

Summary
Aims
To detect specific oculomotor deficits in preclinical stage of spinocerebellar ataxia type 3 (SCA3) and evaluate whether these abnormalities prove useful as potential biomarkers of disease progression.
Methods
A Chinese cohort of 56 patients with SCA3, including 12 preclinical carriers of SCA3 (pre‐SCA3) and 44 manifest SCA3, and 26 healthy control individuals were recruited. We performed a detailed investigation on central oculomotor performance including fixation, gaze, smooth pursuit, prosaccade, and antisaccade using video‐oculography.
Results
Common oculomotor features of pre‐SCA3 included square‐wave jerk during central fixation and gaze holding, impaired vertical smooth pursuit, slow upward saccade, and increased antisaccade error rate. In our SCA3 cohort, all oculomotor parameters were correlated with the score of the Scale for the Assessment and Rating of Ataxia, whilst some of them were correlated with disease duration.
Conclusion
This study showed that a series of neuropathological changes reflected by oculomotor abnormalities appeared preferentially in preclinical stage of SCA3. Accordingly, objective oculomotor preclinical signs may be useful to detect the optimum time‐point for therapeutic interventions in future clinical trials of SCA3. Larger and longitudinal data are warranted to confirm our results.
Keywords: biomarker, oculomotor, preclinical carrier, spinocerebellar ataxia, video‐oculography
1. Introduction
Spinocerebellar ataxias (SCAs) comprise a group of autosomal dominant neurodegenerative progressive ataxias that are classified according to specific genetic mutations. Among them, spinocerebellar ataxia type 3 (SCA3) is the most frequent inherited SCAs throughout the world, which is caused by an abnormal CAG trinucleotide expansion on ATXN3 gene.1 With predictive genetic testing in family members, carriers of mutations in SCA3 and other SCA genes can be detected in a phase in which they do not yet experience any cerebellar ataxic signs. This preclinical stage is attracting increasing research interest, because they could provide a time window for early therapeutic intervention before irreversible brain damage has occurred.2 Currently there is not a cure for SCA3, but several candidate treatments with potential disease‐modifying effects are being explored.3 In this respect, recognition of early signs that develop before onset of ataxia is mandatory to find surrogate biomarkers in future clinical trial.4
Because of the frequent involvement of the cerebellum and brainstem, oculomotor disturbances, including square‐wave jerk (SWJ), gaze‐evoked nystagmus (GEN), impaired smooth pursuit eye movement (SPEM), and dysmetric saccade, are main characteristics of SCA3, some of which have also been recognized as distinguishing features between SCA3 and other SCAs.5, 6, 7, 8, 9, 10 A growing body of evidence indicates that the preclinical stage of SCAs is already characterized by detectable oculomotor signs, including slowing of saccade in preclinical stage of SCA2 and SCA7, saccade dysmetria in SCA6, and impaired SPEM in SCA17.2, 11 Recently, GEN was firstly investigated by clinical observation in preclinical stage of SCA3 (pre‐SCA3).12, 13 However, information is lacking regarding other oculomotor domains that occur as a result of the SCA3 degenerative process in preclinical stage.
For this study, we performed quantitative oculomotor recordings in pre‐SCA3 and manifest SCA3. Some of the central oculomotor domains have not been assessed in SCA3 before. We emphasized the potential value of using oculomotor parameters as surrogate outcome measures in future clinical trials of SCA3 allowing intervention before the appearance of ataxia.
2. Methods
2.1. Participants
In this study, we consecutively recruited 12 participants of pre‐SCA3 (six males) and 44 SCA3 patients (21 males) in the neurogenetic outpatient clinic of the First Affiliated Hospital of Sun Yat‐Sen University, Guangdong, China, from October 2014 to October 2016. All patients were diagnosed clinically and molecularly as SCA3. Healthy normal controls (NC, 26 participants with 14 males) were recruited via our physical examination center and were matched with patients for their age and sex. None of the control subjects had a suspicious family history of ataxia (Table 1).
Table 1.
Demographic, genetic, ataxia, and nonataxia features of participants
| Characteristics | Pre‐SCA3 | SCA3 | NC | P | Post hocs |
|---|---|---|---|---|---|
| No. | 12 | 44 | 26 | NA | NA |
| Male:female | 6:6 | 21:23 | 14:12 | >.05 | NA |
| Age, year | 29.8±7.4 | 39.8±10.9 | 35.7±9.5 | .0091 | SCA3>Pre‐SCA3 |
| Age at onset of gait ataxia, year | NA | 35.2±10.2 | NA | NA | NA |
| Disease duration, year | NA | 4.5±2.2 | NA | NA | NA |
| CAG repeat length | 72.2±3.6 | 74.0±3.1 | NA | >.05 | NA |
| MMSE | 30±0.0 | 29.2±1.4 | 29.6±1.0 | >.05 | NA |
| Barthel index | 100±0.0 | 91.4±19.0 | 100±0.0 | >.05 | NA |
| SARA score | 0.0 (0.0‐1.0) | 8.3 (5.0‐12.9) | 0.0 (0.0‐0.0) | <.001 | SCA3>Pre‐SCA3, NC |
| Nonataxia features | |||||
| Hyperreflexia in lower limbs (%) | 8.3 | 47.7 | 0 | <.001 | NA |
| Hyporeflexia in lower limbs (%) | 8.3 | 20.5 | 0 | .026 | NA |
| Extensor plantar (%) | 8.3 | 47.7 | 0 | <.001 | NA |
| Impaired vibration sense (%) | 0 | 9.1 | 0 | >.05 | NA |
| Extrapyramidal signs (%) | 0 | 9.1 | 0 | >.05 | NA |
| Gaze limitations in vertical direction (%) | 0 | 15.9 | 0 | .047 | NA |
Pre‐SCA3, preclinical SCA3 with mild coordination deficits (SARA<3) and proven ATXN3 gene mutation; SCA3, spinocerebellar ataxia type 3; NC, normal controls; SARA, Scale for the Assessment and Rating of Ataxia; MMSE, Mini‐mental State Examination; NA, Not available.
Demographic and genetic data are given as mean±SD whilst SARA scores are given as median (IQR), percentage of nonataxia features are presented.
We also excluded the following conditions: (i) a lesion in the cerebellum or brainstem on magnetic resonance imaging, except for atrophy; (ii) a history of recent peripheral vestibulopathy and ophthalmic diseases; (iii) medications that could affect vestibular and cerebellar function 72 hours before the test; and (iv) tremor on the head, frequent blink or ptosis that could affect recording.8
The protocol of the experiment was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat‐Sen University [No. (2014) 23]. Before each testing session, a written informed consent was signed by the participant. We performed genetic testing to confirm the diagnosis of SCA3 only in siblings who were older than 18 years.
2.2. Clinical assessment
General information, such as sex, age, age at gait ataxia onset, disease duration, and CAG copies, was collected. Age at gait ataxia onset was determined by a structured interview in which patient and close family members were asked about the age of start of permanent and progressive gait instability.8
To measure the severity of ataxia, we used the Scale for the Assessment and Rating of Ataxia (SARA) that has been validated.6, 14, 15, 16 The sum score of SARA ranges from 0 to 40 with eight domains. SARA was not designed to measure oculomotor deficits.
The strict, objective inclusion criteria of preclinical carrier of SCAs have already been well documented.2 Pre‐SCA3 is now defined as proven SCA3 mutation with mild coordination deficits (SARA<3), because the validation of SARA indicated that a score of 3 or more differentiates controls from SCA patients with manifest ataxia. Abnormal test results, such as oculomotor deficits, might be detected in this stage.
The Mini‐Mental State Examination (MMSE) is a measure of general cognitive function, with scores ranging from 0 (severe impairment) to 30 (no impairment). The Barthel Index was used to quantify the participants' ability of daily living. For both, we used Chinese versions.
2.3. Central oculomotor battery
Eye movements were recorded binocularly using video‐oculography (VOG) (Interacoustics VO425, www.interacoustics.com/VNG) at a sampling rate of 105 Hz. Participants were asked to wear the VOG goggles and put their head on a fixator to avoid any movement. A 5‐seconds rest for eye closing was offered after each test to avoid frequent blinks. Visual stimulation was displayed on a screen (visual angle, 40°×30°) from a fixed viewing distance of 150 cm. Each oculomotor session started with a calibration procedure. All participants underwent a predesigned protocol of eye movement recording described as below to evaluate fixation, gaze, smooth pursuit, and saccades.17, 18
To test fixation stability, a stationary red laser dot was projected to the center of the screen, and participants were asked to fixate the dot without any eye movement or blinks. A 5‐seconds practice trial was performed, followed by a further 20‐seconds test trial. Square‐wave jerk (SWJ) was defined as small saccades of 0.5°‐5° in amplitude, taking the gaze away from the target position, with an intersaccadic interval of 200‐400 milliseconds, followed by another saccade with an amplitude similar to the first, which takes gaze back toward the target position.
Gaze holding was obtained whilst participants looked at the target displaced ±30° horizontally and ±20° vertically away from the center for 20 seconds. Horizontal gaze‐evoked eye movement (GEEM), including SWJ or GEN, could be induced in SCA3 cases.
To perform smooth pursuit eye movement (SPEM), subjects were asked to track the target moving with a constant velocity of 10°/seconds from −10° to 10° and vice versa sinusoidally for 10 turns horizontally and vertically.
The prosaccade task began with a central target for a variable duration (1000‐2000 milliseconds). Immediately after the central target disappeared and a peripheral target pseudo‐randomly elicited and lasted for 800‐1500 milliseconds, subjects were requested to move their eyes to the new one as quickly and correctly as possible to make a prosaccade (horizontal amplitude 20°, vertical 15°). A four‐round practice trial was performed, followed by a further 10‐round test trial.
The antisaccade task began with a central target, then moved randomly to the left (six rounds) or to the right (seven rounds) horizontally at irregular intervals (1000‐2000 milliseconds) with the same amplitude of 20°. After a constant interval (1500 milliseconds), the stimulus returned to the center. When the central target disappeared and a horizontal peripheral target appeared, subjects were asked to voluntarily move to the opposite position with the same amplitude. A six‐round practice trial was performed, followed by a further 13‐round test trial.
2.4. Statistical analysis
All oculomotor data were analyzed by the OtoAccess database software (Interacoustics, http://www.interacoustics.com/otoaccess). All tests were administered in a fixed style, and the recordings with synchronous videos were evaluated by two examiners (CW, JWZ) independently to confirm the presence of oculomotor abnormalities and mark interferential events such as blinking. Normative data were obtained from the age‐matched healthy controls.
The number of SWJ during the fixation period was counted, and the frequency (Hz) and average amplitude (°) of SWJ were measured. Frequent SWJ (9‐16 per minute), also called square‐wave oscillations, is considered pathological.19
The number of GEN or SWJ during the gaze‐holding period was counted, the frequency (Hz) and average amplitude (°) of GEN or SWJ were measured, and the maximum value of the two directions was used as the parameter to reflect the severity.
Mean pursuit gain (mean velocity of participant/velocity of the target×100%) was calculated. A value<mean±2SD of the NC data or abnormal eye drift during the smooth process represents impaired SPEM.
Saccade latency (milliseconds) was calculated as the time between onset of the target and the start of the main saccade. A value>mean±2SD of the NC data represents increased latency. Peak saccade velocity (°/seconds) is the maximum eye velocity between saccade onset and offset, which is very closely related to saccade amplitude. Therefore in the comparison of peak saccadic velocity between groups, saccade amplitude was included as a covariate so that differences in velocity could be analyzed independently from differences in saccadic amplitude. A value<mean±2SD of the NC data represents reduced velocity. Saccadic accuracy (saccade amplitude/target amplitude×100%) was calculated as the difference between the amplitude of the main saccade and the eccentricity of the target. A value>mean±2SD of the NC data represents overshoot (hypermetria), whereas a value<mean±2SD represents undershoot (hypometria).
An antisaccade error is the wrong direction of the first eye movement after eccentric target onset with an amplitude >3°. Total antisaccadic error rate (total antisaccadic errors/number of trials) was calculated.20
For descriptive statistics of demographic, genetic, and measurable oculomotor parameters, means and standard deviations (SD) were obtained. SARA scores are given as median (IQR). Frequencies of nonataxia features were presented. The normality of the distribution for quantitative variables was assessed by the Kolmogorov‐Smirnov test. A χ 2 test was used for categorical data. The ordinary one‐way ANOVA was used to compare oculomotor parameters, age and SARA scores among the pre‐SCA3, SCA3, and NC groups followed by a Tukey's multiple comparisons test. Correlation analysis was performed with the Pearson test. Statistical significance was set at P<.05. Normal values of oculomotor parameters were defined as the mean±2SD. Data analysis was performed using the SPSS 19.0.
3. Results
3.1. Clinical characteristics of participants
The demographic characteristics, SARA score, and key nonataxia symptoms of all participants are shown in Table 1. The pre‐SCA3 group had a median SARA score of 0.0 without any subjective complaint of gait problems. Median SARA score in the SCA3 group was 8.3 (IQR: 5.0‐12.9), which represented that most of the patients could walk independently without external support. Key nonataxia features, such as hyperreflexia in lower limbs and extensor plantar, could be detected in 8.3% (1/12) of pre‐SCA3 carriers and 47.7% (21/44) of the SCA3 patients, whereas none in NC group (P<.001). In addition, 20.5% (9/44) of the SCA3 patients presented hyporeflexia, whereas 8.3% (1/12) in pre‐SCA3 and none in NC group presented hyporeflexia (P=.026). Gaze limitations in vertical direction could only be detected in 15.9% (7/44) of the SCA3 cohort. Impaired vibration sense and extrapyramidal signs were rare in SCA3 group (Table 1).
3.2. Central oculomotor abnormalities in pre‐SCA3
Detail characteristics of twelve pre‐SCA3 carriers, including age, CAG repeats, SARA score, oculomotor performance, key nonataxia features, and awareness of carrier status, are listed in Table 2. Several oculomotor alterations could be detected in pre‐SCA3 as presented in Figure 1. Central fixation was impaired by SWJ (Figure 1A1,B1). Meanwhile, pre‐SCA3 carriers could show GEEM, which could be subtle and might be ignored during clinical observation. Only one carrier (No. 6 carrier in Table 2) showed horizontal and vertical GEN, whereas others presented SWJ during gaze holding with different frequency (Figure 1A2,B2). Carriers could have severe abnormal eye drift during the SPEM process, which affected the gain of SPEM in both vertical and horizontal directions (Figure 1A3‐4,B3‐4). Slow peak upward saccade velocity and increased antisaccade error rate could also be detected (Figure 1C,D).
Table 2.
Characteristics of twelve preclinical SCA3 carriers
| No. | Age (years/sex) | CAG repeats | SARA score | Oculomotor performance | Key nonataxia features | Aware of carrier status |
|---|---|---|---|---|---|---|
| 1 | 35/F | 67/27 | 0 | Frequent SWJ during central fixation and gaze holding, vertical and horizontal SPEM was badly damaged, prosaccade and antisaccade were normal. | Hyporeflexia in lower limbs | No |
| 2 | 41/M | 71/32 | 1 | Frequent SWJ during central fixation, frequency of SWJ increased after gaze holding, impaired vertical and horizontal SPEM. | None | Yes |
| 3 | 31/F | 72/14 | 0 | Frequent SWJ during central fixation and gaze holding, impaired vertical and horizontal SPEM, reduced vertical saccade velocity, increased antisaccade error rate. | None | Yes |
| 4 | 32/F | 74/31 | 1 | Frequent SWJ during central fixation and gaze holding, increased antisaccade error rate. | Hyperreflexia in lower limbs, extensor plantar | Yes |
| 5 | 36/F | 68/18 | 0 | Frequent SWJ during central fixation and gaze holding, impaired vertical SPEM. | None | Yes |
| 6 | 39/M | 71/14 | 1 | Frequent horizontal GEN, vertical GEN during upward gaze holding. | None | Yes |
| 7 | 22/M | 74/14 | 0 | Frequent SWJ during gaze holding, impaired vertical SPEM, reduced vertical saccade velocity. | None | Yes |
| 8 | 20/M | 75/15 | 0 | Paroxysmal SWJ during central fixation and frequency of SWJ increased after gaze holding | None | Yes |
| 9 | 32/M | 75/14 | 1 | Paroxysmal SWJ during central fixation and frequency of SWJ increased after gaze holding, impaired vertical and horizontal SPEM, reduced vertical saccade velocity, increased antisaccade error rate. | None | Yes |
| 10 | 27/F | 67/27 | 0 | Frequent SWJ during central fixation and gaze holding. | None | Yes |
| 11 | 24/F | 73/14 | 0 | Frequent SWJ during central fixation and gaze holding, impaired vertical SPEM. | None | No |
| 12 | 19/M | 79/27 | 0 | Frequent SWJ during central fixation and gaze holding, impaired horizontal and vertical SPEM, increased antisaccade error rate. | None | No |
SWJ, square‐wave jerk; GEN, gaze‐evoked nystagmus; SPEM, smooth pursuit eye movement.
Figure 1.
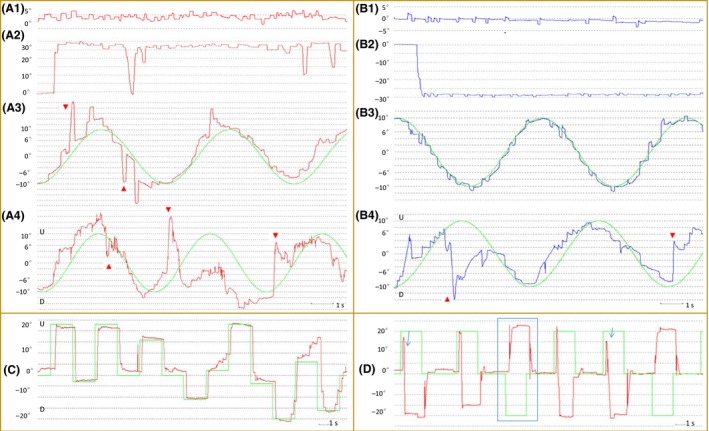
Oculomotor deficits in preclinical SCA3 carriers. A woman aged 35 with SARA=0 and CAG=67 (No. 1 carrier in Table 2) had (A1) frequent SWJ during central fixation. (A2) She also had SWJ during right gaze and with occasionally difficulty in gaze holding. (A3) Abnormal eye drift during the smooth SPEM process could be recorded. (A4) Vertical SPEM was badly damaged. A man aged 41 with SARA=1 and CAG=71 (No. 2 carrier in Table 2) had (B1) frequent SWJ during central fixation. (B2) The frequency of SWJ increased after left gaze holding and (B3) SWJ remained during SPEM. (B4) Vertical SPEM was also badly damaged. (C) Upward saccade in a woman aged 31 with SARA=0 and CAG=72 (No. 3 carrier in Table 2); her upward velocity was 283°/seconds, whereas downward was 339°/seconds within a normal amplitude, which were poorer than NC. (D) Increased antisaccade error rate in a woman aged 32 with SARA=1 and CAG=74 (No. 4 carrier in Table 2). Symbols: blue or red line, left or right eye movement track; 0°, central position; positive or negative number degree, right or left position except in A4, B4, C, where U, upward and D, downward; green line, track of the target; red arrow, abnormal eye drift; blue box, normal antisaccade; blue arrow, wrong antisaccade. SWJ, square‐wave jerk; SPEM, smooth pursuit eye movement; NC, normal controls
Mean and standard deviation performance metrics for the oculomotor parameters of all the pre‐SCA3, SCA3, and NC participants are given in Table 3:
Table 3.
Mean and standard deviation performance metrics for pre‐SCA3, SCA3 and control groups on the oculomotor parameters
| Oculomotor signs | Pre‐SCA3 | SCA3 | NC | P | Post hocs |
|---|---|---|---|---|---|
| Fixation stability | |||||
| Frequency of SWJ (Hz) | 0.54±0.47 | 0.79±0.56 | 0.06±0.13 | <.0001 | NC<Pre‐SCA3, SCA3 |
| Average amplitude of SWJ (°) | 1.15±0.77 | 1.53±0.93 | 0.23±0.43 | <.0001 | NC<Pre‐SCA3, SCA3 |
| Gaze | |||||
| Frequency of horizontal GEEM (Hz) | 0.83±0.5 | 1.65±0.75 | 0.09±0.15 | <.0001 | SCA3>Pre‐SCA3>NC |
| Average amplitude of horizontal GEEM (°) | 1.60±0.66 | 3.40±2.30 | 0.31±0.55 | <.0001 | SCA3>Pre‐SCA3>NC |
| Frequency of horizontal SWJ evoked by upward gaze (Hz) | 0.63±0.52 | 1.03±0.70 | 0.07±0.15 | <.0001 | NC<Pre‐SCA3, SCA3 |
| SPEM | |||||
| Horizontal mean pursuit gain (%) | 81.3±8.0 | 69.4±10.8 | 87.9±4.1 | <.0001 | NC, Pre‐SCA3>SCA3 |
| Vertical mean pursuit gain (%) | 64.4±18.2 | 67.8±15.4 | 81.8±4.5 | .0003 | NC>Pre‐SCA3, SCA3 |
| Prosaccade | |||||
| Upward saccade latency (milliseconds) | 200±27.6 | 205±36.5 | 203±23.2 | >.05 | NC=Pre‐SCA3=SCA3 |
| Horizontal peak saccade velocity (°/seconds) | 529±66.0 | 527±130.3 | 524±56.9 | >.05 | NC=Pre‐SCA3=SCA3 |
| Upward peak saccade velocity (°/seconds) | 424±81.6 | 338±109.3 | 563±100.5 | <.0001 | SCA3<Pre‐SCA3<NC |
| Horizontal saccadic accuracy (%) | 98.4±3.7 | 92.8±16.9 | 98.2±3.5 | >.05 | NC=Pre‐SCA3=SCA3 |
| Upward saccadic accuracy (%) | 93.0±9.0 | 85.1±16.0 | 94.4±7.4 | .0135 | SCA3<NC |
| Antisaccade | |||||
| Total antisaccadic error rate (%) | 36.4±24.1 | 66.8±22.9 | 19.2±14.0 | <.0001 | SCA3>Pre‐SCA3>NC |
Pre‐SCA3, preclinical SCA3; SCA3, spinocerebellar ataxia type 3; NC, normal controls; SWJ, square‐wave jerk; GEEM, gaze‐evoked eye movement; SPEM, smooth pursuit eye movement.
Oculomotor parameters are given as mean±SD.
Pre‐SCA3 compared with NC: Frequency and average amplitude of SWJ during fixation and gaze holding, antisaccade error rate were higher in the pre‐SCA3 group. Vertical SPEM gain and upward saccade velocity were lower in pre‐SCA3 group. Upward saccade latency, horizontal and upward saccade accuracy, and horizontal saccade velocity showed no differences. Horizontal SPEM gain, although with some individual value out of the range of NC, did not statistically differ from the NC group (Table 3).
Pre‐SCA3 compared with SCA3: Frequency and average amplitude of GEEM, antisaccadic error rate were lower in the pre‐SCA3 group, whereas horizontal SPEM gain and upward peak saccade velocity were higher. No statistical difference was found in other oculomotor parameters. However, pre‐SCA3 carriers showed normal horizontal saccade accuracy, whereas SCA3 could present either hypermetric or hypometric saccade individually (Table 3).
3.3. Correlation between oculomotor performance and clinical data in SCA3
We explore the specific relationships between oculomotor parameters and clinical data in SCA3 cohort (both pre‐SCA3 and manifest SCA3 groups together, see Table. 4). There was a positive correlation between SARA score and frequency of SWJ, average amplitude of SWJ, frequency of horizontal GEEM, average amplitude of horizontal GEEM, upward saccade latency and antisaccadic error rate, and a negative correlation between SARA score and horizontal and vertical mean pursuit gain, horizontal and upward peak saccade velocity, horizontal and upward saccadic accuracy. In addition, frequency and average amplitude of horizontal GEEM, horizontal mean pursuit gain, upward saccade latency, peak saccade velocity and accuracy, horizontal saccadic accuracy, and total antisaccadic error rate were correlated with disease duration. Meanwhile, we also found the well‐known correlations between age at onset of gait ataxia and CAG repeat length in expanded allele, and between SARA score and disease duration (Table 4).
Table 4.
Correlations between oculomotor parameters and clinical data in SCA3
| CAG repeat length in expanded allele | Disease duration | SARA score | |
|---|---|---|---|
| Frequency of SWJ | NS | NS | 0.319* |
| Average amplitude of SWJ | NS | NS | 0.287* |
| Frequency of horizontal GEEM | NS | 0.550** | 0.593** |
| Average amplitude of horizontal GEEM | NS | 0.526** | 0.760** |
| Horizontal mean pursuit gain | NS | −0.470** | −0.642** |
| Vertical mean pursuit gain | NS | NS | −0.347** |
| Upward saccade latency | NS | 0.280* | 0.360** |
| Horizontal peak saccade velocity | NS | NS | −0.345** |
| Upward peak saccade velocity | NS | −0.282* | −0.397** |
| Horizontal saccadic accuracy | NS | −0.327* | −0.480** |
| Upward saccadic accuracy | NS | −0.471** | −0.547** |
| Total antisaccadic error rate | 0.330** | 0.360** | 0.533** |
| Age at onset of gait ataxia | −0.835** | NS | NS |
| SARA score | NS | 0.742** | NA |
SCA, spinocerebellar ataxia; SARA, Scale for the Assessment and Rating of Ataxia; SWJ, square‐wave jerk; GEEM, gaze evoked eye movement; SPEM, smooth pursuit eye movement; NS, not significant; NA, not available.
This study included both preclinical and manifest SCA3 together. Pearson correlation coefficients, two‐tailed are given.
*P<.05, **P<.01.
4. Discussion
To our knowledge, this is the first study to have demonstrated that SWJ during central fixation and gaze holding, antisaccade error, slow upward saccade, and impaired vertical SPEM were common features of pre‐SCA3. These oculomotor abnormalities could precede the onset of gait ataxia and other key nonataxia signs and were correlated with disease severity reflected by SARA score.
A growing body of evidence indicates that carriers in pre‐SCAs are already subject to the earliest pathophysiological changes.2 A recent study showed that horizontal GEN was more often detected in SCA3 carriers than in noncarriers [10 (39%) of 26 vs 1 (5%) of 20; P=.013].12 In another cohort of 48 pre‐SCA3, 17% of the participants demonstrated GEN by clinical observation. Carriers with nystagmus at evaluation were closer to reported age at onset than individuals without nystagmus.13 However, these studies were mainly based on clinical observation. With the help of quantitative VOG, our study showed that pre‐SCA3 had a higher frequency of GEEM than previous reported (all pre‐SCA3 presented GEEM). We also confirmed this GEEM mostly as SWJ in pre‐SCA3, only one carrier showed horizontal and vertical GEN. Whereas GEEM mostly showed as GEN in manifest SCA3, with the frequency and average amplitude increased in the course of the disease. The generation of SWJ appears to involve widespread functional networks of neural tissue.19 In the case of SCA3, the cerebellar hypotheses might better explain the transition from SWJ to GEN in manifest cases.
Furthermore, we also screened for several other earliest functional oculomotor alterations for the first time, including SWJ during fixation, impaired SPEM, reduced upward saccade velocity, and increased antisaccade error rate in pre‐SCA3. When compared with NC, these parameters showed significant difference, which are remarkable because in the RISCA study, a study of individuals at risk for SCA1‐3 and 6, oculomotor testing did not identify differences between carriers and noncarriers of each mutation, except a higher rate of horizontal GEN in SCA3 reported previously.12 Compared with Inventory of Non‐Ataxia Symptoms (INAS), a recently designed tool to assess nonataxia and oculomotor symptoms in SCAs which relies on pure clinical observation, noninvasive VOG studies would be more sensitive, quantitative, maneuverable, repeatable, and recordable.21 Our data further implicate that SCA3 mutation carriers, along with SCA2, 6, 7, and 17,2 have manifestations, which was caused by neurodegeneration in different oculomotor neurocircuitries, that could be detected before the clinical onset of gait problem. Future study with expanded pre‐SCA3 numbers should focus on the relationship between appearance and severity of oculomotor findings and age at predicted onset of gait problem to explore the predictive value of oculomotor deficits. In addition, we found that all oculomotor parameters were correlated with the severity of ataxia evaluated by SARA score, whilst some of them were correlated with disease duration in our SCA3 cohort, which implies that progressive alteration in oculomotor neurocircuitries is associated with SCA3 pathogenesis and neurodegeneration. Similar to Huntington's disease and familial Alzheimer's disease,22, 23 potential treatments for SCA3 are being developed, which should lead to prevention or slow progression. Our results suggest oculomotor parameters may work as potential markers in future trials involved preclinical SCA3 carriers.24
In addition, using this central oculomotor battery, we also enriched the oculomotor phenotype of SCA3 by firstly capturing vertical gaze‐evoked SWJ, impaired vertical SPEM, reduced vertical saccade, and increased antisaccade error in both preclinical and symptomatic patients. When we designed this battery, much attention was paid to vertical eye movements due to extensive atrophy of midbrain in an MRI study of SCA3.25 We found that reduced vertical saccade velocity and impaired vertical SPEM could be tested in pre‐SCA3, which might reflect that the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) was preclinically affected. However, cognitive impairment is another important nonataxia symptom of SCA3 in Han Chinese.26 The antisaccade task was introduced to test inhibitory control, which strongly correlated with neuropsychological tests of executive function in multiple diseases20 and was controlled by a well‐described frontoparietal cortical network.27 We found increased antisaccade error rate in both pre‐SCA3 and SCA3 cohorts. The underlying pathophysiological changes of antisaccade in SCA3 should be further studied after applying functional MRI and cognitive battery in the future. Our study also confirmed that cerebellar oculomotor signs, including broken up horizontal SPEM, GEN, and dysmetric saccades, were key clinical features of manifest SCA3.8, 9, 10, 13, 28 However, we showed a much lower prevalence of brainstem oculomotor signs, including horizontal and vertical gaze limitations, and slowing of horizontal saccades than previously reported.6 This discrepancy is probably due to lack of advanced‐stage patients involved in our cohorts (Median SARA score in our SCA3 group is only 8.3).
Several limitations of our study need to be addressed. This is a preliminary study with limited patient number in the pre‐SCA3 group. Thus, our study has an exploratory character and the results should be interpreted with caution. Likewise, the nonsignificance of some results might have been due to this small sample size too. Peripheral vestibular oculomotor abnormalities also should be detected in the future study.8
In summary, we found that oculomotor parameters in several domains could be detected in preclinical SCA3 mutation carriers and showed changes in increasing magnitude across our cohort. Clinical trials that are designed to prevent or delay ataxia may adopt oculomotor parameters as potential surrogate biomarkers to validate the effect of disease‐modifying agents.
Conflict of Interests
The authors declare no conflict of interest.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (30671154, 81500968 http://npd.nsfc.gov.cn/fundingProjectSearchAction.action). We thank Dr. Hongyan Jiang, Min Liu, Xiaoyun Qin, Guangzhi Li, Peng Yang, Beibei Lin (Department of Otorhinolaryngology, First Affiliated Hospital of Sun Yat‐Sen University, Guangzhou, Guangdong, PR China) for the guidance of video‐oculography. The authors sincerely thank the participants for their help and willingness to participate in this study and the anonymous reviewers for improving this manuscript.
Wu C, Chen D‐B, Feng L, et al. Oculomotor deficits in spinocerebellar ataxia type 3: Potential biomarkers of preclinical detection and disease progression. CNS Neurosci Ther. 2017;23:321‐328. 10.1111/cns.12676
References
- 1. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 2. Maas RP, van Gaalen J, Klockgether T, van de Warrenburg BP. The preclinical stage of spinocerebellar ataxias. Neurology. 2015;85:96–103. [DOI] [PubMed] [Google Scholar]
- 3. Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2015;14:985–991. [DOI] [PubMed] [Google Scholar]
- 4. Jacobi H, du Montcel ST, Bauer P, et al. Long‐term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol. 2015;14:1101–1108. [DOI] [PubMed] [Google Scholar]
- 5. Moscovich M, Okun MS, Favilla C, et al. Clinical evaluation of eye movements in spinocerebellar ataxias: a prospective multicenter study. J Neuroophthalmol. 2015;35:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmitz‐Hubsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology. 2008;71:982–989. [DOI] [PubMed] [Google Scholar]
- 7. Jacobi H, Hauser TK, Giunti P, et al. Spinocerebellar ataxia types 1, 2, 3 and 6: the clinical spectrum of ataxia and morphometric brainstem and cerebellar findings. Cerebellum. 2012;11:155–166. [DOI] [PubMed] [Google Scholar]
- 8. Kim JS, Kim JS, Youn J, et al. Ocular motor characteristics of different subtypes of spinocerebellar ataxia: distinguishing features. Mov Disord. 2013;28:1271–1277. [DOI] [PubMed] [Google Scholar]
- 9. Pula JH, Gomez CM, Kattah JC. Ophthalmologic features of the common spinocerebellar ataxias. Curr Opin Ophthalmol. 2010;21:447–453. [DOI] [PubMed] [Google Scholar]
- 10. Burk K, Fetter M, Abele M, et al. Autosomal dominant cerebellar ataxia type I: oculomotor abnormalities in families with SCA1, SCA2, and SCA3. J Neurol. 1999;246:789–797. [DOI] [PubMed] [Google Scholar]
- 11. Globas C, du Montcel ST, Baliko L, et al. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov Disord. 2008;23:2232–2238. [DOI] [PubMed] [Google Scholar]
- 12. Jacobi H, Reetz K, du Montcel ST, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 2013;12:650–658. [DOI] [PubMed] [Google Scholar]
- 13. Raposo M, Vasconcelos J, Bettencourt C, Kay T, Coutinho P, Lima M. Nystagmus as an early ocular alteration in Machado‐Joseph disease (MJD/SCA3). BMC Neurol. 2014;14:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 15. Weyer A, Abele M, Schmitz‐Hubsch T, et al. Reliability and validity of the scale for the assessment and rating of ataxia: a study in 64 ataxia patients. Mov Disord. 2007;22:1633–1637. [DOI] [PubMed] [Google Scholar]
- 16. Tan S, Niu HX, Zhao L, et al. Reliability and validity of the Chinese version of the Scale for Assessment and Rating of Ataxia. Chin Med J (Engl). 2013;126:2045–2048. [PubMed] [Google Scholar]
- 17. Strupp M, Kremmyda O, Adamczyk C, et al. Central ocular motor disorders, including gaze palsy and nystagmus. J Neurol. 2014;261(Suppl 2):S542–S558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shakespeare TJ, Kaski D, Yong KX, et al. Abnormalities of fixation, saccade and pursuit in posterior cortical atrophy. Brain. 2015;138:1976–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lemos J, Eggenberger E. Saccadic intrusions: review and update. Curr Opin Neurol. 2013;26:59–66. [DOI] [PubMed] [Google Scholar]
- 20. Heuer HW, Mirsky JB, Kong EL, et al. Antisaccade task reflects cortical involvement in mild cognitive impairment. Neurology. 2013;81:1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacobi H, Rakowicz M, Rola R, et al. Inventory of Non‐Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum. 2013;12:418–428. [DOI] [PubMed] [Google Scholar]
- 22. Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: cross‐sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the Predict‐HD study. Arch Neurol. 2006;63:883–890. [DOI] [PubMed] [Google Scholar]
- 25. Schulz JB, Borkert J, Wolf S, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. NeuroImage. 2010;49:158–168. [DOI] [PubMed] [Google Scholar]
- 26. Feng L, Chen DB, Hou L, et al. Cognitive impairment in native Chinese with spinocerebellar ataxia type 3. Eur Neurol. 2014;71:262–270. [DOI] [PubMed] [Google Scholar]
- 27. Munoz DP, Everling S. Look away: the anti‐saccade task and the voluntary control of eye movement. Nat Rev Neurosci. 2004;5:218–228. [DOI] [PubMed] [Google Scholar]
- 28. Caspi A, Zivotofsky AZ, Gordon CR. Multiple saccadic abnormalities in spinocerebellar ataxia type 3 can be linked to a single deficiency in velocity feedback. Invest Ophthalmol Vis Sci. 2013;54:731–738. [DOI] [PubMed] [Google Scholar]