

Summary
Introduction
Fungal transversal across the brain microvascular endothelial cells (BMECs) is the essential step for the development of cryptococcal meningoencephalitis. Annexin A2 (AnxA2) is an important signaling protein involved in several intracellular processes such as membrane trafficking, endocytosis, and exocytosis.
Aim
To investigate the roles and mechanism of AnxA2 during cryptococcal transversal of BMECs.
Results
Cryptococcus neoformans infection initiated upregulation of AnxA2 in mouse BMECs. Blockade with anti‐AnxA2 antibody led to a reduction in fungal transcytosis activity but no change in its adhesion efficiency. Intriguingly, AnxA2 depletion caused a significant increase in fungal association activity but had no effect on their transcytosis. AnxA2 suppression resulted in marked reduction in its partner protein S100A10, and S100A10 suppression in BMECs significantly reduced the cryptococcal transcytosis efficiency. Furthermore, AnxA2 dephosphorylation at Tyr23 and dephosphorylation of downstream cofilin were required for cryptococcal transversal of BMECs, both of which might be primarily involved in the association of C. neoformans with host cells.
Conclusions
Our work indicated that AnxA2 played complex roles in traversal of C. neoformans across host BMECs, which might be dependent on downstream cofilin to inhibit fungal adhesion but rely on its partner S100A10 to promote cryptococcal transcytosis.
Keywords: Annexin A2, Blood–Brain Barrier, Cofilin, Cryptococcal meningitis
Introduction
Cryptococcus neoformans is one of the most common fungal pathogens worldwide, capable of causing fatal central nervous system (CNS) infection in both immunocompromised and immunocompetent people 1, 2. It is generally acknowledged that environmental cryptococcal basidiospores or desiccated yeast cells are first inhaled into the respiratory tract in early childhood, resulting in asymptomatic pneumonia or latent infection for months or even decades 1, 3, 4. When host immunity is suppressed or compromised, the pathogen often reactivates and proliferates in the lung, subsequently disseminating through the bloodstream into different organs, especially CNS 1, 5. The remarkable neurotropism of C. neoformans remains an enigma.
To penetrate into the brain, C. neoformans must transverse the blood–brain barrier (BBB), which is an essential step for the development of meningoencephalitis. The BBB consists of several components, including endothelial cells, astrocytes, pericytes, and basal lamina 6. Among them, endothelial cells are the cardinal elements for maintaining brain homeostasis via the formation of brain capillaries and tight junctions. Presumably, cryptococcal transversal across BBB relied on the following three pathways 1, 5, 7. Firstly, the phagocytosis‐mediated pathway utilizes monocytes as a carrier to penetrate the brain 8, 9. Secondly, paracellular migration of C. neoformans relies on mechanical or biochemical disruption of tight junctions 10, 11, 12, 13, 14. Thirdly, the transcellular pathway involves receptor‐mediated adhesion and invasion of C. neoformans into endothelial cells, which is probably the main mechanism responsible for BBB transversal 15, 16, 17. These models have dramatically strengthened our understanding of the pathogenesis of cryptococcal meningitis. However, the molecular events and detailed mechanisms underlying C. neoformans entry and internalization into endothelial cells remain to be fully resolved.
Annexin A2 (AnxA2) is an important protein of annexin family that can bind anionic phospholipids in a Ca2+‐dependent manner 18, 19. AnxA2 is ubiquitous in various cells such as endothelial cells, monocytes, and cancer cells, and it is involved in several biochemical processes such as membrane trafficking, endocytosis, and vesicle transportation 20, 21, 22. AnxA2 has been implicated in cancer progression 23, 24 and host cell infection by a variety of bacteria and viruses 25, 26, 27, 28, 29. Recently, using a proteomic profile assay, AnxA2 in human brain endothelial cells has been found to be significantly upregulated after exposure to cryptococcal cells 30. Furthermore, our previous work has revealed that S100A10, the partner of AnxA2, is involved in the transmigration of C. neoformans across vascular endothelia 31, 32. We found that depletion of S100A10 in mouse brain microvascular endothelial cells (MBMECs) significantly reduced the internalization rate and budding rate of C. neoformans. Hence, we speculated that annexin A2, as an important intracellular signaling protein, might also play an indispensable role in BBB transversal of C. neoformans.
Here, we assessed the role and mechanism of host AnxA2 in the adhesion and transcytosis of C. neoformans across the BBB. AnxA2 suppression by shRNA or PP2 treatment was sufficient to induce cofilin dephosphorylation in brain endothelial cells and thereby enhance fungal adhesion activity. Furthermore, AnxA2 was dependent on the synergy of its partner S100A10 to mediate fungal transcytosis across endothelial cells. This is the first report to demonstrate complex roles of AnxA2 in BBB invasion by a pathogenic microorganism.
Materials and methods
Cells, strains, antibodies, and reagents
MBMEC cell line bEND.3 was purchased from the ATCC (Manassas, VA) and grown in Dulbecco's modified Eagle's medium (DMEM) at 37°C with 5% CO2. C. neoformans var. grubii strain H99 from our laboratory was maintained on YPD agar (1% yeast extract, 2% peptone, 2% dextrose, and 2% agar).
The antibodies and inhibitors used in this study were as follows. Rabbit polyclonal anti‐AnxA2 antibody (Ab) and rabbit monoclonal anti‐phosphorylated tyrosine 23 of AnxA2 Ab were obtained from Santa Cruz Biotechnology, Inc. Rabbit polyclonal anti‐S100A10 antibody and rabbit anti‐Na+/K+‐ATPase were obtained from Abcam Biotechnology, Inc. Rabbit monoclonal anti‐cofilin Ab and rabbit monoclonal anti‐phosphorylated serine 3 of cofilin Ab were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). The agents (PP2 and BAPTA‐AM) were obtained from Selleck Chemicals.
Plasmid construction and cell transfection
Anti‐AnxA2 short hairpin RNA was synthesized (Invitrogen, Inc., Waltham, MA, USA) and then cloned into the lentivirus expression vector pLKO.1 (Addgene, Inc., Cambridge, MA, USA). A scrambled vector was utilized in parallel. Quantitative real‐time PCR and Western blotting were performed to assess the efficacy of AnxA2 knockdown. We constructed the phospho‐defective mutant (Y23A‐AnxA2) in bEND.3 cells as previously described 33. The ANXA2 cDNA fragment was amplified and then cloned into the vector pcdh‐CMV‐MCS‐3FLAG‐EF1‐COPGFP‐T2A‐PURO (Fitgene Inc., Mentone East, VIC, USA). Then, target mutations were generated to create the new vectors pGFP‐AnxA2 Y23A (Tyr changed to Ala) using KOD Plus DNA polymerase (TOYOBO, Kita‐ku, Osaka, Japan). Stable transfectants were analyzed for AnxA2 and green fluorescent protein (GFP) expression by Western blotting and immunofluorescence.
Quantitative real‐time PCR, Western blot analysis, and immunoprecipitation
After exposure to C. neoformans for different time periods, total RNA was extracted from MBMECs using TRIzol reagent (Sigma, Munich, Germany) and then converted to cDNA using the PrimeScript RT‐PCR Kit according to the manufacturer's protocol (Takara, Kusatsu, Shiga, Japan). RT‐PCR analysis was performed to determine the transcription level. All primers are listed in Table S1.
Cells were lysed using lysis buffer as previously reported 34. The ProteinExt Mammalian Membrane Protein Extraction Kit (TransGen, Beijing, China) was used to extract either membrane or cytosolic proteins according to the manufacturer's protocol. Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Densitometric analysis of the protein bands of interest was performed using ImageJ software (National Institute of Health, Bethesda, MD, USA).
For the immunoprecipitation (IP), infected endothelial cells were washed, lysed with IP lysis buffer 34, and then centrifuged at 3000 g for 10 min at 4°C. 50 μL supernatant was used as an input control; the remainder was incubated with Protein A/G Plus–agarose beads and rabbit polyclonal AnxA2 antibody at 4°C overnight. The beads were washed and then processed for Western blotting. To analyze the interaction between AnxA2 and S100A10, the cells were treated with PP2 (10 μM), BAPTA‐AM (5 μM), or DMSO (control) for 1 h before infection with cryptococcal cells, followed by two rinses with warm PBS.
In vitro association and transcytosis assays with MBMECs
The in vitro C. neoformans association and transcytosis assay was performed as previously described 15, 17, 35. 105 bEND.3 cells were incubated in 24‐well plate and grown until confluence. Cryptococcal cells were added to the MBMECs in a 10‐fold inoculum. After 2 h of coincubation, unattached yeast cells were removed by washing four times with 1× PBS buffer. Subsequently, the cells were lysed with 0.5% Triton X‐100, serially diluted, and then plated on YPD agar to count the yeast cell colonies. For the transcytosis assays, MBMECs were seeded into transwell inserts (Corning Costar 3422, CORNING, NY, USA) and grown until confluence. The transendothelial electrical resistance (TEER) was measured using a Millicell‐ERS apparatus (Millipore, Inc., Billerica, MA, USA) to evaluate the integrity of the BBB model. 4 × 105 yeast cells were added to the upper compartment (blood side) of the insert. At different time points, 100 μL of the cultures was collected from the bottom compartment (brain side) and plated on YPD agar to determine the CFU of traversed yeast cells. The bottom compartment contained an equal volume of fresh complete culture medium. Each experiment was performed at least in triplicate.
Statistical analysis
The data are presented as the mean ± SD and were analyzed using the statistical software SPSS 18.0 (IBM, Armonk, NY, USA) Analysis of variance (ANOVA) and Student's t‐test were performed to evaluate the statistical significance between the treatment and control groups. A P value <0.05 was considered statistically significant.
Results
Cryptococcus neoformans exposure upregulated the expression of AnxA2 in mouse brain microvascular endothelial cells
Previous works have suggested that the signaling protein S100A10 is required for C. neoformans transmigration across the host BBB 30, 31. Here, we first examined the expression of its partner protein AnxA2 in MBMECs after exposure to C. neoformans var. grubii (strain H99). As shown in Figure 1A, quantitative real‐time PCR revealed that ANXA2 was gradually upregulated at the transcriptional level with longer cryptococcal exposure, whereas a stable low level was maintained in the mock group (exposure to PBS buffer). By 24 h postexposure, the expression of ANXA2 in the H99 group was enhanced approximately 4‐fold compared with the mock group (P < 0.01). Consistently, the protein blots revealed a similar upregulation of AnxA2 in MBMECs during cryptococcal invasion (Figure 1B). These data suggested that AnxA2 might be involved in the transversal of C. neoformans across MBMECs.
Figure 1.
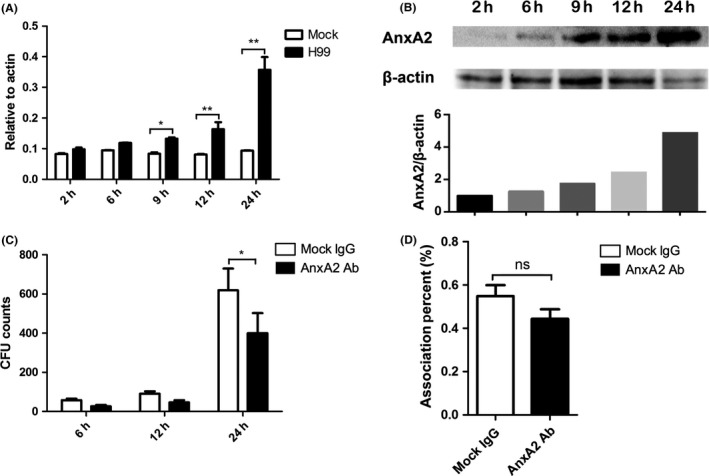
Cryptococcus neoformans exposure initiated upregulation of AnxA2 expression in MBMECs, while the addition of anti‐AnxA2 antibody reduced cryptococcal transcytosis efficiency across endothelial cells. A&B. Quantitative PCR (A) and Western blot (B) assays were performed to evaluate the expression of AnxA2 in MBMECs exposed to C. neoformans at different time points. The above assays were both repeated with similar results. C&D. In vitro transcytosis (C) and association (D; 2 h pi) assay of C. neoformans with MBMECs following treatment with anti‐AnxA2 Ab. Mock IgG was used as a control. ns indicates no statistical significance (P > 0.05). *P < 0.05, **P < 0.01.
Blockade with anti‐AnxA2 antibody led to a reduced transcytosis ability of C. neoformans across MBMECs
We next tested the effects of extracellular anti‐AnxA2 Ab on cryptococcal transversal of the host BBB. Using mock IgG as a control, we conducted transmigration assays with C. neoformans in the presence of anti‐AnxA2 Ab (15 μg/mL) using an in vitro BBB model. The BBB system was established as previously described 15, 17, and its integrity was confirmed by TEER measurement (Figure S1). After treatment with the anti‐AnxA2 Ab, as shown in Figure 1C, the number of C. neoformans cells that transmigrated across the MBMEC monolayers was significantly reduced compared with the transmigration in the control group. Transmigration of C. neoformans was decreased by 35% compared with the control, further indicating that AnxA2 might promote cryptococcal traversal of the BBB. Because the adhesion of fungal cells to MBMEC monolayers is the first step in BBB invasion, association assays were performed during anti‐AnxA2 Ab treatment. Interestingly, the results revealed no significant difference in association efficiency after anti‐AnxA2 Ab treatment compared with the controls (Figure 1D). Our protein blot analysis (Figure S2) further excluded the effect of anti‐AnxA2 Ab on AnxA2 expression during cryptococcal infection. Therefore, anti‐AnxA2 Ab might impede fungal transmigration but did not interfere with the association of C. neoformans with MBMECs, and its inhibitory effect was not associated with AnxA2 expression.
AnxA2 knockdown differentially regulated fungal transcytosis and association with MBMECs
To further investigate the role of AnxA2 during C. neoformans infection of MBMECs, we generated stable AnxA2 knockdown (KD) cell lines using lentiviral small hairpin RNA (LV‐shRNA). Quantitative real‐time PCR displayed that these shRNAs were able to reduce the transcriptional levels of AnxA2 significantly in MBMECs as shown in Figure 2A (P < 0.001). The protein blots displayed highly consistent results, in which full‐length AnxA2 (36 KDa) was detected in untreated or shCtrl cells, but it was only faintly detected in cells transfected with LV‐shRNA vectors, further suggesting a defect in AnxA2 expression in MBMECs (Figure 2B).
Figure 2.
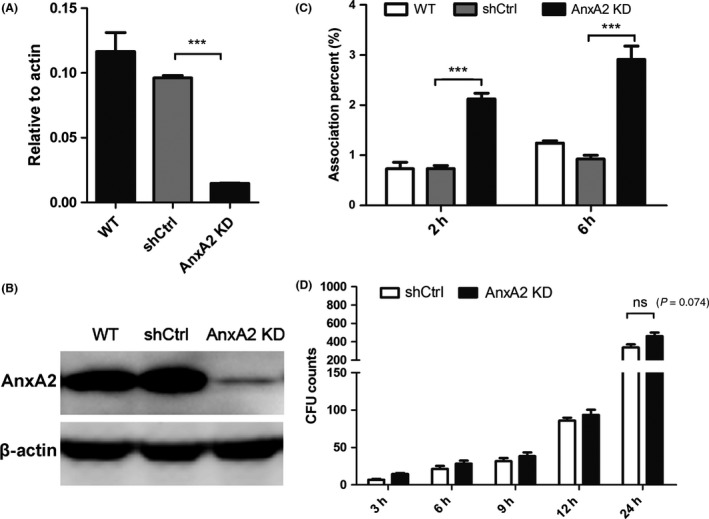
Effects of AnxA2 depletion on the association and transcytosis of Cryptococcus neoformans in MBMECs. A&B. Quantitative real‐time PCR (A) and Western blot analysis (B) were performed to evaluate the expression of ANXA2 in transfected MBMECs. AnxA2 knockdown in MBMECs (bEND.3) was established using a lentiviral shRNA specific for AnxA2 and a scrambled sequence (shCtrl as control). (C) In vitro association assay of C. neoformans with transfected MBMECs at 2 or 6 h. (D) In vitro transcytosis assay of C. neoformans at 3, 6, 9, 12, and 24 h. ns indicates no statistical significance (P > 0.05). ***P < 0.001. WT, wild‐type MBMECs.
We next examined the effect of AnxA2 depletion on the association of C. neoformans with MBMECs. The untreated MBMECs and the cells transfected with empty vector alone (shCtrl) were assessed in parallel as controls. TEER assay suggested that AnxA2 depletion did not disrupt the integrity of BBB system (Figure S1A). However, downregulation of AnxA2 in MBMECs resulted in an approximately 3‐fold increase in the association activity of C. neoformans after 2 or 6 h, whereas no increase was observed in the control groups (P < 0.001) at the same time points (Figure 2C). Therefore, AnxA2 might inversely regulate the association of C. neoformans with MBMECs. We also assessed the effects of AnxA2 downregulation on cryptococcal transmigration. As shown in Figure 2D, time‐dependent transcytosis of C. neoformans across MBMECs was observed in both the AnxA2 KD and control groups. Intriguingly, these two groups displayed no significant differences in transmigration at different time points, in contrast to the results of the association assay. Based on these data, it was tempting to speculate that AnxA2 might play complicated roles in various steps of cryptococcal invasion.
AnxA2 might be dependent on its partner S100A10 to regulate cryptococcal transcytosis across MBMECs
Several biological functions of AnxA2 are achieved during the physical state of the heterotetrameric complex AIIt, and the partner protein S100A10 had been demonstrated to be essential for cryptococcal transversal of the BBB 30, 31. Thus, we evaluated the effect of fungal exposure on the formation of AIIt via immunoprecipitation and immunoblot analysis. As shown in Figure 3A, the expression of S100A10 markedly increased in response to fungal exposure, indicating an enhancement of AIIt expression. Furthermore, AIIt complex formation was significantly blocked by the calcium chelator BAPTA‐AM but appeared to be unaffected by treatment with the Src kinase‐specific inhibitor PP2, which could inhibit Src‐dependent AnxA2 phosphorylation at Tyr23 (Figure 3A) 27.
Figure 3.
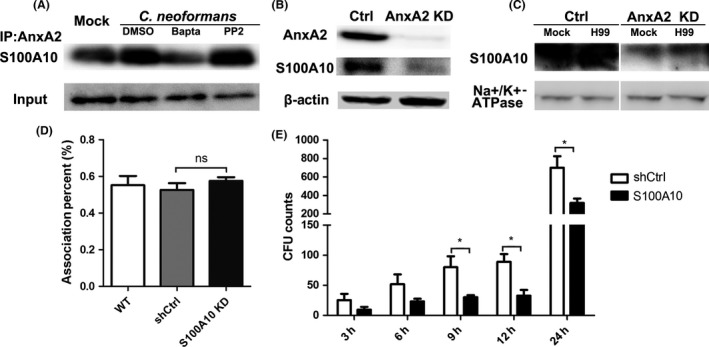
S100A10 was required for AnxA2 to regulate cryptococcal transcytosis across MBMECs. (A) Formation of the AnxA2 complex in MBMECs was evaluated by immunoprecipitation and Western blot analysis after cryptococcal exposure. Endothelial cells were treated with PP2, BAPTA‐AM, or DMSO (control) for 1 h before infection with cryptococcal cells and then rinsed twice with warm PBS. Lysates of infected or noninfected endothelial cells were preincubated with anti‐AnxA2 Ab, and bound proteins were then blotted with anti‐S100A10 Ab. (B & C) Effects of AnxA2 depletion on the expression of total and membrane S100A10. β‐Actin and Na+/K+‐ATPase were utilized as controls for the Western blot analyses. D&E. Effects of S100A10 depletion on the association (D) and transcytosis (E) of C. neoformans in MBMECs. *P < 0.05.
Because AnxA2 has been reported to protect the S100A10 against rapid ubiquitin‐mediated degradation 36, we next examined the effect of AnxA2 deficiency on the expression of S100A10. S100A10 was detected by immunoblot analysis in total proteins and membrane proteins extracted from MBMECs. As shown in Figure 3B,C, S100A10 protein was readily observed in WT MBMECs but not in AnxA2 KD endothelial cells, regardless of C. neoformans infection. We also examined the effects of S100A10 knockdown on the association and transcytosis efficiency of C. neoformans. We stably suppressed S100A10 protein in the MBMEC cell line by RNA interference as previously reported (data not shown), and the integrity of BBB system was not affected by S100A10 depletion (Figure S1B) 31. Our results revealed that S100A10 depletion did not affect cryptococcal association but inhibited cryptococcal transmigration across MBMECs in the late phase (Figure 3D,E). S100A10 depletion in MBMEC resulted in an approximately 2‐ to 3‐fold reduction in the transcytosis activity of C. neoformans at 12 or 24 h (P < 0.05). Therefore, we speculated that AnxA2 might be dependent on its binding to S100A10 to regulate the transmigration of C. neoformans across the BBB.
Src‐dependent AnxA2 phosphorylation at Tyr23 inhibited fungal transversal in MBMECs
Tyr23 phosphorylation in AnxA2 is one of the pivotal modifications involved in the cellular distribution and association with a variety of ligands 19, 37, 38. To ascertain its role in cryptococcal traversal of the BBB, the phosphorylation status of AnxA2 Tyr23 was assessed in MBMECs after fungal exposure. Immunoblotting analysis revealed that AnxA2 Tyr23 was consistently phosphorylated at higher levels in mock MBMECs (Figure 4A). However, AnxA2 phosphorylation was fully blocked during the early stage of fungal infection (from 5 to 30 min pi) and then returned to normal level in endothelial cells at 60 min pi.
Figure 4.
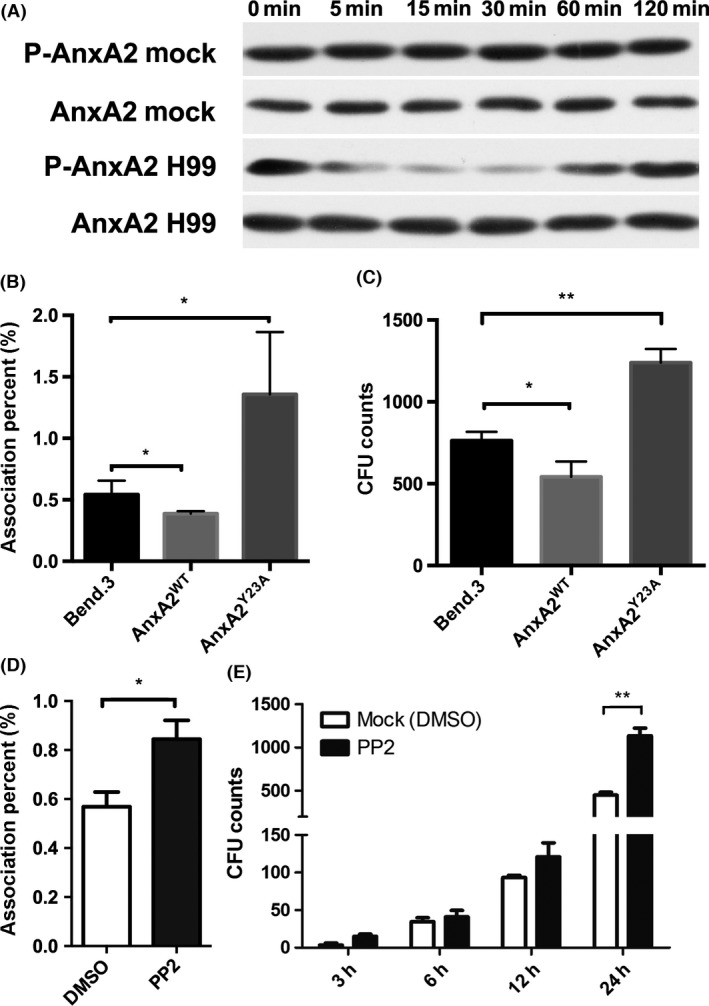
Cryptococcal transversal across brain endothelial cells was controlled by Src‐dependent AnxA2 phosphorylation at Tyr23. (A) Dynamic evaluation of AnxA2 phosphorylation at Tyr23 in endothelial cells during Cryptococcus neoformans invasion. (B & C) Fungal adhesion and transcytosis across endothelial cells were closely associated with the phosphorylation status of AnxA2 Tyr23. We evaluated fungal adhesion (2 h pi) and transcytosis (24 h pi) activity in endothelial cells transfected with WT‐AnxA2 and Y23A‐AnxA2. D&E. Evaluation of fungal association and transcytosis activity in brain endothelial cells after PP2 treatment. *P < 0.05, **P < 0.01.
We questioned whether inhibiting AnxA2 phosphorylation at Tyr23 affects fungal transversal across endothelial cells. MBMECs were transfected with WT‐AnxA2 and Y23A‐AnxA2, in which the Tyr residue was replaced with an Ala, thereby creating a phospho‐defective mutant. Stable transfectants (AnxA2WT and AnxA2Y23A) were confirmed via Western blotting and immunofluorescence (data not shown). As shown in Figure 4B, the group transfected with AnxA2WT displayed an approximately one‐fourth decrease in adhesion activity of C. neoformans compared with the control group (bEND.3, P < 0.05), whereas the phospho‐defective mutation in endothelial cells resulted in an approximately 3‐fold increase in fungal adhesion efficiency (P < 0.05). These transfectants displayed similar alterations in fungal transcytosis activities (Figure 4C). The results indicated that C. neoformans might induce Tyr23 dephosphorylation to promote their transversal of endothelial cells.
Because AnxA2 is a prominent substrate for Src kinase, which can activate the phosphorylation of AnxA2 at Tyr‐23, we also tested its roles in fungal invasion of the CNS. PP2, a potent and selective inhibitor of Src kinases that could lead to AnxA2 dephosphorylation at Tyr‐23 27, was incubated with MBMECs prior to fungal infection. The association activity of PP2‐treated cells was significantly enhanced by 40% compared with mock cells (P < 0.05; Figure 4D). The in vitro transcytosis assay (Figure 4E) showed that PP2 treatment also strengthened the transcytosis ability of MBMECs at 24 h pi, which was approximately more than double that in the mock group (P < 0.01). This result was very similar to those obtained for endothelial cells transfected with AnxA2Y23A (Figure 4B,C). Taken together, our data support that Src‐dependent AnxA2 phosphorylation at Tyr23 played an inhibitory role in C. neoformans transversal of MBMECs.
The effect of AnxA2 on the cryptococcal association with MBMECs relied on dephosphorylation of downstream cofilin
AnxA2 is involved in many different cellular processes including cytoskeletal reorganization, which is a critical event in brain endothelial cells during cryptococcal invasion 10, 30, 39, 40. Cofilin, as an actin‐binding protein, is closely associated with cryptococcal transversal across host brain microvascular endothelial cells 10. We questioned whether AnxA2 requires cofilin to regulate cryptococcal invasion of MBMECs. The phosphorylation status of cofilin was assessed at different time points after cryptococcal exposure. As shown in Figure 5A, cofilin was phosphorylated at 5 min pi but began to undergo dephosphorylation at 15 min pi, while total cofilin was maintained at a stable level. These results implied that fungal exposure initiated the dephosphorylation of both AnxA2 and cofilin in endothelial cells (Figures 4A and 5A); however, the former event occurred earlier but was more transient than the latter one.
Figure 5.
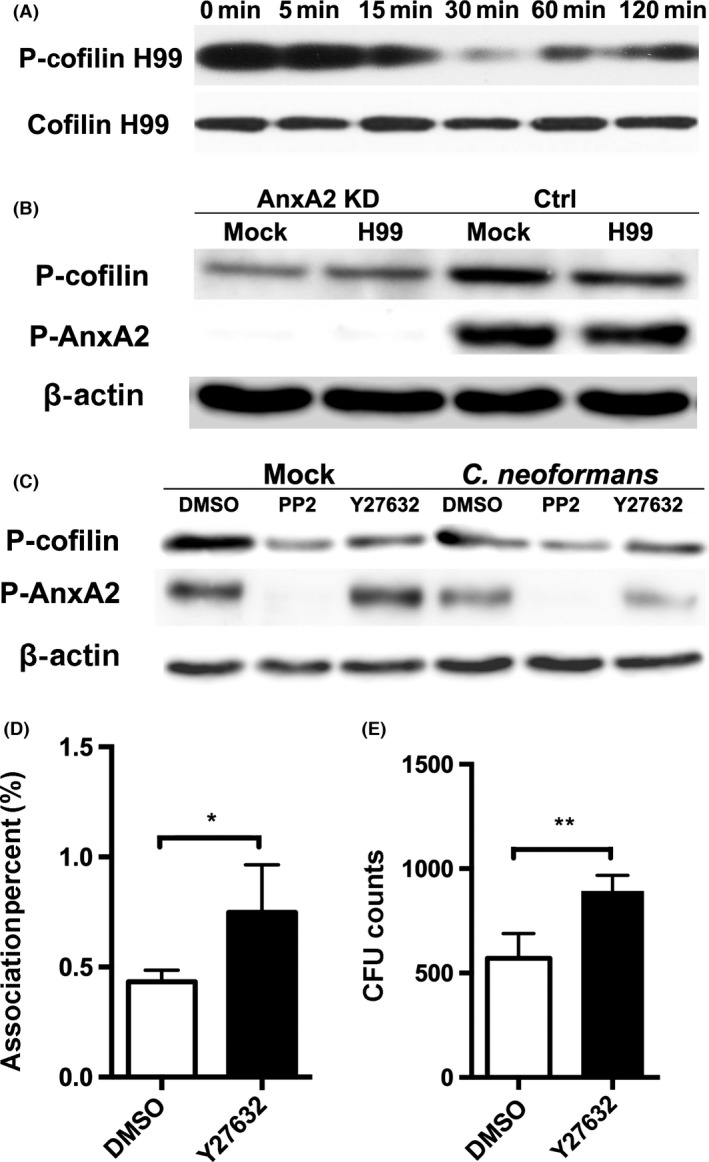
AnxA2‐dependent cofilin dephosphorylation was essential for cryptococcal association with MBMECs. (A) Dynamic evaluation of cofilin phosphorylation in MBMECs during cryptococcal invasion. (B) Effect of AnxA2 depletion on the phosphorylation status of cofilin in host endothelial cells. (C) Evaluation of cofilin phosphorylation in endothelial cells after treatment with PP2 or Y27632 (10 μM). D&E. In vitro transcytosis (D) and association (E) assay of Cryptococcus neoformans with MBMECs following Y27632 treatment. *P < 0.05, **P < 0.01.
To further ascertain the relationship between cofilin and AnxA2, we next examined the phosphorylation status of cofilin in both AnxA2 KD and WT MBMECs. Compared with the control group, AnxA2 depletion led to significant dephosphorylation of cofilin in host endothelial cells with or without cryptococcal infection (Figure 5B), indicating that cofilin functions downstream of AnxA2. As the role of AnxA2 in fungal transversal was closely associated with its phosphorylation status at Tyr23, we also evaluated the functional status of cofilin in MBMECs after treatment with PP2 or DMSO. The results in PP2‐treated cells were similar to those in AnxA2 KD MBMECs, displaying a significant inhibition of cofilin phosphorylation (Figure 5C). These data suggested that cofilin activity was regulated not only by the presence of AnxA2 but also by its phosphorylation status at Tyr23.
Finally, we assessed the effect of cofilin phosphorylation status on fungal invasion of the host BBB. Y27632, a ROCK‐specific inhibitor, was utilized to induce the dephosphorylation of cofilin, as previously described (Figure 5C) 10. As shown in Figure 5D,E, Y27632 treatment significantly enhanced the association of cryptococcus with MBMECs by 74% (P < 0.05), and it had a similar effect on fungal transcytosis of endothelial cells (P < 0.01). The results obtained in Y27632‐treated cells were highly consistent with those in cells that were treated with PP2 (Figure 4D,E). Taken together, these data suggested that AnxA2‐mediated cofilin phosphorylation inhibited the association of cryptococcus with MBMECs.
Discussion
Elucidating the molecular mechanism responsible for fungal traversal across the BBB is crucial to understand the pathogenesis of cryptococcal meningitis. Previous studies have implied that S100A10 is essential for fungal transversal across host endothelial cells 30, 31, 32, but the detailed mechanisms have remained unclear. Because most of the biochemical functions of S100A10 are dependent on its heterotetrameric complex AIIt (AnxA2–S100A10) 18, we focused herein on the roles and mechanism of its partner protein AnxA2 during host CNS invasion by C. neoformans.
Our study first demonstrated that endothelial cell AnxA2 was essential for cryptococcal association of host BBB. Suppression of AnxA2 by shRNA substantially enhanced C. neoformans association activity toward MBMECs (Figure 2), suggesting that AnxA2 inhibited fungal adhesion to endothelial cells. In addition, fungal infection significantly increased the expression of AnxA2 in MBMECs beginning at 9 h pi (Figure 1A,B), consistent with previous findings 30. Therefore, host endothelial cells might initiate the upregulation of AnxA2 to resist fungal invasion.
We also found that fungal adhesion and transcytosis activity were closely related to the phosphorylation status of AnxA2 at Tyr23 in BMECs. After cryptococcal exposure, AnxA2 displayed a transient dephosphorylation at Tyr23 and then returned to normal phosphorylation levels at 1 h pi (Figure 4A). Endothelial cells transfected with a WT AnxA2 displayed a reduced adhesion and transcytosis efficacy of C. neoformans, whereas endothelial cells expressing the Y23A mutant of AnxA2 showed a significant enhancement (Figure 4B). These results indicated that tyrosine dephosphorylation of AnxA2 was essential for fungal attachment and transcytosis to brain endothelia. The addition of exogenous PP2 induced an effect on cryptococcal transversal activity that was similar to the outcome of transfection with the Y23A mutant (Figure 4C), further confirming that Src‐dependent AnxA2 phosphorylation at Tyr23 was essential for host resistance to fungal invasion of brain endothelial cells.
Like other pathogens, C. neoformans can induce cellular cytoskeletal remodeling of host brain endothelial cells, which is regulated by the phosphorylation status of cofilin 10, 15, 41, 42. Cofilin, an actin‐serving protein, is a terminal effector of multiple signaling cascades that is involved in cytokinesis, endocytosis, and other cellular processes 43. In our study, suppression of AnxA2 or PP2 treatment caused significant dephosphorylation of cofilin in endothelial cells (Figure 5B,C), supporting a role for AnxA2 in the regulation of cofilin phosphorylation. Furthermore, cofilin dephosphorylation caused by the ROCK/cofilin pathway inhibitor agent Y27632 could significantly enhance fungal adhesion and transcytosis activity across endothelial cells, which is consistent with a previous report 10. These results implied that the tyrosine phosphorylation switch of AnxA2 might trigger cofilin‐dependent and actin‐mediated changes in the cellular morphology of endothelia associated with fungal adhesion.
Although AnxA2 depletion in host endothelial cells led to an enhanced association activity of C. neoformans, intriguingly, the fungal transcytosis efficiency did not differ from that in the control group (Figure 2D). Therefore, AnxA2 might play different roles in the adhesion and transcytosis steps of fungal invasion. Three separate observations strongly suggest that AnxA2 might rely on the synergy of its partner S100A10 to promote fungal transcytosis across brain endothelial cells. First, cryptococcal exposure significantly enhanced the formation of the AnxA2–S100A10 complex in host endothelial cells (Figure 3A). Second, suppression of AnxA2 resulted in significant depletion of S100A10 in both membrane and cytosolic proteins of MBMECs with or without cryptococcal exposure (Figure 3B,C), implying that AnxA2 was required for maintenance of the homeostasis of S100A10 in brain endothelial cells. Our findings are highly consistent with those of He et al. 36 demonstrating that AnxA2 can prevent polyubiquitin‐mediated degradation of intracellular S100A10 to stabilize its intracellular levels in vascular endothelial cells. Third, we also observed that S100A10 suppression in endothelial cells by RNA interference led to a drastic reduction in fungal transcytosis activity but no alterations in their association efficiency. We hypothesized that AnxA2 knockdown weakened host defense but also inhibited cryptococcal transmigration due to S100A10 suppression, and thus, no functional difference was detected in transcytosis of C. neoformans across MBMECs upon altering the amount of AnxA2 (Figure 2). These results strongly supported the notion that S100A10 was required for AnxA2 to mediate the transcytosis of C. neoformans across the brain endothelium. Previous studies have reported that S100A10 in cancer cells and tumor‐associated phagocytes can bind plasminogen and plasminogen activator to stimulate the generation of plasmin, which is involved in the invasion and metastasis of cancer cells 18, 44, 45. Recently, host plasmin was verified to contribute to cryptococcal invasion of the BBB by the penetration of endothelial cells and the underlying matrix 14, 46. Therefore, AnxA2 might be an indirect contributor to the generation of plasmin by stabilizing S100A10 in endothelial cells and then regulating fungal transcytosis of the BBB, although this hypothesis requires further confirmation.
We were surprised to find that the antibody against AnxA2 inhibited the transcytosis of C. neoformans but seemingly did not affect adhesion activity across endothelial cells (Figure 1C,D). The anti‐AnxA2 antibody is directed against amino acids 1 to 50 of the N‐terminal domain of AnxA2, which contains the S100A10 binding site (the first 12 residues) 44, 47. Therefore, it appears that the antibody–AnxA2 interaction impedes the formation or function of the AIIt complex, but it does not interfere with Tyr23 phosphorylation and expression of AnxA2. Because PP2 appeared to have no effect on the formation of the AnxA2–S100A10 complex (Figure 3A), AnxA2 Tyr23 phosphorylation might not directly participate in the process of fungal transcytosis across endothelial cells. It is plausible that AnxA2 might rely on these two distinct mechanisms to regulate the fungal adhesion and transcytosis steps, respectively (Figure 6). On the one hand, Src‐dependent AnxA2 phosphorylation induces the phosphorylation of downstream cofilin in brain endothelial cells, which is essential for host resistance to cryptococcal adhesion. On the other hand, AnxA2 may depend on binding to its partner S100A10 to mediate fungal transcytosis across endothelial cells.
Figure 6.
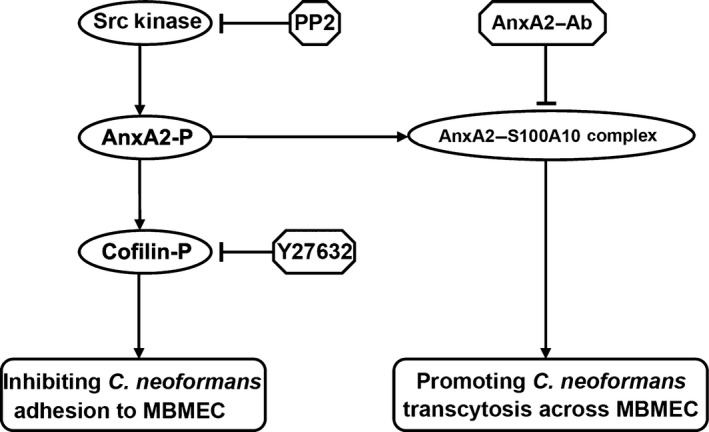
Current model of the role of AnxA2 in Cryptococcus neoformans traversal across the blood–brain barrier. On the one hand, Src‐dependent AnxA2 phosphorylation induces the phosphorylation of downstream cofilin in brain microvascular endothelial cells, which could inhibit C. neoformans adhesion to BBB. PP2 (Src kinase inhibitor) and Y27632 (indirect inducer of cofilin dephosphorylation) pretreatment could enhance the efficiency of cryptococcal association with MBMEC. On the other hand, AnxA2 might rely on binding to its partner S100A10 to promote cryptococcal transcytosis across MBMEC. AnxA2‐Ab, the antibody against AnxA2, might impede the formation or function of the AnxA2–S100A10 complex and thus interfere with cryptococcal transcytosis across brain endothelial cells.
In summary, our studies demonstrate that host AnxA2 plays critical roles in fungal traversal of the BBB. AnxA2 is upregulated to enhance host defense against C. neoformans leading to decreased association and transmigration (Figures 1 and 4). C. neoformans seems to override AnxA2's protective function by inducing AnxA2 and cofilin dephosphorylation and promoting AIIt formation. However, our work needs further confirmation by in vivo experiments. Furthermore, it is unclear whether other CNS pathogens utilize a similar or different mechanism to cross the BBB, such as Neisseria meningitidis, Mycobacterium tuberculosis, and Herpes simplex. Further studies investigating the upstream and downstream molecular programs that control and mediate the function of AnxA2 may facilitate the development of novel strategies for therapeutic intervention in infective meningoencephalitis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. TEER measurements of in vitro BBB system.
Figure S2. Anti‐AnxA2 Ab treatment had no effect on AnxA2 expression during cryptococcal infection.
Table S1. Primers used in this study.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81271799, 81401651, and 81471926), the National Key Basic Research Programs of China (2013CB531604 and 2013CB531601), and the Shanghai Municipal Natural Science Foundation (14495800500 and 14DZ2272900).
The first three authors contributed equally to this study.
Contributor Information
Ju‐Lin Gu, Email: wujgjl@126.com.
Wan‐Qing Liao, Email: liaowanqing@sohu.com.
References
- 1. Heitman J, Kozel TR, Kwon‐Chung KJ, Perfect JR, Casadevall A. Cryptococcus: From human pathogen to model yeast. Washington, DC: ASM Press, 2011. [Google Scholar]
- 2. Fang W, Fa Z, Liao W. Epidemiology of Cryptococcus and cryptococcosis in China. Fungal Genet Biol 2015;78:7–15. [DOI] [PubMed] [Google Scholar]
- 3. Velagapudi R, Hsueh YP, Geunes‐Boyer S, Wright JR, Heitman J. Spores as infectious propagules of Cryptococcus neoformans. Infect Immun 2009;77:4345–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldman DL, Khine H, Abadi J, et al. Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics 2001;107:E66. [DOI] [PubMed] [Google Scholar]
- 5. Kronstad JW, Attarian R, Cadieux B, et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol 2011;9:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abbott NJ, Rnc L. Hansson E. Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci 2006;7:41–53. [DOI] [PubMed] [Google Scholar]
- 7. Liu TB, Perlin DS, Xue C. Molecular mechanisms of cryptococcal meningitis Virulence 2012;3:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chretien F, Lortholary O, Kansau I, Neuville S, Gray F, Dromer F. Pathogenesis of cerebral Cryptococcus neoformans infection after fungemia. J Infect Dis 2002;186:522–530. [DOI] [PubMed] [Google Scholar]
- 9. Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect Immun 2009;77:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen SH, Stins MF, Huang SH, et al. Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol 2003;52(Pt 11):961–970. [DOI] [PubMed] [Google Scholar]
- 11. Olszewski MA, Noverr MC, Chen GH, et al. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am J Pathol 2004;164:1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Charlier C, Chretien F, Baudrimont M, Mordelet E, Lortholary O, Dromer F. Capsule structure changes associated with Cryptococcus neoformans crossing of the blood‐brain barrier. Am J Pathol 2005;166:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu CY, Zhu HM, Wu JH, Wen H, Liu CJ. Increased permeability of blood‐brain barrier is mediated by serine protease during Cryptococcus meningitis. J Int Med Res 2014;42:85–92. [DOI] [PubMed] [Google Scholar]
- 14. Stie J, Fox D. Blood‐brain barrier invasion by Cryptococcus neoformans is enhanced by functional interactions with plasmin. Microbiology 2012;158(Pt 1):240–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang YC, Stins MF, McCaffery MJ, et al. Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood‐brain barrier. Infect Immun 2004;72:4985–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabiiti W, May RC. Capsule independent uptake of the fungal pathogen Cryptococcus neoformans into brain microvascular endothelial cells. PLoS One 2012;7:e35455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jong A, Wu CH, Gonzales‐Gomez I, et al. Hyaluronic acid receptor CD44 deficiency is associated with decreased Cryptococcus neoformans brain infection. J Biol Chem 2012;287:15298–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bharadwaj A, Bydoun M, Holloway R, Waisman D. Annexin A2 heterotetramer: Structure and function. Int J Mol Sci 2013;14:6259–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deora AB, Kreitzer G, Jacovina AT, Hajjar KA. An annexin 2 phosphorylation switch mediates p11‐dependent translocation of annexin 2 to the cell surface. J Biol Chem 2004;279:43411–43418. [DOI] [PubMed] [Google Scholar]
- 20. Hitchcock JK, Katz AA, Schäfer G. Dynamic reciprocity: The role of annexin A2 in tissue integrity. J Cell Commun Signal 2014;8:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee DB, Jamgotchian N, Allen SG, Kan FW, Hale IL. Annexin A2 heterotetramer: Role in tight junction assembly. Am J Physiol Renal Physiol 2004;287:F481–F491. [DOI] [PubMed] [Google Scholar]
- 22. Urbanska A, Sadowski L, Kalaidzidis Y, Miaczynska M. Biochemical characterization of APPL endosomes: The role of annexin A2 in APPL membrane recruitment. Traffic 2011;12:1227–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma MR, Rothman V, Tuszynski GP, Sharma MC. Antibody‐directed targeting of angiostatin's receptor annexin II inhibits Lewis Lung Carcinoma tumor growth via blocking of plasminogen activation: Possible biochemical mechanism of angiostatin's action. Exp Mol Pathol 2006;81:136–145. [DOI] [PubMed] [Google Scholar]
- 24. Sharma M, Ownbey RT, Sharma MC. Breast cancer cell surface annexin II induces cell migration and neoangiogenesis via tPA dependent plasmin generation. Exp Mol Pathol 2010;88:278–286. [DOI] [PubMed] [Google Scholar]
- 25. Jolly C, Winfree S, Hansen B, Steele‐Mortimer O. The Annexin A2/p11 complex is required for efficient invasion of Salmonella Typhimurium in epithelial cells. Cell Microbiol 2014;16:64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li R, Tan S, Yu M, Jundt MC, Zhang S, Wu M. Annexin A2 Regulates Autophagy in Pseudomonas aeruginosa Infection through the Akt1‐mTOR‐ULK1/2 Signaling Pathway. J Immunol 2015;195:3901–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dziduszko A, Ozbun MA. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol 2013;87:7502–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derry MC, Sutherland MR, Restall CM, Waisman DM, Pryzdial EL. Annexin 2‐mediated enhancement of cytomegalovirus infection opposes inhibition by annexin 1 or annexin 5. J Gen Virol 2007;88(Pt 1):19–27. [DOI] [PubMed] [Google Scholar]
- 29. Ryzhova EV, Vos RM, Albright AV, Harrist AV, Harvey T. Gonz¨¢lez‐Scarano F. Annexin 2: A novel human immunodeficiency virus type 1 Gag binding protein involved in replication in monocyte‐derived macrophages. J Virol 2006;80:2694–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vu K, Eigenheer RA, Phinney BS, Gelli A. Cryptococcus neoformans promotes its transmigration into the CNS by inducing molecular and cellular changes in brain endothelial cells. Infect Immun 2013;81:3139–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Y, Chen J, Wen H, et al. S100A10 downregulation inhibits the phagocytosis of Cryptococcus neoformans by murine brain microvascular endothelial cells. Microb Pathog 2011;51:96–100. [DOI] [PubMed] [Google Scholar]
- 32. Wang XJ, Zhu YJ, Cui JG, et al. Proteomic analysis of human umbilical vein endothelial cells incubated with Cryptococcus neoformans var. neoformans. Mycoses 2011;54:e336–e343. [DOI] [PubMed] [Google Scholar]
- 33. Jung H, Kim JS, Kim WK, et al. Intracellular annexin A2 regulates NF‐κB signaling by binding to the p50 subunit: Implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis 2015;6:e1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaboord B, Perr M. Isolation of proteins and protein complexes by immunoprecipitation. Methods Mol Biol 2008;424:349–364. [DOI] [PubMed] [Google Scholar]
- 35. Huang SH, Wu CH, Chang YC, Kwon‐Chung KJ, Brown RJ, Jong A. Cryptococcus neoformans‐derived microvesicles enhance the pathogenesis of fungal brain infection. PLoS One 2012;7:e48570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He KL, Deora AB, Xiong H, et al. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J Biol Chem 2008;283:19192–19200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng L, Foley K, Huang L, et al. Tyrosine 23 phosphorylation‐dependent cell‐surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS One 2011;6:e19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morel E, Gruenberg J. Annexin A2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J Biol Chem 2009;284:1604–1611. [DOI] [PubMed] [Google Scholar]
- 39. Rescher U, Ludwig C, Konietzko V, Kharitonenkov A, Gerke V. Tyrosine phosphorylation of annexin A2 regulates Rho‐mediated actin rearrangement and cell adhesion. J Cell Sci 2008;121(Pt 13):2177–2185. [DOI] [PubMed] [Google Scholar]
- 40. Hayes MJ, Shao D, Bailly M, Moss SE. Regulation of actin dynamics by annexin 2. EMBO J 2006;25:1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen YH, Chen SH, Jong A, et al. Enhanced Escherichia coli invasion of human brain microvascular endothelial cells is associated with alternations in cytoskeleton induced by nicotine. Cell Microbiol 2002;4:503–514. [DOI] [PubMed] [Google Scholar]
- 42. Eugène E, Hoffmann I, Pujol C, Couraud PO, Bourdoulous S, Nassif X. Microvilli‐like structures are associated with the internalization of virulent capsulated Neisseria meningitidis into vascular endothelial cells. J Cell Sci 2002;115(Pt 6):1231–1241. [DOI] [PubMed] [Google Scholar]
- 43. Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal 2013;25:457–469. [DOI] [PubMed] [Google Scholar]
- 44. Madureira PA, O'Connell PA, Surette AP, Miller VA, Waisman DM. The biochemistry and regulation of S100A10: A multifunctional plasminogen receptor involved in oncogenesis. J Biomed Biotechnol 2012;2012:353687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phipps KD, Surette AP, O'Connell PA, Waisman DM. Plasminogen receptor S100A10 is essential for the migration of tumor‐promoting macrophages into tumor sites. Cancer Res 2011;71:6676–6683. [DOI] [PubMed] [Google Scholar]
- 46. Stie J, Bruni G, Fox D. Surface‐associated plasminogen binding of Cryptococcus neoformans promotes extracellular matrix invasion. PLoS One 2009;4:e5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Becker T, Weber K, Johnsson N. Protein‐protein recognition via short amphiphilic helices; a mutational analysis of the binding site of annexin II for p11. EMBO J 1990;9:4207–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. TEER measurements of in vitro BBB system.
Figure S2. Anti‐AnxA2 Ab treatment had no effect on AnxA2 expression during cryptococcal infection.
Table S1. Primers used in this study.