

Summary
Aims
This study was to investigate whether cell proliferation and adult neurogenesis are affected at early neurodegenerative stage when neuron loss has not begun to display.
Methods and Results
Forebrain‐specific nicastrin (NCT) conditional knockout (cKO) mice were generated by crossing NCT f/f with CaMKIIα‐Cre Tg mice. BrdU was used as a lineage tracer to label proliferating neural progenitor cells (NPCs). Immunohistochemistry (IHC) on BrdU indicated that the total number of BrdU positive (+) cells was increased in NCT cKO mice. IHC on doublecortin (DCX) showed that the total number of DCX+ cells was also increased in NCT cKO mice. NCT cKO mice displayed significant astrogliosis as well. However, NCT cKO mice at 3 months did not show significant neuronal death or synaptic loss.
Conclusions
NCT‐dependent γ‐secretase activity plays an important role in cell proliferation and immature neuron generation. Enhanced neurogenesis and astrogliosis may be early cellular events prior to the occurrence of neuronal death in neurodegenerative disease.
Keywords: γ‐secretase, astrogliosis, cell proliferation, neurodegeneration, neurogenesis
1. INTRODUCTION
Neurogenesis is a process of generating functional neurons from neural stem cells in the brain of mammals.1 Adult neurogenesis takes place in certain regions of postnatal brain, including the subgranular zone (SGZ) of the dentate gyrus (DG) and the subventricular zone (SVZ) around the lateral ventricles.2 It is believed that adult neurogenesis is not only important for the repair of the damaged nerve cells after brain injury 3 but also for hippocampal synaptic plasticity and learning and memory.4, 5 Neurogenesis involves several neurogenic processes, including NPC proliferation, immature neuron migration, the survival of newborn neurons, and the integration of new neurons into the network of the central nervous system (CNS).2
Alzheimer's disease (AD) is the most common form of dementia. AD is characterized by massive neuron loss and the formation of amyloid plaques and neurofibrillary tangles.6 Recent evidence has shown that mutations on presenilin 1/2 (PS1/2) are the major cause for familial AD.6 PS is a subunit for the γ‐secretase complex, which is a membrane‐embedded protease that cleaves amyloid precursor protein (APP) and Notch receptors.7 The other three subunits include NCT, anterior pharynx‐defective 1 (Aph1) and presenilin enhancer 2 (PEN2).8 It has been proposed that PS mutations may cause AD in a loss of function mechanism.9, 10 Consistent with this, several groups have shown that conditional inactivation of PS1/2 leads to AD‐like neurodegeneration in mice.11, 12, 13, 14, 15 Moreover, loss of NCT in the forebrain also causes age‐dependent neurodegeneration.16, 17, 18 The above findings suggest that PS/NCT‐dependent γ‐secretase activity is required for neuronal survival.
Studies on adult neurogenesis in AD have led to opposite conclusions. First, early work using brain tissues of Alzheimer patients revealed increased protein levels of DCX, suggesting increased neurogenesis in AD.19 However, another study showed that there is increased glial proliferation but not neurogenesis in AD.20 Second, results from different lines of APP transgenic (Tg) mice are somehow contradictory. Work by Jin et al21 showed that the PDGF‐APP Sw,Ind mouse exhibits increased neurogenesis in the SGZ and the SVZ. Similar findings were obtained by several other groups.22, 23, 24 In contrast, Haughey et al25, 26 were the first to show decreased adult hippocampal neurogenesis in an APP Tg mouse model of AD. Moreover, decreased neurogenesis has been reported in several different lines of APP Tg mice 27, 28, 29 and the 3 × Tg mouse model of AD.30, 31, 32
Abundant evidence has shown that conditional inactivation of PS1/2 causes age‐dependent AD‐like neurodegeneration,12, 14, 15, 33, 34 making it an open question whether loss of γ‐secretase activity impairs neurogenesis. Interestingly, a recent study has nicely shown that forebrain‐specific PS1/2 double cKO mice aged at 7‐9 and 18‐20 months exhibit increased cell proliferation and increased generation of new neurons in the hippocampus,34 suggestive of enhanced adult neurogenesis at early or late stage of neurodegeneration. However, it has remained unknown whether loss of γ‐secretase activity affects neurogenesis before neuronal death has taken place. In this study, we examined cell proliferation and the number of immature neurons in NCT cKO mice at 3 months, an age when neuron death and neuron loss were not detected. We observed increased cell proliferation and immature neuron generation, highlighting a critical role of NCT‐dependent γ‐secretase activity in adult neurogenesis.
2. MATERIALS AND METHODS
2.1. The animals
Floxed NCT mice (NCT f/f) and CaMKIIα‐Cre Tg mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA).16 Mice were bred in an SPF (specific pathogen free) room of the core animal facility of the Model Animal Research Center at Nanjing University. The room temperature was 25 ± 1°C, and the light‐cycle was automatically controlled (12 hours for light and 12 hours for dark). Mice had free access to food and water. The originally generated NCT f/+ mouse had the mixed genetic background (129/C57BL/6), but it had been backcrossed to BL/6 for >10 generations.18 Whereas NCT f/f ;CaMKIIa‐Cre was used as NCT cKO, its age‐matched littermates, NCT f/f and NCT f/+ ;CaMKIIa‐Cre, served as the control in this study.
2.2. Nissl staining
The mice were euthanized with CO2, perfused with PBS, fixed in 4% paraformaldehyde overnight at 4°C, dehydrated using graded ethanol. After paraffin embedding, each block which contained 4 hemi‐brains including 2 controls and 2 cKOs was sectioned sagittally (10 μm) using a microtome. On each slide, there were 4 sagittal brain sections (2 controls and 2 cKOs), which were on identical stereotaxic plane. Brain sections were deparaffinized, ethanol rehydrated, and then rinsed for 5 minutes using distilled water. Sections were treated with 0.1% cresyl‐violet for 1 min and then rinsed with water. Sections were dried naturally and then sealed using neutral resin.
2.3. Immunohistochemistry
A total of 6‐8 slides (spaced at 200 μm), on which each brain section contained intact hippocampal formation including CA1, CA3, and DG areas, were used for IHC and FIHC on BrdU, DCX, or GFAP. Sagittal brain sections were deparaffinized, ethanol rehydrated. After retrieval of antigen (boiled in 0.01M pH6.0 sodium citrate buffer solution for 25 minutes and then cooled to room temperature), sections were blocked using hydrogen peroxide (30% H2O2 diluted 10:1 in methanol for 30 minutes), incubated with BSA (5% bovine serum albumin in PBS for 30 minutes) and were incubated overnight at 4°C with the following antibodies including GFAP (1:500; Sigma‐Aldrich, St. Louis, MO, USA), BrdU (1:200; Abcam, Cambridge, UK), DCX (1:200; Santa Cruz Biotechnology, Santa Cruz, Dallas, TX, USA), NeuN (1:500; Millipore, Billerica, MA, USA), MAP2 (1:200; Sigma‐Aldrich), or SVP38 (1:500; Sigma‐Aldrich), respectively. After incubation with secondary antibodies diluted in PBS (phosphate buffer solution), the sections were developed using the avidin‐biotin peroxidase complex method (ABC kit). Sections were then dehydrated using graded ethanol and sealed using neutral resin. For fluorescence immunostaining, brain sections were incubated with either Alexa Fluor 488 goat anti‐mouse/anti‐rabbit or Alexa Fluor 594 goat anti‐mouse/anti‐rabbit secondary antibodies (Invitrogen, Waltham, MA, USA), and then analyzed with a Leica SP5 confocal laser‐scanning microscope.
2.4. Western blotting
Mice cortices were dissected after CO2 euthanasia and PBS perfusion. Tissues were homogenized in cold radio immunoprecipitation assay lysis buffer containing protease and phosphatase inhibitors (Thermo). Lysates were cleared by centrifugation (13 400 g for 30 minutes, 4°C). Normalized volume of samples (40 μg total protein) was resolved in 10% SDS‐PAGE (invitrogen), transferred to nitrocellulose membrane. After blocking with 5% (w/v) of nonfat milk solution, membranes were probed with primary antibodies overnight and detected using infrared dye‐coupled secondary antibodies. Membranes were scanned using Odyssey Infrared Imaging System (Li‐Cor). Primary antibodies used were as following: anti‐NCT (1:200; Sigma‐Aldrich), anti‐actin (1:10000; SAB, College Park, MA, USA), GFAP (1:500; Sigma‐Aldrich), NeuN (1:500; Millipore), anti‐SVP38 (1:1000; Sigma‐Aldrich), anti‐PSD95 (1:1000; CST, Danvers, MA, USA), and anti‐MAP2 (1:500; Sigma‐Aldrich).
2.5. Quantitative real‐time PCR
Total RNA was extracted by using the TRIzol (Invitrogen) according to the instructions. PrimeScript RT reagent Kit (Takara, Kusatsu, Japan) was used to get first‐strand cDNA. Real‐time PCR was performed in an ABI StepOne Plus machine. To examine relative GFAP mRNA levels, primers including GTTACCAGGAGGCACTTGCT (forward, 5′‐>3′) and ACAGGAATGGTGATGCGGTT (reverse, 5′‐>3′) were used.
2.6. BrdU injection
For the proliferation study, mice received intraperitoneal injection of BrdU (Sigma) once a day for 3 consecutive days at the concentration of 100 mg/kg, and were sacrificed at the fourth day. Paraffin embedded brain sections were then prepared. Incorporated BrdU was detected by immunostaining using an antibody against BrdU. The total number of BrdU+ cells was counted using a stereological method.
2.7. TUNEL staining
Brain sections were blocked using 5% BSA for 30 minutes followed by the treatment of Fluorescein (Vazyme, Nanjing, China) at 37°C for an hour. TUNEL staining was analyzed using a Leica confocal laser‐scanning microscope.
2.8. Cell counting
IHC images for BrdU, DCX and GFAP staining were captured for the cortex and the DG using the Olympus BX53‐CellSens Standard system. Images were taken under the 10 × objective lens of the Olympus BX53 microscope. Whereas the paraffin sectioning, the IHC and the FIHC experiments were conducted by one person (LT), cell counting was performed by a different person (YXL) who was blinded to the genotype. The total number of BrdU+ cells or DCX+ cells in the granule cell layer (GCL) was counted and then averaged across sections. The total number of GFAP+ cells in the DG area was counted and then averaged.
2.9. Statistical analysis
Data were presented as the mean ± SEM. Two‐tailed Student's t test for pairwise comparisons was performed to examine the difference between control and cKO mice. P < .05 (*) was considered statistically significant. For Western analysis, at least four pairs of control and cKO mice were used. For cell‐counting analysis, at least three pairs of control and cKO mice were included.
3. RESULTS
3.1. Increased cell proliferation in neuron‐specific NCT cKO mice
We generated neuron‐specific NCT cKO mice by crossing NCT f/f with CaMKIIα‐Cre 35 mice, which began to express Cre recombinase specifically in excitatory neurons of the forebrain since the age of 6‐8 weeks.36 To confirm the expression pattern of Cre, we bred CaMKIIα‐Cre with a reporter line, Rosa26 mTmG 37 and obtained CaMKIIα‐Cre;mTmG (Figure 1A). Using cortical samples of mice aged at 3 months, we performed biochemical analysis and observed significantly reduced protein levels of NCT in cKO mice (Figure 1B). We then conducted Nissl staining and found no detectable change in brain morphology of NCT cKO animals (Figure 1C). We have previously reported that this line of NCT cKO mice exhibit about 50% reduction in the total number of NeuN+ cell in the cortex aged at 13 months.16
Figure 1.
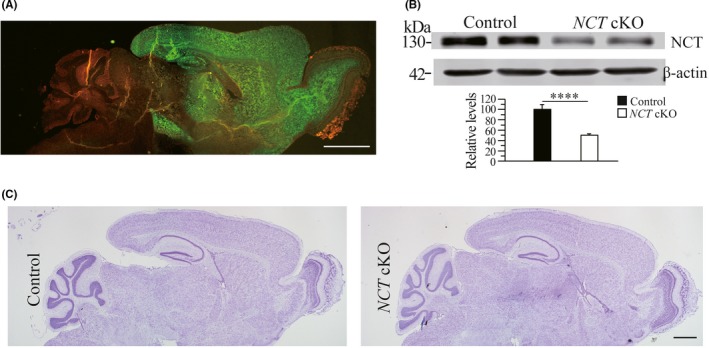
Conditional inactivation of NCT in the forebrain of NCT cKO mice. A, The expression pattern of Cre recombinase. Sagittal brain section was obtained from CaMKIIα‐Cre;mTmG mouse. Green fluorescence (by GFP) is clearly seen in the cortex, the hippocampus, the olfactory bulb and the striatum but not cerebellum. Scale bar = 500 μm. B, Western blotting on NCT using cortical samples of 3‐month NCT cKO mice. There was significant difference on protein levels of NCT between NCT cKO mice and age‐matched littermate controls (control = 100% ± 9.4%, cKO = 50.2% ± 3.0%; n = 3 per group; ****, P < .001). C, Nissl staining. Comparable brain morphology was detected between control and NCT cKO mice. Scale bar = 500 μm
To investigate whether adult neurogenesis was affected in NCT cKO mice prior to the occurrence of neurodegeneration, we used BrdU as a lineage tracer to label proliferating NPCs. Each mouse received intraperitoneal injection of BrdU. Brain sections spaced at 200 μm were collected to conduct IHC for BrdU (Figure 2A). BrdU+ cells in the GCL, the hilus of the DG, and the cortex were counted separately. The averaged number of BrdU+ cells in the GCL of NCT cKO mice was significantly more than that in control mice (Figure 2B‐C: control = 10.7 ± 0.7, cKO = 17.9 ± 1.0; n = 4 mice per group; P < .01; two‐tailed Student's t test). In contrast, the averaged number of BrdU+ cells in the cortex was not different between control and NCT cKO mice (data not shown). Therefore, loss of NCT caused increased cell proliferation in the GCL of the hippocampus.
Figure 2.
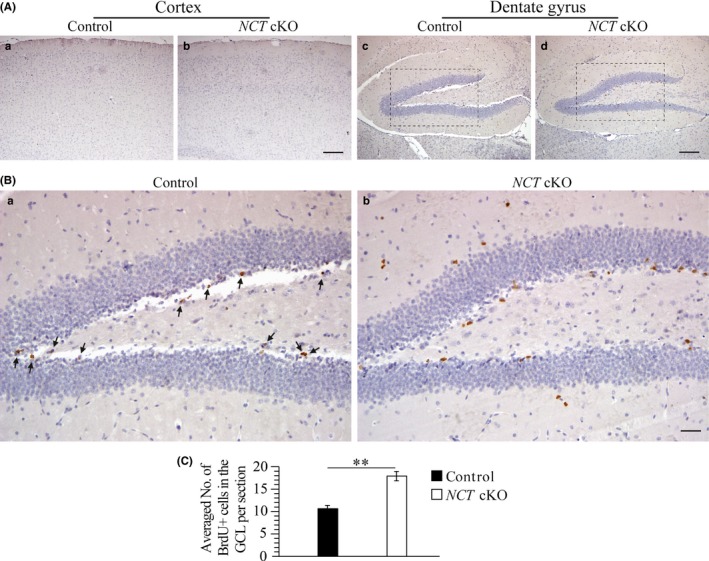
Increased cell proliferation in NCT cKO mice at 3 mos. A, IHC on BrdU with counter staining of Nissl at low magnification. Representative images for BrdU immunostaining were taken for the cortex (a‐b) and the DG (c‐d) of control and NCT cKO mice. BrdU+ cells were shown in Brown. The Blue+ cells were Nissl+. Scale bar = 100 μm. B, The boxed areas in Figure 2A‐c, d were enlarged here. There were more BrdU+ cells in the GCL of NCT cKO mice (b) than in controls (a). BrdU+ cells in NCT cKO mice were indicated by black arrows. Scale bar = 25 μm. C, Cell‐counting results for BrdU+ cells in the GCL. There was a significant difference on the averaged number of BrdU+ cells per section between control and NCT cKO mice (**, P < .01)
3.2. Increased number of immature neurons in NCT cKO mice
To investigate whether increased cell proliferation affected adult neurogenesis, we conducted IHC for DCX, a marker for immature neurons (Figure 3A). We observed increased number of DCX+ cells in NCT cKO mice (Figure 3B). Cell counting results confirmed this finding in NCT cKO mice (Figure 3C: control = 60.4 ± 4.1, cKO = 86.3 ± 9.1; n = 4 mice per group; P < .05). There was no difference on the averaged number of DCX+ cells in the cortex between control and NCT cKO mice (data not shown). Overall, adult neurogenesis was increased in NCT cKO mice.
Figure 3.
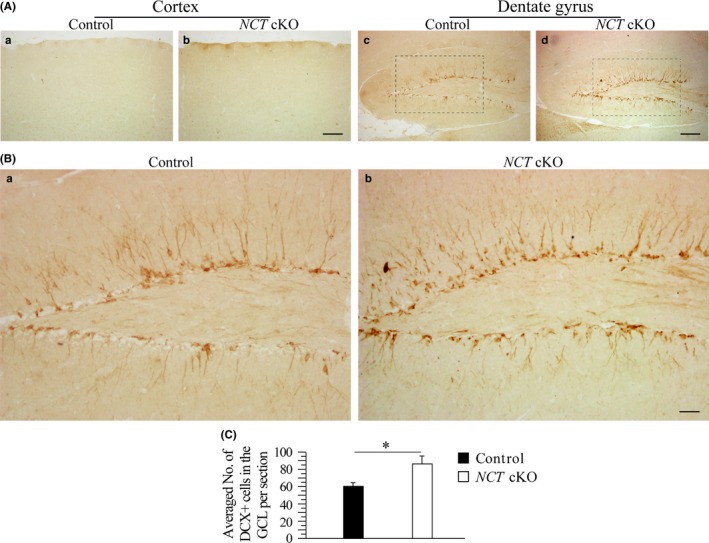
Increased number of immature neurons in NCT cKO mice at 3 mos. A, IHC on DCX. Representative images for DCX IHC were taken for the cortex and the DG of control and NCT cKO mice. DCX+ cells were shown in Brown. There were more DCX+ cells in the DG of NCT cKO mice than in controls. Scale bar = 100 μm. B, The boxed areas in Figure 3A‐c,d were enlarged here. There were more DCX+ cells in the DG of NCT cKO mice than in controls. Scale bar = 25 μm. C, Cell‐counting results for DCX+ cells in the GCL. There was a significant difference on the averaged number of DCX+ cells per section between control and NCT cKO mice (*, P < .05)
3.3. Enhanced astrocytosis in NCT cKO mice
To investigate whether there was change on neuroinflammatory responses in NCT cKO mice at early adulthood, we performed IHC for GFAP. Increased immunoreactivity of GFAP was detected in the cortex and the hippocampus of NCT cKO mice as compared to control animals (Figure 4A). The total number of GFAP+ cells in the DG of NCT cKO mice was larger than that in control animals (Figure 4B: control = 69.8 ± 4.1, n = 4; cKO = 103 ± 3.4, n = 6; P < .001). In the cortex, cell counting results showed significant difference on the averaged number of GFAP+ cells per 100 × 100 μm field between NCT cKO and control mice as well (control = 1.4 ± 0.4; cKO = 7.2 ± 0.7; P < .001). To confirm these morphological changes, we performed Western blotting and quantitative real‐time RT‐PCR analysis for GFAP. First, GFAP protein levels were significantly increased in NCT cKO mice as compared to controls (Figure 4C: control = 100 ± 11.7; cKO = 147.4 ± 35.2; n = 4/group; P < .05). Second, relative GFAP mRNA levels were significantly higher in NCT cKO mice than in controls (Figure 4D: control = 100 ± 6.5, n = 4; cKO = 185.9 ± 26.1, n = 3; P < .05). Overall, the above results confirmed significant reactive astrocytosis.
Figure 4.
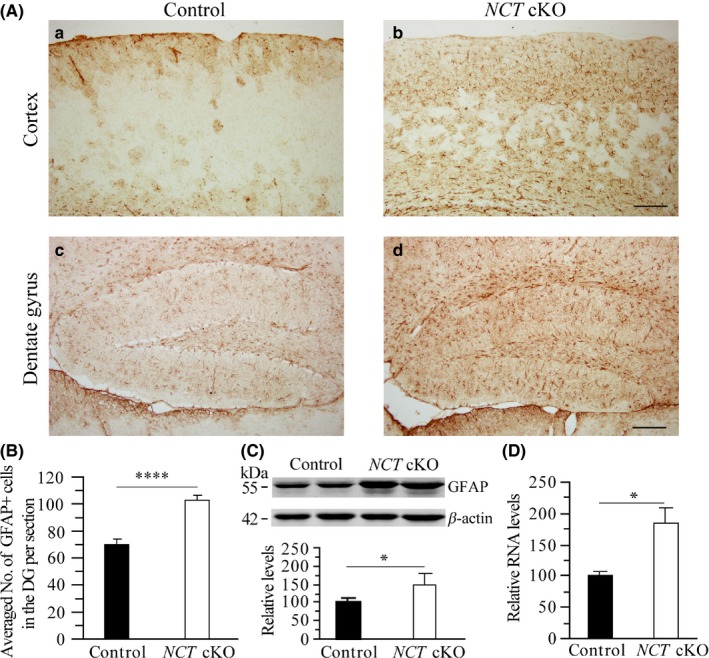
Reactive astrocytosis in NCT cKO mice at 3 mos. A, IHC on GFAP. Representative images for GFAP IHC were taken for the cortex and the DG of control and NCT cKO mice. GFAP+ cells were shown in Brown. There were more GFAP+ cells in the DG and the cortex of NCT cKO mice than in controls. Scale bar = 100 μm. B, Cell‐counting results for GFAP+ cells in the DG. The total number of GFAP+ cells in the DG was counted for control and NCT cKO mice. There was a highly significant difference on the averaged number of GFAP+ cells between two groups (****, P < .001). C, Western blotting on GFAP. Protein levels of GFAP were significantly increased in NCT cKO mice as compared to controls (*, P < .05). D, Quantitative real‐time PCR results for GFAP. Relative mRNA levels of GFAP were significantly increased in NCT cKO mice (*, P < .05)
3.4. Neither neuron loss nor synaptic loss in young NCT cKO mice
To determine whether loss of NCT affected the total neuron number at 3 months, we performed IHC for NeuN, a marker for mature neurons. First, we did not find significant changes on NeuN immune‐reactivity in the cortex, the hippocampus and the cerebellum of NCT cKO mice (Figure 5A). The total number of NeuN+ cells was not different between NCT cKO and age‐matched littermate controls (data not shown). Second, Western analysis confirmed no reduction in total protein levels of NeuN in NCT cKO mice as compared to controls (Figure 5B). Third, to examine whether there was apoptotic neuron death at this age, we performed the terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labeling (TUNEL) experiment. TUNEL+ cells were not observed in NCT cKO mice and controls (Figure 5C). Overall, the above data suggest that there was no evident neuron loss in NCT cKO mice at 3 months.
Figure 5.
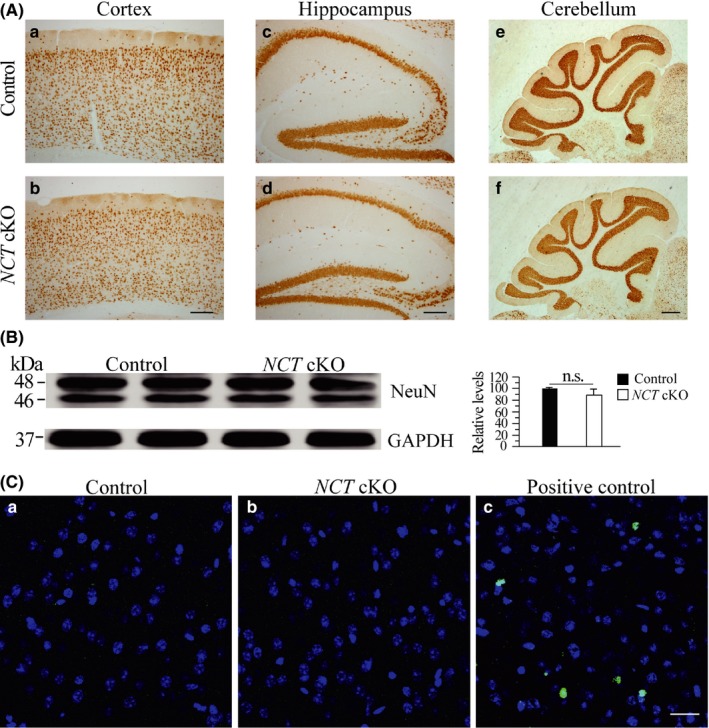
No neuron loss in NCT cKO mice at 3 mos. A, IHC on NeuN in control and NCT cKO mice. No detectable changes on NeuN immunoreactivity were observed in the cortex (b), the hippocampus (d) and the cerebellum (f) of NCT cKO mice. Scale bar = 200 μm. B, Western analysis of NeuN. There was no significant difference in NeuN protein levels between control and NCT cKO mice at 3 mos of age (control = 100 ± 1.7%, cKO = 88.6 ± 10.3%; n = 3‐4/group; n.s.: not significant, P > .2). C, TUNEL staining in NCT cKO mice at 3 mos. A positive control from a 4‐month‐old Dicer cKO mouse was used (c). TUNEL+ cells were shown in Green, and DAPI+ cells were in Blue. No TUNEL+ cells were observed in control (a) and NCT cKO mice (b). Scale bar = 25 μm
We next conducted IHC on synaptophysin (SVP38), a marker for presynaptic terminals. No changes on SVP38 immune‐reactivity were found in the cortex and the hippocampus of NCT cKO mice as compared to controls (Figure 6A), suggesting no significant synaptic loss. To analyze whether loss of NCT affected dendritic morphology, we performed IHC on MAP2, a marker for dendrite. There were no changes on MAP2 immunoreactivity in the cortex and the hippocampus of NCT cKO mice as compared to control animals (Figure 6B). Our Western analyses showed that there were no significant changes on protein levels of SVP38, MAP2, and PSD95 (postsynaptic density protein 95) between control and NCT cKO mice (Figure 6C), suggesting no evident synaptic or dendritic loss in young NCT cKO mice.
Figure 6.
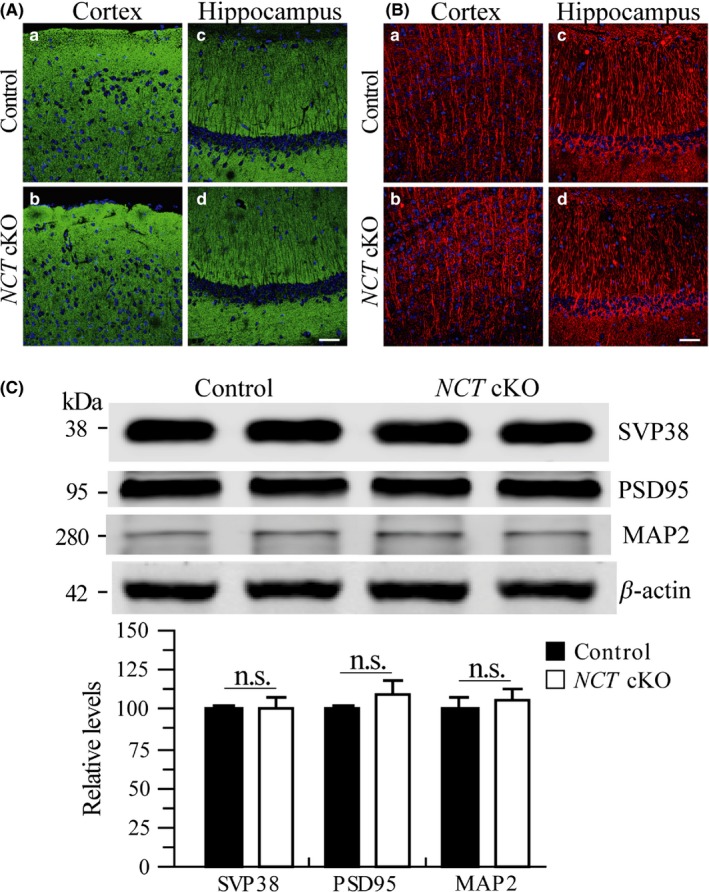
Neither synaptic loss nor dendritic degeneration in NCT cKO mice at 3 mos. A, Immunostaining of SVP38 in NCT cKO mice at 3 months. DAPI+ cells were in Blue, and SVP38 + cells were in Green. There was no detectable difference on SVP38 immunoreactivity in the cortex (a,b) and the hippocampus (c,d) of control and NCT cKO mice. Scale bar = 50 μm. B, Immunostaining of MAP2 in NCT cKO mice at 3 mos. DAPI+ cells were in Blue, and MAP2 + dendrites were in Red. There was no qualitative difference on MAP2 immunoreactivity in the cortex (a,b) and the hippocampus (c,d) of control and NCT cKO mice. Scale bar = 50 μm. C, Western analyses on SVP38, PSD95, and MAP2. There were no significant differences on relative protein levels between control and NCT cKO mice at 3 mos (SVP38: control = 100 ± 3.1%, cKO=101.4 ± 5.5%; PSD95: control = 100 ± 2.7%, cKO = 110.1 ± 7.3%; MAP2: control = 100 ± 8.4%, cKO = 106.8 ± 5.4%; n = 4/group; n.s.: not significant, P > .2)
4. DISCUSSION
Recent work has revealed a complex relationship between neurogenesis and AD.38 Interestingly, it remained largely uninvestigated whether neurogenesis is affected prior to neuronal death in neurodegenerative disease. To address this question, we used forebrain‐specific NCT cKO mice (Figure 1), as the latter exhibit age‐related neurodegeneration. The following novel observations have been obtained. First, cell proliferation is increased in NCT cKO mice (Figure 2). Second, more immature neurons are generated in NCT cKO mice than in controls (Figure 3). Third, astrocytosis is enhanced in NCT cKO mice as compared to controls (Figure 4). Finally, loss of NCT does not cause apoptosis, neuron loss or synaptic loss in mice at 3 months (Figures 5 and 6).
Previous studies on neurogenesis using APP mouse models of AD have yielded opposite findings. For example, some groups reported increased proliferation of NPCs but others showed impaired neurogenesis in different lines of APP Tg mice.38 As APP Tg models of AD overexpress different types/numbers of human mutated APP genes in the brain, this could cause the discrepancy.38 In this study, we employed a neurodegenerative model to study adult neurogenesis. The NCT cKO mouse used in this study16 differs from APP Tg mice in that it displays remarkable age‐related cortical neurodegeneration without Aβ deposition. Here, we have examined whether cell proliferation and adult neurogenesis are affected at an age when neuron death has not started. We have shown that both cell proliferation and the generation of immature neurons are increased in young NCT cKO mice. Interestingly, a previous study has demonstrated that cell proliferation, neuron differentiation, and the survival of newborn neurons are increased at both early and late stages of neurodegeneration in PS1/2 double cKO mice.34 These findings have thus raised a possibility that loss of γ‐secretase activity may enhance adult neurogenesis.
In this study, we have also shown that 3‐month‐old NCT cKO mice do not display neuronal death but exhibit abnormal astrocytosis. The first possibility to explain increased neurogenesis in NCT cKO mice is that reactive astrocytosis caused by loss of NCT may promote neurogenesis. The following evidence may provide some indirect support to this explanation. First, reactive astrocytes have been reported to prevent tissue loss after acute brain injury.39 Second, after brain injury, astrocytes release fibroblast growth factor 2 to stimulate cell proliferation in rat brain.40, 41 Third, it has been shown that sonic hedgehog (SHH) signaling is required for the maintenance of NPCs.42 After brain injury, reactive astrocytes produce SHH43 which stimulates NPCs to generate new neurons in vitro and in vivo.44
The second possibility is that NCT may negatively regulate neurogenesis via the Notch signaling. It is well known that Notch is required for the cell fate determination of NPCs.45 Previous studies have shown that deletion of PS1 in the whole body46 or conditional inactivation of PS1/2 in NPCs causes premature neuron differentiation.47 As inactivation of NCT or PS1/2 directly affects γ‐secretase activity, the expression of Notch targets is inhibited. Consistent with this, neurogenesis is enhanced either in young NCT cKO mice displaying no neuron death16 or in PS1/2 double cKO mice at early or late stage of neurodegeneration.34 Overall, our findings highlight a critical role of NCT‐dependent γ‐secretase activity in adult neurogenesis.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank Yingqian Xia for her contribution to this study. This work was supported by grants from the National Natural Science Foundation of China (31271123),the National Basic Research Program of Ministry of Science and Technology of China (2014CB942804), the Fundamental Research Funds for the Central Universities (14380012 and 14380322) and the Key Research and Development Program of Nanjing Municipal Health Bureau (ZKX16031).
Liu T‐T, Ye X‐L, Zhang J‐P, et al. Increased adult neurogenesis associated with reactive astrocytosis occurs prior to neuron loss in a mouse model of neurodegenerative disease. CNS Neurosci Ther. 2017;23:885–893. 10.1111/cns.12763
The first two authors contributed equally to this work.
Contributor Information
Gui‐Quan Chen, Email: chenguiquan@nju.edu.cn.
Zhen‐Yu Yin, Email: zhenyuyin68@163.com.
REFERENCES
- 1. Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223‐250. [DOI] [PubMed] [Google Scholar]
- 3. Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol. 2005;75:321‐341. [DOI] [PubMed] [Google Scholar]
- 4. Shors TJ, Miesegaes G, Beylin A, et al. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372‐376. [DOI] [PubMed] [Google Scholar]
- 5. Leuner B, Mendolia‐Loffredo S, Kozorovitskiy Y, et al. Learning enhances the survival of new neurons beyond the time when the hippocampus is required for memory. J Neurosci. 2004;24:7477‐7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. [DOI] [PubMed] [Google Scholar]
- 7. De Strooper B. Aph‐1, Pen‐2, and Nicastrin with Presenilin generate an active gamma‐Secretase complex. Neuron. 2003;38:9‐12. [DOI] [PubMed] [Google Scholar]
- 8. Kimberly WT, LaVoie MJ, Ostaszewski BL, et al. Gamma‐secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph‐1, and Pen‐2. Proc Natl Acad Sci USA. 2003;100:6382‐6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104:403‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Strooper B. Loss‐of‐function presenilin mutations in Alzheimer disease ‐ Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xia D, Watanabe H, Wu B, et al. Presenilin‐1 knockin mice reveal loss‐of‐function mechanism for familial Alzheimer's disease. Neuron. 2015;85:967‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kallhoff‐Munoz V, Hu L, Chen X, Pautler RG, Zheng H. Genetic dissection of gamma‐secretase‐dependent and‐independent functions of presenilin in regulating neuronal cell cycle and cell death. J Neurosci. 2008;28:11421‐11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP. Loss of presenilin function causes Alzheimer's disease‐like neurodegeneration in the mouse. J Neurosci Res. 2008;86:1615‐1625. [DOI] [PubMed] [Google Scholar]
- 14. Saura CA, Choi SY, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron. 2004;42:23‐36. [DOI] [PubMed] [Google Scholar]
- 15. Feng R, Wang H, Wang J, et al. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin‐1 and presenilin‐2. Proc Natl Acad Sci USA. 2004;101:8162‐8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hou JX, Cheng SS, Chen L, et al. Astroglial activation and tau hyperphosphorylation precede to neuron loss in a neurodegenerative mouse model. CNS Neurosci Ther. 2016;22:244‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sesele K, Thanopoulou K, Paouri E, et al. Conditional inactivation of nicastrin restricts amyloid deposition in an Alzheimer's disease mouse model. Aging Cell. 2013;12:1032‐1040. [DOI] [PubMed] [Google Scholar]
- 18. Tabuchi K, Chen G, Sudhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age‐related neurodegeneration. J Neurosci. 2009;29:7290‐7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:343‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular‐associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis. 2006;24:1‐14. [DOI] [PubMed] [Google Scholar]
- 21. Jin K, Galvan V, Xie L, et al. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF‐APPSw, Ind) mice. Proc Natl Acad Sci USA. 2004;101:13363‐13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mirochnic S, Wolf S, Staufenbiel M, Kempermann G. Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus. 2009;19:1008‐1018. [DOI] [PubMed] [Google Scholar]
- 23. Gan L, Qiao SH, Lan X, et al. Neurogenic responses to amyloid‐beta plaques in the brain of Alzheimer's disease‐like transgenic (pPDGF‐APP(SW, Ind)) mice. Neurobiol Dis. 2008;29:71‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lopez‐Toledano MA, Shelanski ML. Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J Alzheimers Dis. 2007;12:229‐240. [DOI] [PubMed] [Google Scholar]
- 25. Haughey NJ, Nath A, Chan SL, et al. Disruption of neurogenesis by amyloid beta‐peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509‐1524. [DOI] [PubMed] [Google Scholar]
- 26. Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid β‐peptide. NeuroMol Med. 2002;1:125‐135. [DOI] [PubMed] [Google Scholar]
- 27. Pan H, Wang D, Zhang X, et al. Amyloid β is not the major factor accounting for impaired adult hippocampal neurogenesis in mice overexpressing amyloid precursor protein. Stem Cell Rep. 2016;7:707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C. Alzheimer's‐type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neurosci. 2007;27:6771‐6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Donovan MH, Yazdani U, Norris RD, et al. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol. 2006;495:70‐83. [DOI] [PubMed] [Google Scholar]
- 30. Hamilton LK, Aumont A, Julien C, et al. Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer's disease. Eur J Neurosci. 2010;32:905‐920. [DOI] [PubMed] [Google Scholar]
- 31. Demars M, Hu YS, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer's disease in transgenic mice. J Neurosci Res. 2010;88:2103‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodriguez JJ, Jones VC, Tabuchi M, et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS ONE. 2008;3:e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wines‐Samuelson M, Schulte EC, Smith MJ, et al. Characterization of age‐dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS ONE. 2010;5:e10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Q, Nakajima A, Choi SH, et al. Adult neurogenesis is functionally associated with AD‐like neurodegeneration. Neurobiol Dis. 2008;29:316‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA. 2003;100:4855‐4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheng S, Zhang C, Xu C, et al. Age‐dependent neuron loss is associated with impaired adult neurogenesis in forebrain neuron‐specific Dicer conditional knockout mice. Int J Biochem Cell. 2014;57:186‐196. [DOI] [PubMed] [Google Scholar]
- 37. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis. 2007;45:593‐605. [DOI] [PubMed] [Google Scholar]
- 38. Lazarov O, Marr RA. Neurogenesis and Alzheimer's disease: at the crossroads. Exp Neurol. 2010;223:267‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427‐434. [DOI] [PubMed] [Google Scholar]
- 40. Mudo G, Bonomo A, Di Liberto V, et al. The FGF‐2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. J Neural Transm. 2009;116:995‐1005. [DOI] [PubMed] [Google Scholar]
- 41. Frinchi M, Bonomo A, Trovato‐Salinaro A, et al. Fibroblast growth factor‐2 and its receptor expression in proliferating precursor cells of the subventricular zone in the adult rat brain. Neurosci Lett. 2008;447:20‐25. [DOI] [PubMed] [Google Scholar]
- 42. Machold R, Hayashi S, Rutlin M, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937‐950. [DOI] [PubMed] [Google Scholar]
- 43. Amankulor NM, Hambardzumyan D, Pyonteck SM, et al. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury‐related inflammation. J Neurosci. 2009;29:10299‐10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiao J, Chen DF. Induction of neurogenesis in nonconventional neurogenic regions of the adult central nervous system by niche astrocyte‐produced signals. Stem Cells. 2008;26:1221‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471‐490. [DOI] [PubMed] [Google Scholar]
- 46. Handler M, Yang X, Shen J. Presenilin‐1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593‐2606. [DOI] [PubMed] [Google Scholar]
- 47. Kim WY, Shen J. Presenilins are required for maintenance of neural stem cells in the developing brain. Mol Neurodegener. 2008;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]