (1:5000, LI‐COR Biosciences, Lincoln, NE, USA). The images were captured by the Odyssey infrared imaging system (LI‐COR Bioscience).
2.9. Statistical analysis
Data were presented as means ± SEM. For analysis, a Kruskal‐Wallis test followed by Dunn's post hoc test and one‐way analysis of variance (ANOVA) followed by Bonferroni post hoc test were used to determine nonparametric data and continuous variables, respectively. P values <0.05 were considered statistically significant. Data were analyzed with SPSS 21.0K for Windows (SPSS, Chicago, IL, USA).
3. RESULTS
3.1. Dysregulation of β‐arrestin‐1 in spinal cord of experimental auto‐immune encephalomyelitis mice
It is commonly believed that accumulation and overactivation of microglia could aggravate the severity of symptoms and demyelination in EAE mice.22 Recently, there is evidence that highlighted the importance of β‐arrestin‐1 in the progression of EAE.15 Therefore, whether the expression of β‐arrestin‐1 in microglia is changed during EAE needs to be explored. Our results revealed the mRNA and protein levels of β‐arrestin‐1 were significantly increased in spinal cord of EAE mice (Figure 1A,B). Double immunostaining for β‐arrestin‐1 (red spots) and the CD45‐positive cells (green spots) confirmed that the number of β‐arrestin‐1 and CD45‐positive cells was significantly increased in spinal cord of EAE mice (Figure 1C), suggesting that the expression of β‐arrestin‐1 in microglia is increased during EAE.
Figure 1.
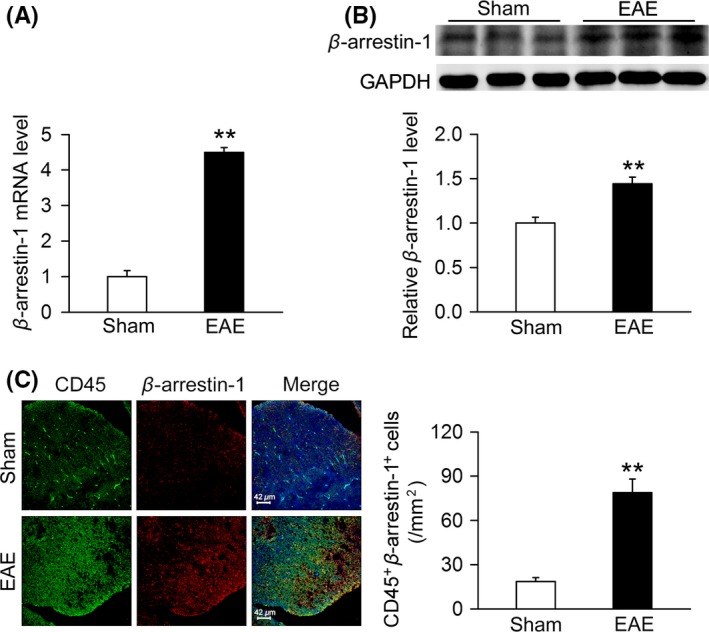
β‐arrestin‐1 expression in spinal cord of the sham and experimental auto‐immune encephalomyelitis (EAE) mice. (A,B) Total RNA and protein in spinal cord of the sham and EAE mice were collected, and the mRNA and protein levels of β‐arrestin‐1 were analyzed using real‐time PCR (RT‐PCR) and Western blot. Compared with the sham group, both mRNA and protein levels of β‐arrestin‐1 were elevated in EAE mice (n = 6 per group). **P < 0.01 vs sham. (C) Spinal cords of the sham and EAE mice were fixed in 4% (w/v) paraformaldehyde overnight and then paraffin‐embedded and sectioned. The colocalization of β‐arrestin‐1 and CD45‐positive cells was stained by immunofluorescence assay. The colocalization of β‐arrestin‐1 (red) and CD45‐positive cells (green) was significantly increased in spinal cord of EAE mice compared with the sham mice (n = 6 per group). **P < 0.01 vs sham
3.2. Activation of α7nAChR decreases the expression of β‐arrestin‐1 in vivo and in vitro
Given that the role of β‐arrestin‐1 in the pathological mechanism of EAE and activation of α7nAChR alleviates neuroinflammation in EAE model,9 we therefore explored the influence of α7nAChR activation on the expression of β‐arrestin‐1 in EAE mice. We found that treatment with PNU282987, a selective α7nAChR agonist, significantly decreased β‐arrestin‐1 in spinal cord from EAE mice when compared with the sham mice (Figure 2A). In comparison with WT mice, the expression of β‐arrestin‐1 is significantly increased in spinal cord from α7nAChR KO EAE mice (Figure 2B). Furthermore, PNU282987 treatment dose‐dependently inhibited the expression of β‐arrestin‐1 in BV2 microglia stimulated with LPS/ATP (Figure 2C). Taken together, these results suggest that α7nAChR negatively regulates the expression of β‐arrestin‐1 both in vivo and in vitro.
Figure 2.

α7 nicotinic acetylcholine receptor (α7nAChR) regulated the expression of β‐arrestin‐1 in experimental auto‐immune encephalomyelitis (EAE) mice and LPS/ATP‐stimulated BV2 microglia. Spinal cords of the sham and EAE mice as well as BV2 microglia were collected and lysated, and the expression of β‐arrestin‐1 was measured using Western blot. (A) Compared with the vehicle group, treatment with PNU282987 significantly decreased the expression of β‐arrestin‐1 in spinal cords of EAE mice (n = 6 per group). PNU, PNU282987. **P < 0.01 vs sham; ## P < 0.01 vs vehicle. (B) The expression of β‐arrestin‐1 in spinal cords of α7nAChR KO mice was increased compared with those of WT mice (n = 6 per group). **P < 0.01 vs sham; ## P < 0.01 vs WT. (C) PNU282987 dose‐dependently inhibited the expression of β‐arrestin‐1 in BV2 microglia challenged by LPS/ATP (n = 6 per group). PNU, PNU282987. **P < 0.01 vs control; ## P < 0.01 vs vehicle
3.3. α7nAChR negatively regulates NLRP3 inflammasome activation in vivo and in vitro
Recent studies have revealed that hyperactivation of NLRP3 inflammasome in myeloid cells was associated with the auto‐immune diseases including MS and EAE.23, 24 Thus, we tested whether activating α7nAChR could inhibit NLRP3 inflammasome in spinal cord from EAE mice and LPS/ATP‐stimulated BV2 microglia. The results showed that treatment with PNU282987 (0.1 mg/kg) significantly decreased NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 in spinal cord from EAE mice (Figure 3A,B). When compared with WT mice, the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 were significantly increased in α7nAChR KO mice (Figure 3C,D). In addition, treatment with PNU282987 dose‐dependently decreased NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 in BV2 microglia stimulated with LPS/ATP (Figure 4A‐E). PNU282987 also decreased the mRNA levels of Casp‐1 and IL‐1β in BV2 microglia stimulated with LPS/ATP (Figure 4F‐G). The combined data suggest that α7nAChR negatively regulates NLRP3 inflammasome activation both in vivo and in vitro.
Figure 3.

α7 nicotinic acetylcholine receptor (α7nAChR) regulates NLRP3 inflammasome activation in experimental auto‐immune encephalomyelitis (EAE) mice. Spinal cords were isolated from the sham and EAE mice on day 12 PI and then lysated with buffer. The levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 were analyzed using Western blot. (A‐B) Treatment with PNU282987 (0.1 mg/kg) significantly decreased the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 compared with the vehicle group (n = 6 per group). PNU, PNU282987. **P < 0.01 vs sham; ## P < 0.01 vs vehicle. (C‐D) Compared with the WT mice, the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 were significantly increased in α7nAChR KO mice (n = 6 per group). **P < 0.01 vs sham; # P < 0.05 vs WT; ## P < 0.01 vs WT
Figure 4.
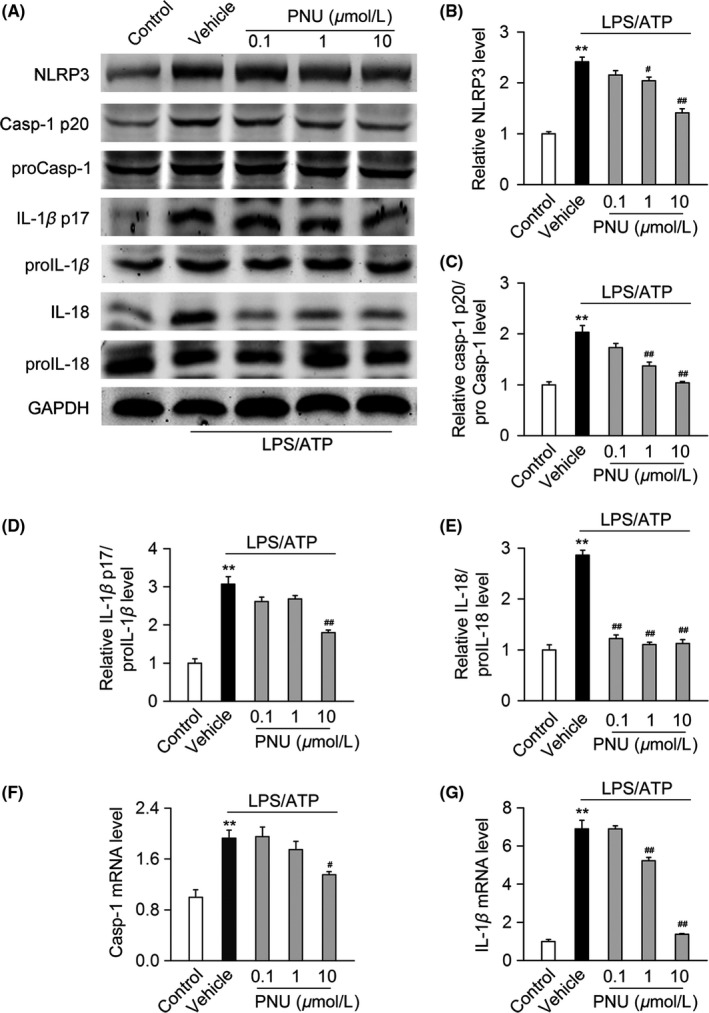
α7 nicotinic acetylcholine receptor (α7nAChR) regulates NLRP3 inflammasome activation in LPS/ATP‐stimulated BV2 microglia. BV2 microglia were pretreated with vehicle or PNU282987 (0.1, 1 or 10 μmol/L) for 10 minutes before stimulated with LPS (100 ng/mL) and ATP (1 mmol/L) for 12 hours. (A‐E) Cells were lysed, and the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 were analyzed using Western blot. Compared with the vehicle group, treatment with PNU282987 significantly decreased the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 (n = 6 per group). PNU, PNU282987. **P < 0.01 vs control; # P < 0.05 vs vehicle; ## P < 0.01 vs vehicle. (F‐G) Cells were collected, and mRNA levels of Casp‐1 and IL‐1β were analyzed by RT‐PCR. Compared with the vehicle group, PNU282987 significantly decreased the mRNA levels of Casp‐1 and IL‐1β (n = 6 per group). PNU, PNU282987. **P < 0.01 vs control; # P < 0.05 vs vehicle; ## P < 0.01 vs vehicle
3.4. β‐arrestin‐1 mediates the inhibitory effect of α7nAChR activation on NLRP3 inflammasome in LPS/ATP‐stimulated BV2 microglia
Recent studies have reported that β‐arrestin‐1 plays a critical role in the assembly and activation of NLRP3 and NLRC4 inflammasomes.16 To identify the role of β‐arrestin‐1 on the inhibitory effects of α7nAChR on NLRP3 inflammasome activation, BV2 microglia were transfected with the control lentivirus (LV‐GFP) or β‐arrestin‐1 overexpression lentivirus (LV‐β‐arr‐1) and then pretreatment with vehicle or PNU282987 before LPS/ATP stimulation. We found that overexpression of β‐arrestin‐1 significantly attenuated the inhibitory effects of PNU282987 on NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 (Figure 5), suggesting that β‐arrestin‐1 at least partially mediates the inhibitory effect of α7nAChR activation on NLRP3 inflammasome.
Figure 5.

β‐arrestin‐1 mediates the inhibitory effects of α7 nicotinic acetylcholine receptor (α7nAChR) on the NLRP3 inflammasome activation. BV2 microglia were transfected with the control lentivirus (LV‐GFP) or β‐arrestin‐1 overexpression lentivirus (LV‐β‐arr‐1) for 72 hours and then treated with vehicle or PNU282987 (10 μmol/L) for 10 minutes followed by the stimulation with LPS (100 ng/mL) and ATP (1 mmol/L) for 12 hours. BV2 microglia were lysated, and expression of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β and IL‐18/proIL‐18 was analyzed using Western blot. (A‐E) Compared with LV‐GFP group, overexpression of β‐arrestin‐1 significantly attenuated the inhibitory effects of PNU282987 on the levels of NLRP3 protein, Casp‐1 p20/proCasp‐1, IL‐1β p17/proIL‐1β, and IL‐18/proIL‐18 (n = 6 per group). PNU, PNU282987. **P < 0.01 vs control; ## P < 0.01 vs LPS/ATP
3.5. Activation of α7nAChR inhibits the bind of β‐arrestin‐1 with NLRP3 protein in LPS/ATP‐stimulated BV2 microglia
To further explore the molecular mechanism that β‐arrestin‐1 mediates the effect of activating α7nAChR on NLRP3 inflammasome, we detected the colocalization and interaction of β‐arrestin‐1 and NLRP3 protein using immunofluorescence staining and IP. Our results showed that LPS/ATP stimulation induced the colocalization of NLRP3 protein (red spots) and β‐arrestin‐1 (green spots) in BV2 microglia. Pretreatment with PNU282987 significantly decreased the colocalization (Figure 6A,B). Furthermore, we found that LPS/ATP stimulation induced the interaction between β‐arrestin‐1 and NLRP3 protein in BV2 microglia, which was prevented by pre‐incubated with PNU282987 (Figure 6C). Collectively, these data suggest that activating α7nAChR might prevent the interaction of β‐arrestin‐1 and NLRP3 protein and subsequently inhibit the NLRP3 inflammasome activation Figure 7.
Figure 6.

PNU282987 suppresses the interaction of β‐arrestin‐1 and NLRP3 protein in LPS/ATP‐stimulated BV2 microglia. BV2 microglia were treated with vehicle or PNU282987 (10 μmol/L) for 10 minutes and then were left without stimulation or stimulated with LPS (100 ng/mL) and ATP (1 mmol/L) for 12 hours. (A‐B) β‐arrestin‐1, NLRP3 protein, and nuclei (with DAPI) were stained and then observed with confocal laser scanning microscope. LPS/ATP stimulation increased the colocalization of NLRP3 protein (red) and β‐arrestin‐1 (green), which was significantly inhibited by treatment with PNU282987 (n = 6 per group). PNU, PNU282987. *P < 0.05 vs control; ## P < 0.01 vs LPS/ATP. (C) BV2 microglia were lysated and then the interaction of β‐arrestin‐1 and NLRP3 protein was detected by immunoprecipitation. LPS/ATP stimulation increased the bind of NLRP3 protein and β‐arrestin‐1, which was inhibited by treatment with PNU282987. Three independent experiments were repeated. PNU, PNU282987
Figure 7.

Schematic illustration of α7 nicotinic acetylcholine receptor (α7nAChR) inhibits NLRP3 inflammasome activation. Upon LPS/ATP stimulation, β‐arrestin‐1 binds with NLRP3 protein and facilitates the formation and activation of NLRP3 inflammasome complex. Activation of α7nAChR with PNU282987 not only could decrease the expression of β‐arrestin‐1, but also inhibit the bind of β‐arrestin‐1 with NLRP3 protein, which subsequently suppresses the NLRP3 inflammasome activation
4. DISCUSSION
So far, the alleviative and protective role of α7nAChR in MS or EAE has been demonstrated by several studies, for which activating α7nAChR through the specific agonist has been regarded as a potential therapy in the treatment of MS.9, 25, 26 However, the underlying mechanisms remain unclarified, which to a large extent limits the development of new drugs taking advantage of this process. This is the first study demonstrating that activating α7nAChR contributes to the inhibition of NLRP3 inflammasome through the β‐arrestin‐1‐mediated manner in EAE, thus uncovering a potential mechanism underlying the protective effect of α7nAChR in EAE. In our current study, we observed an increasing expression of β‐arrestin‐1 and an increasing colocalization of β‐arrestin‐1 and CD45‐positive cells in spinal cord from EAE mice, which was attenuated by pretreatment with PNU282987, a specific α7nAChR agonist. We then reported that activating α7nAChR by PNU282987 significantly inhibited the activation of NLRP3 inflammasome and thus decreased the production of NLRP3 inflammasome‐related cytokines including IL‐1β and IL‐18 both in BV2 microglia stimulated with LPS/ATP in vitro and spinal cord from EAE mice in vivo. Activating α7nAChR could suppress the interaction between β‐arrestin‐1 and NLRP3 protein, which led to NLRP3 inflammasome inhibition and thus alleviated EAE.
Several studies have reported the antiinflammatory effects of α7nAChR in different kinds of diseases.27, 28, 29, 30, 31 For example, it was shown by Jurado‐Coronel et al30 that the administration of nicotine contributed to the attenuation of the severity of Parkinson's diseases (PD) in PD patients through the activation of α7nAChR expressed in glial cells, thus downregulating the inflammatory reaction in CNS and protecting dopaminergic neurons against degeneration. Furthermore, Kimura et al31 reported that central insulin action could regulate the inflammatory response in Kupffer cells through the mediation of α7nAChR‐regulated suppression of IL‐6/STAT3 signaling pathway, thus controlling the pathogenesis of chronic hepatic inflammation in obesity. Several studies from our laboratory also demonstrated the antiinflammatory effects of α7nAChR.5, 32, 33, 34 For example, Liu et al5 showed a beneficial effect of anisodamine on arterial pressure and secretion of tumor necrosis factor‐α and IL‐1β in LPS‐induced shock through triggering the α7nAChR‐dependent antiinflammatory pathway. In addition, Xu et al34 reported that anisodamine/neostigmine combination led to a cholinergic antiinflammatory pathway mediated by α7nAChR, thus contributing to the alleviation of acute lethal crush syndrome. Consistent with those previous studies, here we reported that activating α7nAChR led to an antiinflammatory effect in microglia in vitro and spinal cord from EAE mice in vivo.
The NLRP3 inflammasome is so far the best characterized inflammasome, which belongs to the innate immune system.35 As previously reviewed by us,36 the NLRP3 inflammasome is associated with the initiation and progression of various kinds of diseases, including metabolic diseases, multiple sclerosis, inflammatory bowel disease and other auto‐immune and auto‐inflammatory disorders. Thus, targeting NLRP3 inflammasome has been increasingly considered as an effective or potential therapy in the treatment of several kinds of diseases. For example, it has been demonstrated that the administration of certain small‐molecule inhibitors of NLRP3 inflammasome played an important role in the attenuation of the inflammatory reaction, thus contributing to the alleviation of EAE, pointing out a potential target for the treatment of MS or EAE.23, 37 As discussed above, activating α7nAChR produces an antiinflammatory effect through the induction of “cholinergic antiinflammatory pathway.” It was recently reported by Lu et al13 that activating α7nAChR contributed to the inhibition of NLRP3 inflammasome in peritoneal mouse macrophages and dendritic cells as well as preventing the release of mitochondrial DNA. In accordance with those previous data, in our current study, we further demonstrated the inhibitory effect of activating α7nAChR on microglia as well as EAE mice, showing its therapeutic potential and value in the treatment of inflammation‐ and immune‐related disorders in CNS.
β‐arrestins belong to the family of small molecular proteins, functioning as an endocytic adaptor and mediating trafficking of a variety of cell‐surface receptors, including seven transmembrane receptors.14, 38 It has been recently demonstrated by Lee et al39 that β‐arrestin‐1 KO mice led to the attenuation of severity and symptoms in clinical signs, gross pathology, and histopathology of the colon isolated from experimental colitis mice model through the decreasing production and secretion of inflammatory cytokines including IL‐6, IL‐12, and IL‐22. In addition, it was also reported that overexpression of β‐arrestin‐1 enhanced the severity of rheumatoid arthritis in mice model through the upregulating of inflammatory reaction.40 Those studies imply the roles of β‐arrestin‐1 in the regulation of inflammation. For the association between β‐arrestin‐1 and inflammasome, it was previously showed that β‐arrestin‐1 was critical for the assembly and activation of the NLRP3 and NLRC4 inflammasome.16 Here in our present study, we found that in spinal cord of EAE mice or BV2 microglia challenged with LPS/ATP, the mRNA and protein levels of β‐arrestin‐1 were increased, which was attenuated by pretreatment with PNU282987, a specific α7nAChR agonist. Furthermore, overexpression of β‐arrestin‐1 attenuated the inhibitory effects of activating α7nAChR on the NLRP3 inflammasome. In addition, with the stimulation of LPS/ATP, the interaction of β‐arrestin‐1 and NLRP3 protein was triggered, while the administration of PNU282987 abolished this effect. Taken together, our results demonstrate that β‐arrestin‐1 signaling is involved in the inhibitory effect of α7nAChR on the NLRP3 inflammasome.
Here in our current study, we found that activating α7nAChR through the administration of PNU282987 inhibited the interaction between β‐arrestin‐1 and NLRP3 protein. However, the underlying mechanisms are quite complicated and remain to be explored. A previous study from our laboratory demonstrated that autophagy might be probably involved in the antiinflammatory effects of activating α7nAChR through the administration of PNU282987 in the challenge of LPS loading.41 In that study, we found that the levels of the NLRP3 inflammasome‐related inflammatory cytokines including IL‐1β and IL‐18 were significantly decreased with the activation of α7nAChR and the blockade of autophagy largely attenuated those effects. In combination with our current findings, it is reasonable for us to deduce that autophagy might serve as one of the mechanisms in the inhibitory effects of activating α7nAChR on the interaction between β‐arrestin‐1 and NLRP3 protein. In addition, it was previously reported that the reactive oxygen species (ROS) could activate the NLRP3 inflammasome42 and overexpression of β‐arrestin‐1 contributed to increased mitochondrial ROS generation.43 Furthermore, the activation of α7nAChR was reported to protected against oxidative stress under the inflammatory challenge.44 As the ROS has been demonstrated to be connected with the α7nAChR, NLRP3 inflammasome, and β‐arrestin‐1, the ROS might be another potential mechanism in the process. All in all, to uncover the specific mechanisms in this process, further studies are demanded on this issue.
5. CONCLUSIONS
In this study, we for the first time demonstrated that activating α7nAChR contributed to the inhibition of NLRP3 inflammasome activation through the β‐arrestin‐1‐mediated manner in EAE, thus uncovering a potential and promising therapeutic strategy in the treatment of MS or EAE. However, as the pathogenesis of MS or EAE is quite complicated, further studies are needed to explore the specific mechanisms and novel therapeutic strategies are demanded for the fight against MS.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENT
This work was supported by grants from the National Natural Science Foundation of China (No.81670260) and Natural Science Foundation of Zhejiang Province (No.LQ17C180001).
Ke P, Shao B‐Z, Xu Z‐Q, Chen X‐W, Wei W, Liu C. Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β‐arrestin‐1. CNS Neurosci Ther. 2017;23:875‐884. 10.1111/cns.12758
The first two authors contributed equally to this work.
Contributor Information
Wei Wei, Email: weiw8426@163.com.
Chong Liu, Email: wanlc2004@aliyun.com.
REFERENCES
- 1. Marck CH, Neate SL, Taylor KL, Weiland TJ, Jelinek GA. Prevalence of comorbidities, overweight and obesity in an international sample of people with multiple sclerosis and associations with modifiable lifestyle factors. PLoS One. 2016;11:e0148573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shao BZ, Wei W, Ke P, et al. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther. 2014;20:1021‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rossi B, Constantin G. Live imaging of immune responses in experimental models of multiple sclerosis. Front Immunol. 2016;7:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu C, Su D. Nicotinic acetylcholine receptor alpha7 subunit: a novel therapeutic target for cardiovascular diseases. Front Med. 2012;6:35‐40. [DOI] [PubMed] [Google Scholar]
- 5. Liu C, Shen FM, Le YY, et al. Antishock effect of anisodamine involves a novel pathway for activating alpha7 nicotinic acetylcholine receptor. Crit Care Med. 2009;37:634‐641. [DOI] [PubMed] [Google Scholar]
- 6. Chen L, Liu DH, Zhang X, et al. Baroreflex deficiency aggravates atherosclerosis via alpha7 nicotinic acetylcholine receptor in mice. Vascul Pharmacol. 2016;87:92‐99. [DOI] [PubMed] [Google Scholar]
- 7. Guo JM, Zhang L, Niu XC, et al. Involvement of arterial baroreflex and nicotinic acetylcholine receptor alpha7 subunit pathway in the protection of metformin against stroke in stroke‐prone spontaneously hypertensive rats. Eur J Pharmacol. 2017;798:1‐8. [DOI] [PubMed] [Google Scholar]
- 8. Yu JG, Song SW, Shu H, et al. Baroreflex deficiency hampers angiogenesis after myocardial infarction via acetylcholine‐alpha7‐nicotinic ACh receptor in rats. Eur Heart J. 2013;34:2412‐2420. [DOI] [PubMed] [Google Scholar]
- 9. Nizri E, Irony‐Tur‐Sinai M, Lory O, et al. Activation of the cholinergic anti‐inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol. 2009;183:6681‐6688. [DOI] [PubMed] [Google Scholar]
- 10. Qiao C, Yin N, Gu HY, et al. Atp13a2 deficiency aggravates astrocyte‐mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther. 2016;22:451‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malhotra S, Rio J, Urcelay E, et al. NLRP3 inflammasome is associated with the response to IFN‐beta in patients with multiple sclerosis. Brain. 2015;138:644‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta‐amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu B, Kwan K, Levine YA, et al. alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014;20:350‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shenoy SK, Lefkowitz RJ. beta‐Arrestin‐mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi Y, Feng Y, Kang J, et al. Critical regulation of CD4+ T cell survival and autoimmunity by beta‐arrestin 1. Nat Immunol. 2007;8:817‐824. [DOI] [PubMed] [Google Scholar]
- 16. Mao K, Chen S, Wang Y, et al. beta‐arrestin1 is critical for the full activation of NLRP3 and NLRC4 inflammasomes. J Immunol. 2015;194:1867‐1873. [DOI] [PubMed] [Google Scholar]
- 17. Mansilla MJ, Selles‐Moreno C, Fabregas‐Puig S, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther. 2015;21:222‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moraes AS, Paula RF, Pradella F, et al. The suppressive effect of IL‐27 on encephalitogenic Th17 cells induced by multiwalled carbon nanotubes reduces the severity of experimental autoimmune encephalomyelitis. CNS Neurosci Ther. 2013;19:682‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sone M, Morone N, Nakamura T, et al. Hybrid cellular metabolism coordinated by Zic3 and esrrb synergistically enhances induction of naive pluripotency. Cell Metab. 2017;25:e1106. [DOI] [PubMed] [Google Scholar]
- 20. Guo JM, Liu AJ, Zang P, et al. ALDH2 protects against stroke by clearing 4‐HNE. Cell Res. 2013;23:915‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song DD, Chen Y, Li ZY, et al. Protein tyrosine phosphatase 1B inhibits adipocyte differentiation and mediates TNFalpha action in obesity. Biochim Biophys Acta. 2013;1831:1368‐1376. [DOI] [PubMed] [Google Scholar]
- 22. Ta HM, Le TM, Ishii H, et al. Atf6alpha deficiency suppresses microglial activation and ameliorates pathology of experimental autoimmune encephalomyelitis. J Neurochem. 2016;139:1124‐1137. [DOI] [PubMed] [Google Scholar]
- 23. Coll RC, Robertson AA, Chae JJ, et al. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Torre‐Minguela C, Mesa Del Castillo P, Pelegrin P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol. 2017;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hao J, Simard AR, Turner GH, et al. Attenuation of CNS inflammatory responses by nicotine involves alpha7 and non‐alpha7 nicotinic receptors. Exp Neurol. 2011;227:110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang W, St‐Pierre S, Roy P, et al. Infiltration of CCR2 + Ly6Chigh proinflammatory monocytes and neutrophils into the central nervous system is modulated by nicotinic acetylcholine receptors in a model of multiple sclerosis. J Immunol. 2016;196:2095‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dash PK, Zhao J, Kobori N, et al. Activation of alpha 7 cholinergic nicotinic receptors reduce blood‐brain barrier permeability following experimental traumatic brain injury. J Neurosci. 2016;36:2809‐2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi S, Liang D, Bao M, et al. Gx‐50 Inhibits neuroinflammation via alpha7 nAChR activation of the JAK2/STAT3 and PI3K/AKT pathways. J Alzheimers Dis. 2016;50:859‐871. [DOI] [PubMed] [Google Scholar]
- 29. Skok M, Lykhmus O. The role of alpha7 nicotinic acetylcholine receptors and alpha7‐specific antibodies in neuroinflammation related to alzheimer disease. Curr Pharm Des. 2016;22:2035‐2049. [DOI] [PubMed] [Google Scholar]
- 30. Jurado‐Coronel JC, Avila‐Rodriguez M, Capani F, et al. Targeting the nicotinic acetylcholine receptors (nAChRs) in astrocytes as a potential therapeutic target in parkinson's disease. Curr Pharm Des. 2016;22:1305‐1311. [DOI] [PubMed] [Google Scholar]
- 31. Kimura K, Tanida M, Nagata N, et al. Central insulin action activates kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep. 2016;14:2362‐2374. [DOI] [PubMed] [Google Scholar]
- 32. Qian J, Zhang JM, Lin LL, et al. A combination of neostigmine and anisodamine protects against ischemic stroke by activating alpha7nAChR. Int J Stroke. 2015;10:737‐744. [DOI] [PubMed] [Google Scholar]
- 33. Liu AJ, Zang P, Guo JM, et al. Involvement of acetylcholine‐alpha7nAChR in the protective effects of arterial baroreflex against ischemic stroke. CNS Neurosci Ther. 2012;18:918‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu ZQ, Shao BZ, Ke P, et al. Combined administration of anisodamine and neostigmine rescued acute lethal crush syndrome through alpha7nAChR‐dependent JAK2‐STAT3 signaling. Sci Rep. 2016;6:37709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eigenbrod T, Dalpke AH. Bacterial RNA: an underestimated stimulus for innate immune responses. J Immunol. 2015;195:411‐418. [DOI] [PubMed] [Google Scholar]
- 36. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med. 2015;21:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steen A, Larsen O, Thiele S, Rosenkilde MM. Biased and g protein‐independent signaling of chemokine receptors. Front Immunol. 2014;5:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee T, Lee E, Irwin R, et al. beta‐Arrestin‐1 deficiency protects mice from experimental colitis. Am J Pathol. 2013;182:1114‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li P, Cook JA, Gilkeson GS, et al. Increased expression of beta‐arrestin 1 and 2 in murine models of rheumatoid arthritis: isoform specific regulation of inflammation. Mol Immunol. 2011;49:64‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shao BZ, Ke P, Xu ZQ, et al. Autophagy plays an important role in anti‐inflammatory mechanisms stimulated by alpha7 nicotinic acetylcholine receptor. Front Immunol. 2017;8:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Philip JL, Razzaque MA, Han M, et al. Regulation of mitochondrial oxidative stress by beta‐arrestins in cultured human cardiac fibroblasts. Dis Model Mech. 2015;8:1579‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Navarro E, Buendia I, Parada E, et al. Alpha7 nicotinic receptor activation protects against oxidative stress via heme‐oxygenase I induction. Biochem Pharmacol. 2015;97:473‐481. [DOI] [PubMed] [Google Scholar]