

Summary
Aims
β‐amyloid (Aβ) aggregation and deposition play a central role in the pathogenic process of Alzheimer's disease (AD). α‐Mangostin (α‐M), a polyphenolic xanthone, have been shown to dissociate Aβ oligomers. In this study, we further investigated the effect of α‐M on Aβ production and its molecular mechanism.
Methods
The Aβ and soluble amyloid precursor protein α (sAPPα) in culture medium of cortical neurons were measured by ELISA. The activities of α‐, β‐, and γ‐secretases were assayed, and the interaction between α‐M and β‐ or γ‐secretases was simulated by molecular docking.
Results
α‐M significantly decreased Aβ40 and Aβ42 production. α‐M did not affect the expression of enzymes involved in nonamyloidogenic and amyloidogenic pathways, but significantly decreased the activities of β‐secretase and likely γ‐secretase with IC 50 13.22 nmol·L−1 and 16.98 nmol·L−1, respectively. Molecular docking demonstrated that α‐M interacted with β‐site amyloid precursor protein cleaving enzyme 1 and presenilin 1 to interfere with their active sites.
Conclusions
Our data demonstrate that α‐M decreases Aβ production through inhibiting activities of β‐secretase and likely γ‐secretase in the amyloidogenic pathway. The current data together with previous study indicated that α‐M could be a novel neuroprotective agent through intervention of multiple pathological processes of AD.
Keywords: α‐mangostin, β‐amyloid, β‐secretase, γ‐secretase, Alzheimer's disease
1. Introduction
Alzheimer's disease (AD) is characterized by progressive and irreversible neurodegeneration accompanied by β‐amyloid (Aβ) deposition and neurofibrillary tangles. Currently, there are mainly two types of the drugs available for the treatment of AD, namely acetylcholinesterase inhibitors and N‐methyl‐D‐aspartate receptor antagonists.1, 2, 3 However, these drugs have limited efficiency in improving cognition, delaying the progression and reversing the course of the disease as well as various side effects.2, 3, 4, 5 Development of specific treatment is still mainly focused on Aβ cascade hypothesis of AD, which states that peptides play a central role in the pathogenic process of AD.1, 6, 7 Monomeric Aβ peptides aggregate as oligomers, which are highly neurotoxic, and deposit in cell membranes and dendrites of neurons to cause progressive neurodegeneration.8, 9, 10
Three amyloid precursor protein (APP) processing enzymes α‐, β‐, and γ‐secretases dominate the production of Aβ. Aβ1‐40 and Aβ1‐42 are generated by the cleavage of APP by β‐ and γ‐secretases, which is amyloidogenic.11, 12 APP could also be cleaved by nonamyloidogenic α‐secretase within the Aβ sequence and preclude the formation of Aβ and release the C‐terminal soluble ectodomain of APP, known as soluble amyloid precursor protein α (sAPPα), which has been shown to exert neurotrophic and neuroprotective properties.13 Therefore, drugs targeting these three proteases with the principle of inhibiting β‐ or γ‐secretases while activating α‐secretase are under extensive investigation. Inhibitors or modulators of β‐secretase (such as LY2886721,14, 15 MK‐8931,16, 17 and E2609 18) or γ‐secretase (such as semagacestat 19, 20 and NIC51‐5 18) have been reported, and some of them have already entered clinical trials.17, 18, 21 However, adverse effects have been reported with some of these modulators and inhibitors.16, 22
The side effects and toxicity after the application of the synthetic drugs described above have led to an increased interest in plant derived natural products. α‐Mangostin (α‐M), a polyphenolic xanthone derivative from mangosteen, has long been used as a traditional medicine and has been shown to have a broad range of bioactivities, including antioxidative, anti‐inflammation, and anticancerogenic activities.23, 24, 25 We have reported previously that α‐M can dissociate Aβ oligomers and thus attenuates Aβ oligomers‐induced neurotoxicity.26 However, whether α‐M affects Aβ production is still unclear. In this study, we investigated the effects of α‐M on Aβ40, Aβ42 production in primary cortical neurons. Our results showed that α‐M could decrease the production of Aβ40 and Aβ42 by inhibiting β‐secretase and likely γ‐secretase activities.
2. Materials and Methods
2.1. Reagents
α‐M was purchased from Shanghai Sunny Biotech co, ltd, purity >99% by HPLC and dissolved in DMSO to 5 mmol·L‐1, stored at −20°C. All the cell culture reagents were purchased from GIBCO (Grand Island, NY, USA).
2.2. Cell culture
Primary rat cerebral cortical neurons were prepared as previously described [26]. Briefly, the cortices of the embryos at embryonic day 17 (E17) (Sprague Dawley, Slac Laboratories, Chinese Academy of Sciences) were dissected out and dissociated with 0.05% trypsin‐EDTA. After centrifugation, the cells were resuspended in complete MEM medium and plated on poly‐L‐lysine precoated plates. Cells were cultured in a 5% CO2 humidified atmosphere at 37°C for 2 hours, and then, the medium was replaced with neurobasal medium plus 2% B27 and 2 mmol·L‐1 GlutaMAX supplements. The purity of neurons is around 90% as indicated by Hoechst staining for nucleus and β‐tubulin staining for neural cell body and neurites.
2.3. Elisa
Primary cultured cortical neurons were plated in 96‐well plates at the density of 2.5 × 105 cells/well and cultured for 7 days, then treated with various concentrations of α‐M for another 24 hours. Cell culture medium was collected and centrifuged, and supernatant was stored in −80°C for further test. Aβ40 and Aβ42 levels from culture media were determined using monoclonal and horseradish peroxidase‐conjugated antibody‐based human/rat Aβ40 and Aβ42 ELISA Kits which detect Aβ1‐40 and Aβ1‐42 as well as truncated or modified N‐terminus Aβ40 and Aβ42 (Wako, Osaka, Japan). sAPPα levels were measured using mouse/rat sAPPα ELISA Kit (IBL‐America, Minneapolis, MN, USA) according to the manufacturer's instructions. OD450 was determined in triplicates using ELISA plate reader (Varioskan Flash, Thermo Fisher, Waltham, MA, USA).
2.4. Real‐time PCR
Total RNA was extracted using total RNA isolation kit from Omega (Norcross, GA, USA). RNA was reverse transcribed to synthesize cDNA at the concentration of 50 ng/uL using PrimeScript® RT reagent kit (Takara, Shiga, Japan). Real‐time PCR was performed with SYBR® Premix Ex TaqTM (Takara). 18 seconds was used as the normalization control. The relative mRNA levels were calculated by a comparative Cp value. The primers used were as follows: ADAM9: forward: AGTACCAACCTATGCCATCAAGCAG, reverse: GAGCTATATAAAGGCGGTGCAGGA; ADAM10: forward: GCACCTGTGCCAGCTCTGAT, reverse: TCCGACCATTGAACTGCTTGT; ADAM17: forward: CCTGAACAACGACACCTGCTG, reverse: CTTCTGGGCCGTCTCAAACTG; BACE1: forward: TTGTCACGGCAGACATGGAA, reverse: CATGAGGCAGAGTGGCAACA; PS1: forward: GCGATGATGGTGGCTTCAG, reverse: TCCTGGACAGCAGCTCTTGA; NEP: forward: GGAAGCCATTCAGCTGGT, reverse: TGGAGCATAAACAACCACTTCT; IDE: forward: AAAGAAACTCTCTGCAGA, reverse: TTATGAATCACCTCAGGT; 18s: forward: GAGAGGGAGCCTGAGAAACG, reverse: GGCCTCGAAAGAGTCCTGTA.
2.5. Western blot
After treatment, cortical neurons were extracted with lysis buffer (20 mmol·L‐1 Tris‐HCl, pH 7.5, 150 mmol·L‐1 NaCl, 1 mmol·L‐1 Na2EDTA, 1 mmol·L‐1 EGTA, 1% TritonX‐100, 2.5 mmol·L‐1 sodium pyrophosphate, 1 mmol·L‐1 β‐glycerophosphate, 1 mmol·L‐1 Na3VO4, 1 μg/mL leupeptin, and 1 × protease inhibitor cocktail (Sigma, Saint Louis, MO, USA)). After centrifuging at 12 000 × g for 5 minutes, sample loading buffer was added to the supernatants and boiled for 5 minutes. Approximately 30‐40 μg of protein was subjected to SDS‐PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% milk and then incubated with primary antibodies against APP (22C11, 1:1 000, Millipore), IDE (1:1 000, Cell Signaling Technology, Danvers, MA, USA), BACE1 (1:1 000, Cell Signaling Technology), PS1 (1:1 000, Cell Signaling Technology), and β‐actin (1:5 000, Cell Signaling Technology) overnight at 4°C. Membranes were then incubated with goat anti‐rabbit/mouse HRP‐conjugated secondary antibody (1:10 000) for 1 hours at room temperature, visualized with SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical, Rockford, IL, USA), and analyzed by Image J (National Institutes of Health) software.
2.6. Secretase activity assay
α‐Secretase activity measurements were performed using SenoLyte® 520 TACE (α‐secretase) activity assay kit *Fluorimetric* (Ana Spec, Fremont, CA, USA). β‐Secretase activity measurements were performed using β‐secretase activity assay kit (Biovision, Milpitas, CA, USA). γ‐Secretase activity measurements were performed using γ‐secretase substrate, fluorogenic which is a peptide substrate containing the C‐terminal APP amino acid sequence that is cleaved by γ‐secretase (Millipore) according to manufacturer's instructions. Briefly, primary cortical neurons were seeded in 6‐well plates at 5 × 105 cells/well; 7 days later, cells were treated with vehicle or various concentrations of α‐M for another 24 hours. Cells were then washed with cold PBS and lysed with 100 μL of extraction buffer for 30 minutes on ice. Lysate was centrifuged and supernatant was incubated with substrate and reaction buffer, mixed gently and incubate at 37°C for 1 hours, fluorescence was measured using PE LS45 (PerkinElmer, Waltham, MA, USA) plate reader with λex 490 nm, λem 520 nm for α‐secretase, λex 345 nm, λem 505 nm for β‐secretase, and λex 355 nm, λem 440 nm for γ‐secretase activities.
2.7. In Vitro β‐secretase (BACE1) activity assay
In Vitro inhibitory activity of α‐M on β‐secretase was measured by a FRET based β‐secretase–β‐site amyloid precursor protein cleaving enzyme 1 (BACE1) activity assay kit (Sigma) according to manufacturer's instructions. Briefly, 50 μmol·L−1 BACE1 substrate, 0.3 unit/μL BACE1 enzyme solutions and α‐M solutions were added to a fluorometer 96 well plate, mixed well by gentle pipetting and incubated at 37°C for 2 hours. Fluorescence was read immediately and 2 hours later after adding BACE1 enzyme by PE LS45 (PerkinElmer, Waltham, MA, USA) plate reader with λex 320 nm, λem 405 nm.
2.8. Molecular docking
The crystal structures of BACE1 and main‐component of γ‐secretase complex–presenilin 1 (PS1) were retrieved from protein data bank (PDB) (ID: 1FKN and 5A63, respectively) 27, 28 and prepared by Molecular Operating Environment (MOE, Canada). Transmembrane segment (TM) 2 of PS1 was unidentified due to highly flexibility and was homology modeled in the docking session. Before docking, protonation was carried out using Protonate 3D tools because of little or no hydrogen coordinate data contained in most macromolecular crystal structures of BACE1 and PS1. Energy minimization up to 0.05 Gradient using Amber 99 force field followed. The best ligand pose of α‐M was identified as described previously.26 MOE's automatic docking algorithm was employed to dock α‐M into BACE1 and PS1. Potential energy and ligand interaction were calculated, and then hydrophobic surface and interaction diagram were rendered.
2.9. Statistical analysis
Data were presented as mean ± standard error of the mean (SEM). The results were analyzed by one‐way analysis of variances (ANOVA) followed by Dunnett's test to compare with control using GraphPad Prism. P<.05 was considered statistically significant.
3. Results
3.1. α‐M decreased Aβ40 and Aβ42 production in primary cultured cortical neurons
We first tested the effects of α‐M on Aβ levels in mouse primary neuronal cells. Primary cultured cortical neurons were incubated with α‐M (6.25, 12.5 and 25 nmol·L−1) for 24 hours, and Aβ40 and Aβ42 levels were measured using ELISA. α‐M treatment decreased both Aβ40 and Aβ42 levels, reaching its maximum effect at 25 nmol·L−1 (Figure 1A,B). In vehicle‐treated neurons, Aβ40 levels were 197.26 ± 21.69 pmol/L. 25 nmol·L−1 α‐M reduced Aβ40 levels to 116.83±25.36 pmol/L (P<.01). Aβ42 levels were 40.91±11.36 pmol/L in control group, whereas 25 mol·L−1 α‐M reduced it to 18.65 ± 3.97 pmol/L (P<.01). Furthermore, sAPPα levels in supernatant were detected. As shown in Figure 1C, α‐M treatment did not increase sAPPα levels. These data suggest that α‐M decreased Aβ levels in cultured neurons, probably through affecting amyloidogenic pathway.
Figure 1.
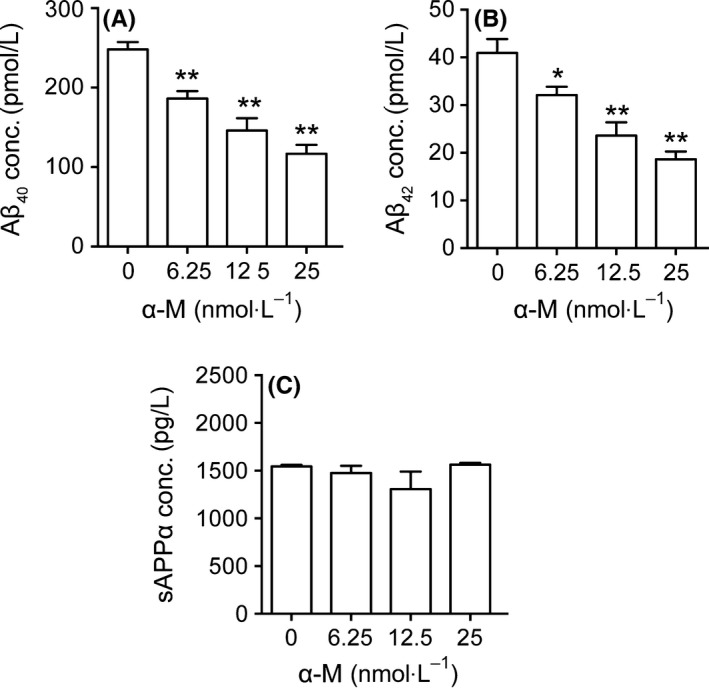
α‐M significantly decreased Aβ40, Aβ42 levels in primary cortical neurons. Aβ40 (A), Aβ42 (B), and sAPPα (C) levels were detected in supernatant from cultured cortical neurons incubated with various concentration of α‐M for 24 h. Data represent mean±SEM from at least three independent experiments performed in triplicate. *P<.05, **P<.01, compared with 0 nmol·L−1 of α‐M
3.2. α‐M did not affect the expression of enzymes involved in nonamyloidogenic and amyloidogenic pathways and APP maturation in primary cultured cortical neurons
Aβ was generated during the sequential cleavage of APP by β‐secretase–β‐site amyloid precursor protein cleaving enzyme 1 (BACE1) and γ‐secretase complex that includes presenilin 1 (PS1).18 Aβ levels in CNS were balanced by its production and clearance, which was mediated by passive diffusion into blood stream, degradation by degrading enzymes such as neprilysin (NEP) and insulin‐degrading enzyme (IDE).29, 30 The enzymes possessing α‐secretase activity to cleave APP include several members of a disintegrin and metalloproteinase (ADAM) family, ADAM9, ADAM10 and ADAM17.31 We measured the effects of α‐M on mRNA and protein expressions of these enzymes. As shown in Figure 2, mRNA expressions of α‐secretase ADAM9, ADAM10, ADAM17, β‐secretase BACE1 and γ‐secretase PS1 were not changed, suggesting that α‐M did not decrease the Aβ levels through modulating enzymes involved in nonamyloidogenic and amyloidogenic pathways. IDE, NEP mRNA expressions were also determined, and found that 6.25, 25 nmol·L−1 α‐M significantly increased IDE mRNA expression after 12 hours treatment (Figure 2). The protein expressions of IDE were increased to some extent without statistical significance after α‐M treatment for 24 hours (Figure 3A,C). These data indicated that the expressions of enzymes involved in the Aβ secretion and degradation were not significantly affected by α‐M.
Figure 2.
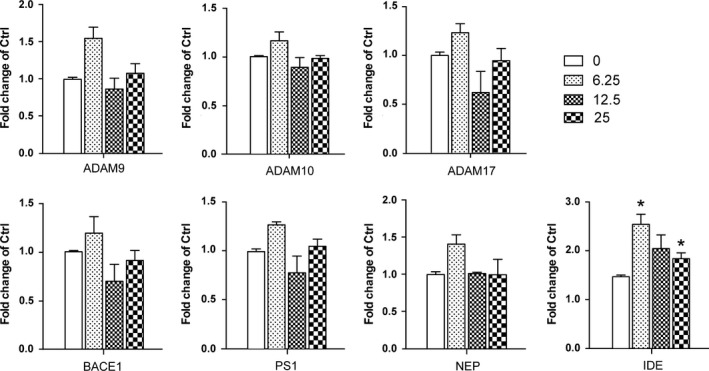
The effect of α‐M on mRNA expression of enzymes involved in amyloid precursor protein(APP) metabolism and Aβ degradation in primary cortical neurons. α‐Secretase ADAM9, ADAM10, ADAM17, β‐secretase BACE1, γ‐secretase PS1 and Aβ degrading enzyme neprilysin(NEP) and insulin‐degrading enzyme(IDE) mRNA expression were detected in cultured cortical neurons incubated with various concentration of α‐M for 12 h by quantitative real‐time PCR and was normalized to that of 18 s gene. Data represent mean ± SEM of at least three independent experiments performed in triplicate. *P<.05, **P<.01, compared with 0 nmol·L−1 of α‐M
Figure 3.
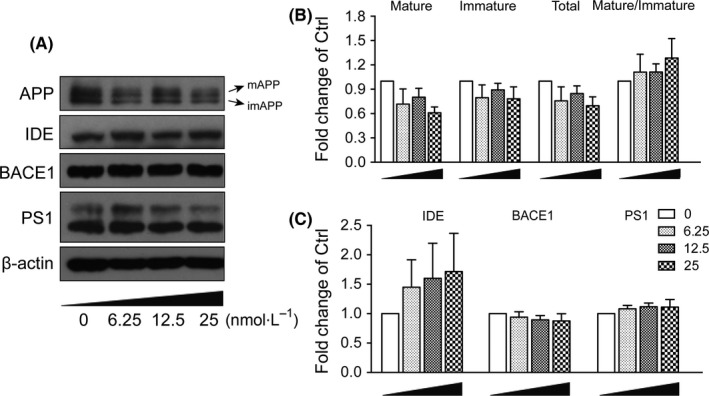
α‐M did not affect protein expression of secretases and amyloid precursor protein(APP) metabolism in primary cortical neurons. APP (mAPP, imAPP), IDE, BACE1 and PS1 protein expressions were immunoblotted (A) and quantified (B for mAPP, imAPP and mAPP:imAPP ratio, and C for IDE, BACE1, PS1) from cultured cortical neurons incubated with various concentration of α‐M for 24 h. Data represent mean ± SEM of at least three independent experiments
Mature APP (mAPP, N‐ and O‐glycosylated form) localizes in the late protein secretory pathway such as trans‐Golgi and plasma membrane and is able to be successively cleaved by β‐, and γ‐ or α‐secretases, whereas immature APP (imAPP, N‐glycosylated form) locates in endoplasmic reticulum and cis‐Golgi where it cannot be accessed by metabolic enzymes.32 So we also measured whether α‐M affected APP maturation. As shown in Figure 3A,B, α‐M at 25 nmol·L−1 decreased the levels of total APP, mAPP and imAPP to some extent, which did not reach statistical significance. Furthermore, α‐M had no effect on mAPP:imAPP ratio.
3.3. α‐M inhibited β‐secretase and likely γ‐secretase activities
To further investigate the role of secretases in α‐M's effects on Αβ production, the activities of α‐, β‐ and γ‐secretases in primary cultured cortical neurons were measured. Compared with vehicle‐treated group, α‐secretase activity was not affected by α‐M treatment (Figure 4A). However, α‐M (6.25, 12.5 and 25 nmol·L−1) reduced β‐secretase activities to 95.31 ± 6.58%, 80.31 ± 6.58% (P<.01) and 64.24 ± 5.26% (P<.01) of vehicle‐treated group (Figure 4A). Meanwhile, determined using a peptide substrate containing the C‐terminal APP amino acid sequence that is cleaved by γ‐secretase, α‐M (6.25, 12.5 and 25 nmol·L−1) reduced γ‐secretase activities to 64.86 ± 15.26% (P<.01), 60.38 ± 16.41% (P<.01) and 43.70 ± 25.89% (P<.01) of vehicle‐treated group (Figure 4). IC50 of α‐M against β‐ and γ‐secretases were 13.22 nmol·L−1 and 16.98 nmol·L−1. These data suggest that α‐M specifically inhibits amyloidogenic pathway enzyme activities, which may account for the reduced Αβ levels in cortical neurons after α‐M treatment.
Figure 4.
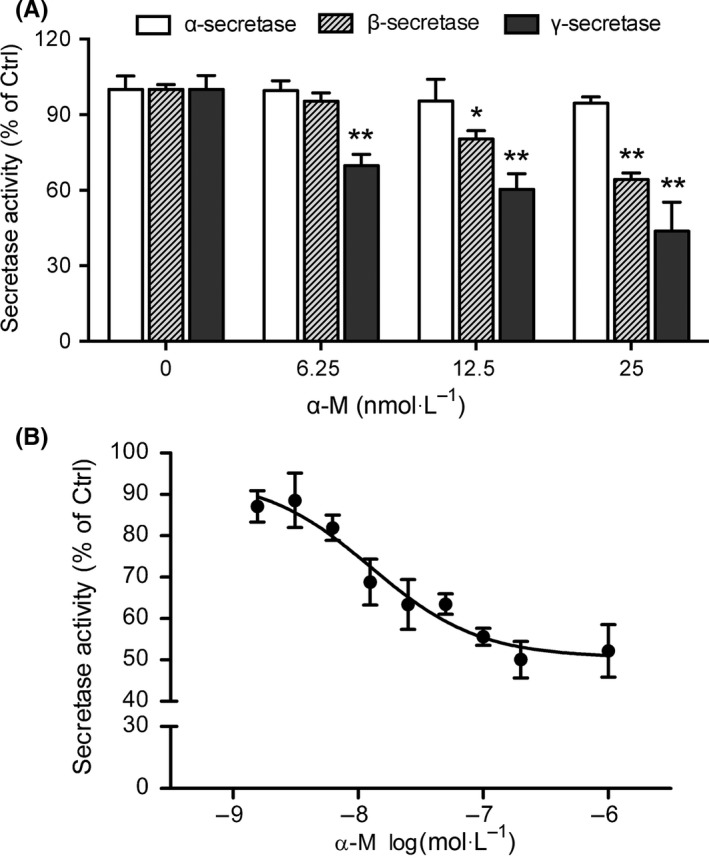
α‐M inhibited β‐secretase and likely γ‐secretase activities. (A) The activities of α‐, β‐ and γ‐secretases were measured from cortical neurons incubated with various concentrations of α‐M for 24 h. *P<.05, **P<.01, compared with 0 nmol·L−1 of α‐M. (B) α‐M inhibited β‐secretase (BACE1) activity by in vitro FRET assay. Data represent mean ± SEM of at least three independent experiments performed in triplicate
Further in vitro FRET based assay of β‐secretase (BACE1) activity was performed. As shown in Figure 4B, α‐M dose‐responsively inhibited BACE1 activity. The IC50 value of α‐M was 12.63 nmol·L−1.
3.4. α‐M potentially bound to BACE1 and PS1
Molecular docking analysis was further performed to investigate their binding modes. As shown in Figure 5A, α‐M fitted well into the binding cavity of BACE1 catalytic site. As shown in details (Figure 5B), α‐M interacted with BACE1 at Asp32, one residue of an important catalytic diad (Asp32/Asp228) in the S1 and S2’ pocket 27, 33 and at Phe108 of N‐terminal lobe through hydrogen bonds. Furthermore, the α‐M was surrounded by polar residues and a few aromatic residues. Interactions between α‐M and Tyr71, Thr72, Gln73 of BACE1 suggest α‐M could occupy the flap and intervene BACE1 catalytic cycle. Moreover, the methoxyl and adjacent phenolic hydroxyl group of α‐M interacted with Lys107, Ile110 in the S1 pocket of BACE1. In addition, Tyr198, Thr232, Thr231 and Gly230 in S3 active pocket of BACE1 may participate in the interaction network with α‐M. For PS1, as shown in Figure 6A,B, α‐M docked into its catalytic subunit and bridged two important segments TM2 (Gln112, Leu113, Ile114, Thr119, and Glu120) and TM6 (Thr245, Leu248, Ile249).
Figure 5.
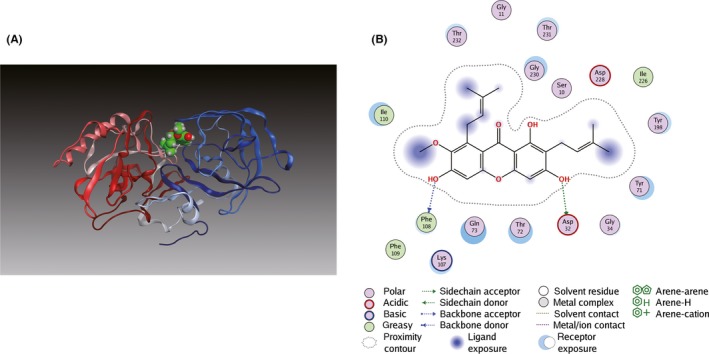
Binding mode of α‐M to BACE1. (A) α‐M was docked into crystal structure of BACE1 (PDB ID: 1FKN). The N‐terminal lobe and the C‐terminal lobe of BACE1 are blue and red, respectively. (B) Key hydrogen bonding interactions between α‐M and BACE1 at the catalytic residue Asp32 and at Phe108 of N‐terminal lobe are indicated with green and blue dashed lines
Figure 6.
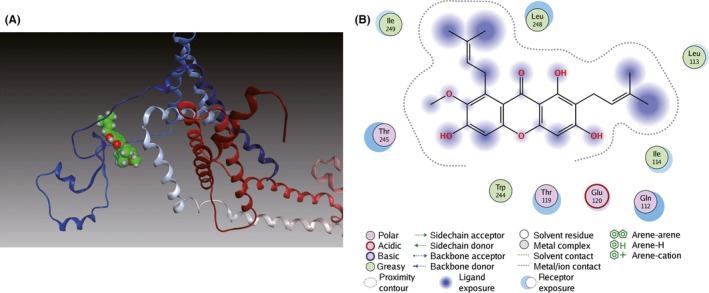
Binding mode of α‐M to PS1. (A) Ribbon diagram of PS1 (PDB ID: 5A63) in complex with α‐M. TM2 and TM6 of PS1 are blue and gray. (B) Interactions between α‐M and residues of PS1
4. Discussion
Aβ peptides are important components of senile plaque of AD brain. When the balance of Aβ synthesis and clearance is broken, Aβ can aggregate and deposit in neurons. Aβ42 peptides aggregate and deposit early and easily as it is more hydrophobic than Aβ40. 34 Our previous study showed that α‐M could attenuate Aβ oligomers‐induced neurotoxicity by inhibiting amyloid aggregation.26 The present study demonstrated that α‐M could decrease the Aβ40 and Aβ42 levels. These results indicate that α‐M could attenuate the Aβ toxicity through both decreasing its production and reducing its aggregation.
APP can be cleaved by α‐secretase to prevent Aβ generation. Hence, activation of α‐secretase could be an effective approach to decrease Aβ and to promote generation of neurotrophic sAPPα.35 We found that α‐M did not significantly increase sAPPα. Moreover, the gene expression of enzymes ADAM9, ADAM10 and ADAM17 and α‐secretase activity were not changed by α‐M. These results suggested that the nonamyloidogenic pathway was not involved in the effects of α‐M on decreasing Aβ levels.
Aβ levels could also be decreased by inhibiting the expressions or activities of β‐ and γ‐secretases.1, 9, 18, 36 BACE1, belongs to the group of aspartyl proteases and is the limiting step of generating neurotoxic Aβ.37 γ‐Secretase is a transmembrane multisubunit protease complex and responsible for the final cleavage of APP.1, 18, 19 Its proteolytic activity is determined by PS1.38 NEP could degrade Aβ both in monomeric form and the pathological oligomeric form and its deficiency resulted in defect of the metabolic suppression of the endogenous Aβ levels.39, 40 IDE was a thiol metalloendopeptidase that could degrade small peptides and IDE deficiency resulted in a >50% decrease in Aβ degradation.41, 42 Our results showed that the mRNA or protein levels of these enzymes were not significantly affected by α‐M, suggesting that α‐M does not directly affect the expressions of enzymes of amyloidogenic pathway. Instead, our study showed that α‐M inhibited activities of β‐secretase and likely γ‐secretase without affecting the activity of α‐secretase. Molecular docking further supports the interference of α‐M in the active site of β‐ and γ‐secretases.
It is reported that β‐secretase can be inhibited by interfering the hydrogen bonding network essential for its proteolytic activity and γ‐secretase can be inhibited by occupying its active site.43 In the 3D structure of BACE1 which was a long cleft made up of NH2‐ and COOH‐terminal lobes of polypeptide chain,27 α‐M was located in its active site and was tightly covered by the flap of BACE1. Furthermore, α‐M was shown to occupy the center of the BACE1 binding pockets and directly interacted with catalytic‐site Asp32. These data suggest that α‐M could inhibit BACE1, which is consistent with the activity assay. The atomic model of human γ‐secretase was reported and TM2 and TM6 in PS1 were shown to exhibit considerable flexibility that yielded a plastic active site.28 Molecular docking showed that α‐M interacted with the interface of TM2 and TM6, suggesting that α‐M interfere with the plastic active site of PS1.
Although inhibition of β‐secretase or γ‐secretase has been marked as potential AD therapies, BACE1 and γ‐secretase have substrates other than APP, which may cause undesirable effects.44, 45 Except APP, additional BACE1 substrates are shown to be involved in cell signaling, immune and inflammatory responses.45 But partial inhibition of β‐secretase activity seems beneficial.46 Decrease of BACE1 by chicoric acid did not affect the motor activities of lipopolysaccharide‐injected C57BL/6J mice.47 No physiological activity abnormalities were reported when the level of BACE1 was decreased by hydrogen sulfide in APP/PS1 transgenic mice.48 Both chicoric acid and hydrogen sulfide do not have dramatic effects on BACE1. While, the major obstacle for γ‐secretase inhibitors is the fact that γ‐secretase is an essential part of the Notch signaling. As such, γ‐secretase inhibitor LY‐411, 575 is shown to delay lymphocyte development and alter intestine morphology.49 Whether α‐M can cause physiological changes due to its inhibition of β‐ and γ‐secretases has not been investigated. However, in vivo evaluation of potential toxicity of α‐M shows that oral gavage with α‐M pure compound or extracts of mangosteen at doses as high as 1 000 mg/kg body weight does not produce any changes of physiological status and general behavior in rodents.50, 51 But dietary of α‐M may exacerbate existing colonic inflammation,52, 53 whether such effect is related to its inhibition of β‐ and γ‐secretases needs to be further investigated.
α‐M is a polyphenolic xanthone. There are also some other polyphenols have been reported to be involved in APP metabolism. Epigallocatechin‐3‐gallate (EGCG) and octyl gallate enhance the nonamyloidogenic processing of APP by activation of ADAM10 through estrogen receptor‐mediated mechanism.54, 55 Curcumin is able to lower Aβ levels by attenuating the maturation of APP in the secretory pathway.56 Curcumin derivative CU6 could down‐regulate intracellular APP trafficking by inducing expression of the endoplasmic reticulum chaperone glucose‐regulated protein 78, however, it does not inhibit neither β‐ nor γ‐secretase activity.57 Myricetin inhibits BACE1.58 Our results showed that α‐M inhibited β‐ and γ‐secretases activities in the amyloidogenic processing of APP by interacting with BACE1 and PS1 to disable them and then reducing Aβ formation. This is the first report of the dual inhibition of both β‐ and γ‐secretases by polyphenols.
Combined with our previously reports showing that α‐M could dissociate Aβ oligomers and accelerate Aβ1‐42 clearance,26, 59 α‐M could influence Aβ in multiple aspects. However, whether α‐M selectively targets Aβ is still uncertain. Whether α‐M targets other proteins relevant to AD, such as tau, warrants further investigations.
5. Conclusions
In summary, the current study demonstrates that α‐M decreases the Aβ levels through the inhibition of the activities of β‐secretase and likely γ‐secretase in the amyloidogenic pathway. Collectively, the study, together with previous published data showing its ability to dissociate toxically aggregated Aβ oligomers, showed α‐M could attenuate Aβ neurotoxicity through intervening in multiple processes of Aβ production and aggregation. Thus α‐M can be a promising candidate for treating AD‐related β‐amyloid pathology.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgements
This research was supported by National Natural Science Foundation of China (81473217, 81173044), International Science & Technology Cooperation Program of China (2011DFA33180).
Zhao L‐X, Wang Y, Liu T, et al. α‐Mangostin decreases β‐amyloid peptides production via modulation of amyloidogenic pathway. CNS Neurosci Ther.2017;23:526–534. 10.1111/cns.12699
The first two authors contributed equally to this work.
Contributor Information
Jian‐Rong Xu, Email: janker.xu@gmail.com.
Yu Qiu, Email: yu_qiu@hotmail.com.
References
- 1. Awasthi M, Singh S, Pandey VP, Dwivedi UN. Alzheimer's disease: an overview of amyloid beta dependent pathogenesis and its therapeutic implications along with in silico approaches emphasizing the role of natural products. J Neurol Sci. 2016;361:256‐271. [DOI] [PubMed] [Google Scholar]
- 2. Daulatzai MA. Pharmacotherapy and Alzheimer's disease: the M‐drugs (melatonin, minocycline, modafinil, and memantine) approach. Curr Pharm Des. 2016;22:2411‐2430. [DOI] [PubMed] [Google Scholar]
- 3. Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702‐716. [DOI] [PubMed] [Google Scholar]
- 4. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011;377:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 5. Bullock R. Efficacy and safety of memantine in moderate‐to‐severe Alzheimer disease: the evidence to date. Alzheimer Dis Assoc Disord. 2006;20:23‐29. [DOI] [PubMed] [Google Scholar]
- 6. De Strooper B, Karran E. The cellular Phase of Alzheimer's disease. Cell. 2016;164:603‐615. [DOI] [PubMed] [Google Scholar]
- 7. Kowalska A. The beta‐amyloid cascade hypothesis: a sequence of events leading to neurodegeneration in Alzheimer's disease. Neurol Neurochir Pol. 2004;38:405‐411. [PubMed] [Google Scholar]
- 8. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245‐4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schonherr C, Bien J, Isbert S, et al. Generation of aggregation prone N‐terminally truncated amyloid beta peptides by meprin beta depends on the sequence specificity at the cleavage site. Mol Neurodegener. 2016;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sandberg A, Luheshi LM, Sollvander S, et al. Stabilization of neurotoxic Alzheimer amyloid‐beta oligomers by protein engineering. Proc Natl Acad Sci U S A. 2010;107:15595‐15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katsurabayashi S, Kawano H, Ii M, et al. Overexpression of Swedish mutant APP in aged astrocytes attenuates excitatory synaptic transmission. Physiol Rep. 2016;4:e12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Veeraraghavalu K, Zhang C, Zhang X, Tanzi RE, Sisodia SS. Age‐dependent, non‐cell‐autonomous deposition of amyloid from synthesis of beta‐amyloid by cells other than excitatory neurons. J Neurosci. 2014;34:3668‐3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Esch FS, Keim PS, Beattie EC, et al. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122‐1124. [DOI] [PubMed] [Google Scholar]
- 14. May PC, Willis BA, Lowe SL, et al. The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci. 2015;35:1199‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lahiri DK, Maloney B, Long JM, Greig NH. Lessons from a BACE1 inhibitor trial: off‐site but not off base. Alzheimers Dement. 2014;10:S411‐S419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13:319‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kothare PA, Bateman KP, Dockendorf M, et al. An integrated strategy for implementation of dried blood spots in clinical development programs. AAPS J. 2016;18:519‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Godyn J, Jonczyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol Rep. 2016;68:127‐138. [DOI] [PubMed] [Google Scholar]
- 19. Mikulca JA, Nguyen V, Gajdosik DA, et al. Potential novel targets for Alzheimer pharmacotherapy: II. update on secretase inhibitors and related approaches. J Clin Pharm Ther. 2014;39:25‐37. [DOI] [PubMed] [Google Scholar]
- 20. Roher AE, Maarouf CL, Kokjohn TA, et al. Neuropathological and biochemical assessments of an Alzheimer's disease patient treated with the gamma‐secretase inhibitor semagacestat. Am J Neurodegener Dis. 2014;3:115‐133. [PMC free article] [PubMed] [Google Scholar]
- 21. Cacabelos R, Torrellas C, Carrera I, et al. Novel therapeutic strategies for dementia. CNS Neurol Disord Drug Targets. 2016;15:141‐241. [DOI] [PubMed] [Google Scholar]
- 22. Kuhn PH, Koroniak K, Hogl S, et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31:3157‐3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franceschelli S, Pesce M, Ferrone A, et al. A novel biological role of alpha‐mangostin in modulating inflammatory response through the activation of sirt‐1 signaling pathway. J Cell Physiol. 2016;231:2439‐2451. [DOI] [PubMed] [Google Scholar]
- 24. Kritsanawong S, Innajak S, Imoto M, Watanapokasin R. Antiproliferative and apoptosis induction of alpha‐mangostin in T47D breast cancer cells. Int J Oncol. 2016;48:2155‐2165. [DOI] [PubMed] [Google Scholar]
- 25. Fang Y, Su T, Qiu X, et al. Protective effect of alpha‐mangostin against oxidative stress induced‐retinal cell death. Sci Rep. 2016;6:21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Xia Z, Xu JR, et al. Alpha‐mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates beta‐amyloid oligomers‐induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology. 2012;62:871‐881. [DOI] [PubMed] [Google Scholar]
- 27. Hong L, Koelsch G, Lin X, et al. Structure of the protease domain of memapsin 2 (beta‐secretase) complexed with inhibitor. Science. 2000;290:150‐153. [DOI] [PubMed] [Google Scholar]
- 28. Bai XC, Yan C, Yang G, et al. An atomic structure of human gamma‐secretase. Nature. 2015;525:212‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shankar GM, Leissring MA, Adame A, et al. Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leissring MA, Farris W, Chang AY, et al. Enhanced proteolysis of beta‐amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087‐1093. [DOI] [PubMed] [Google Scholar]
- 31. Lammich S, Kojro E, Postina R, et al. Constitutive and regulated alpha‐secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922‐3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saito Y, Akiyama M, Araki Y, et al. Intracellular trafficking of the amyloid beta‐protein precursor (APP) regulated by novel function of X11‐like. PLoS ONE. 2011;6:e22108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorfe AA, Caflisch A. Functional plasticity in the substrate binding site of beta‐secretase. Structure. 2005;13:1487‐1498. [DOI] [PubMed] [Google Scholar]
- 34. Sandebring A, Welander H, Winblad B, Graff C, Tjernberg LO. The pathogenic abeta43 is enriched in familial and sporadic Alzheimer disease. PLoS ONE. 2013;8:e55847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Marum RJ. Current and future therapy in Alzheimer's disease. Fundam Clin Pharmacol. 2008;22:265‐274. [DOI] [PubMed] [Google Scholar]
- 36. Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. Gamma‐Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828:2898‐2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seubert P, Oltersdorf T, Lee MG, et al. Secretion of beta‐amyloid precursor protein cleaved at the amino terminus of the beta‐amyloid peptide. Nature. 1993;361:260‐263. [DOI] [PubMed] [Google Scholar]
- 38. Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma‐secretase. J Alzheimers Dis. 2011;25:3‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113‐116. [DOI] [PubMed] [Google Scholar]
- 40. Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550‐1552. [DOI] [PubMed] [Google Scholar]
- 41. Qiu WQ, Walsh DM, Ye Z, et al. Insulin‐degrading enzyme regulates extracellular levels of amyloid beta‐protein by degradation. J Biol Chem. 1998;273:32730‐32738. [DOI] [PubMed] [Google Scholar]
- 42. Farris W, Mansourian S, Chang Y, et al. Insulin‐degrading enzyme regulates the levels of insulin, amyloid beta‐protein, and the beta‐amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162‐4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lakey‐Beitia J, Berrocal R, Rao KS, Durant AA. Polyphenols as therapeutic molecules in Alzheimer's disease through modulating amyloid pathways. Mol Neurobiol. 2015;51:466‐479. [DOI] [PubMed] [Google Scholar]
- 44. Wolfe MS. Gamma‐secretase inhibitors and modulators for Alzheimer's disease. J Neurochem. 2012;120(Suppl 1):89‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kandalepas PC, Vassar R. Identification and biology of beta‐secretase. J Neurochem. 2012;120(Suppl 1):55‐61. [DOI] [PubMed] [Google Scholar]
- 46. Ghosh AK, Osswald HL. BACE1 (beta‐secretase) inhibitors for the treatment of Alzheimer's disease. Chem Soc Rev. 2014;43:6765‐6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Q, Chen Y, Shen C, et al. Chicoric acid supplementation prevents systemic inflammation‐induced memory impairment and amyloidogenesis via inhibition of NF‐kappaB. FASEB J. 2017;31:1494‐1507. [DOI] [PubMed] [Google Scholar]
- 48. Liu Y, Deng Y, Liu H, Yin C, Li X, Gong Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: a novel mechanism mediated by the activation of Nrf2. Pharmacol Biochem Behav. 2016;150–151:207‐216. [DOI] [PubMed] [Google Scholar]
- 49. Wong GT, Manfra D, Poulet FM, et al. Chronic treatment with the gamma‐secretase inhibitor LY‐411,575 inhibits beta‐amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876‐12882. [DOI] [PubMed] [Google Scholar]
- 50. Ibrahim MY, Hashim NM, Mohan S, et al. α‐Mangostin from Cratoxylum arborescens: an in vitro and in vivo toxicological evaluation. Arabian J Chem. 2015;8:129‐137. [Google Scholar]
- 51. Bunyong R, Chaijaroenkul W, Plengsuriyakarn T, Na‐Bangchang K. Antimalarial activity and toxicity of Garcinia mangostana Linn. Asian Pac J Trop Med. 2014;7:693‐698. [Google Scholar]
- 52. Gutierrez‐Orozco F, Thomas‐Ahner JM, Berman‐Booty LD, et al. Dietary alpha‐mangostin, a xanthone from mangosteen fruit, exacerbates experimental colitis and promotes dysbiosis in mice. Mol Nutr Food Res. 2014;58:1226‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gutierrez‐Orozco F, Thomas‐Ahner JM, Galley JD, et al. Intestinal microbial dysbiosis and colonic epithelial cell hyperproliferation by dietary alpha‐mangostin is independent of mouse strain. Nutrients. 2015;7:764‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandez JW, Rezai‐Zadeh K, Obregon D, Tan J. EGCG functions through estrogen receptor‐mediated activation of ADAM10 in the promotion of non‐amyloidogenic processing of APP. FEBS Lett. 2010;584:4259‐4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang SQ, Sawmiller D, Li S, et al. Octyl gallate markedly promotes anti‐amyloidogenic processing of APP through estrogen receptor‐mediated ADAM10 activation. PLoS ONE. 2013;8:e71913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang C, Browne A, Child D, Tanzi RE. Curcumin decreases amyloid‐beta peptide levels by attenuating the maturation of amyloid‐beta precursor protein. J Biol Chem. 2010;285:28472‐28480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kotani R, Urano Y, Sugimoto H, Noguchi N. Decrease of amyloid‐beta levels by curcumin derivative via modulation of amyloid‐beta protein precursor trafficking. J Alzheimers Dis. 2017;56:529‐542. [DOI] [PubMed] [Google Scholar]
- 58. Chakraborty S, Kumar S, Basu S. Conformational transition in the substrate binding domain of beta‐secretase exploited by NMA and its implication in inhibitor recognition: BACE1‐myricetin a case study. Neurochem Int. 2011;58:914‐923. [DOI] [PubMed] [Google Scholar]
- 59. Yao L, Gu X, Song Q, et al. Nanoformulated alpha‐mangostin ameliorates Alzheimer's disease neuropathology by elevating LDLR expression and accelerating amyloid‐beta clearance. J Control Release. 2016;226:1‐14. [DOI] [PubMed] [Google Scholar]