

Summary
Aims
Vascular dementia (VaD) is a heterogeneous brain disorder for which there are no effective approved pharmacological treatments available. We aimed to evaluate the effect of calmodulin inhibitor, DY‐9836, and its loaded nanodrug carrier system on cognitive impairment and gain a better understanding of the protective mechanisms in mice with bilateral carotid artery stenosis (BCAS).
Results
DY‐9836 (0.5 or 1 mg/kg) or DY‐9836 (0.25 mg/kg)‐encapsulated polysialic acid‐octadecylamine (PSA‐ODA) micelles (PSA‐ODA/DY) were given to BCAS mice for 4 weeks. Administration of DY‐9836 or PSA‐ODA/DY reduced escape latency in space exploration and working memory test compared with vehicle group. Vehicle‐treated mice showed reduced phospho‐CaMKII (Thr286/287) levels in the hippocampus, whereas partially restored by DY‐9836 (1 mg/kg) or PSA‐ODA/DY (0.25 mg/kg) treatment. In accordance with the pharmacological profile of DY‐9836 observed during behavioral studies, experimental molecular and biochemical markers induced by BCAS, such as protein tyrosine nitration, Nod‐like receptor protein 3 (NLRP3), caspase‐1, and interleukin‐1β, were reduced by DY‐9836 and PSA‐ODA/DY treatment.
Conclusions
These data disclose novel findings about the therapeutic potential of DY‐9836, and its encapsulated nanodrug delivery system significantly enhanced the cognitive function via inhibitory effect on nitrosative stress and NLRP3 signaling in VaD mice.
Keywords: calmodulin inhibitor, cognitive impairment, inflammasome, nitrosative stress, vascular dementia
1. BACKGROUND
Cerebrovascular disease is a common cause of vascular dementia (VaD), a heterogeneous brain disorder, which attributes to cognitive decline.1, 2, 3 VaD remains a devastating and socioeconomic burden with the worldwide occurrence of ~5% in aging population.4, 5, 6, 7 To date, there are no effective approved pharmacological treatments available.
The neurological diseases progressed through activation of calcium/calmodulin (Ca2+/CaM)‐dependent enzymes, including neuronal nitric oxide synthase (NOS), spectrin, and calcineurin.8, 9, 10, 11 Ca2+/CaM‐dependent protein kinase II (CaMKII) is widely distributed in the brain and a major mediator of physiological excitatory glutamate signals underlying long‐term potentiation induction and maintenance.12, 13, 14 The activation of CaMKII signaling contributes to the improvement in hypoperfusion‐induced learning and memory deficits.15, 16 No pharmacological therapy is available to specifically target the large subsets of VaD patients.
DY‐9836 (3‐{2‐[4N‐(2‐methyl‐3‐chlorophenyl)‐1N‐piperazinyl] ethyl}‐5, 6‐dimethoxy‐yl‐1H‐indazole oxalate) is one of the calmodulin inhibitors, derived from DY9760e.17 DY9760e has been reported to preserve the blood‐brain barrier integrity during cerebral edema caused by microsphere embolism.18 Polysialic acid (PSA) is a biodegradable natural carbohydrate polymer, found also on the surface of both bacterial and mammalian cells.19, 20 Currently, the patterns of PSA expression have also been observed in the brain with plasticity and in injured neural tissues.20 PSA with octadecylamine (ODA) ‐based micelles possess similar properties to those of PEG‐lipid nanoparticles and widely used as a drug carrier in brain disorders.21, 22 The complicated degenerative process of VaD suggests that new successful treatment strategies need to be multifaceted and combination designed. Based on the knowledge about DY‐9836 and polymer micelle drug delivery system, the DY‐9836 was further encapsulated in PSA‐ODA micelles, and their efficacy as well as mechanisms was tested in bilateral carotid artery stenosis (BCAS) mice model 23, 24 in this study.
We, here, demonstrated that either DY‐9836 (1 mg/kg) treatment or DY‐9836 (0.25 mg/kg)‐encapsulated PSA‐ODA micelle mitigates the hippocampus‐dependent spatial cognition dysfunction in a mouse model of VaD. Moreover, this pharmacological effect was associated with a marked decrease in peroxynitrite formation, which correlated with downregulation of NLRP3/caspase‐1/IL‐1β signaling in BCAS mice.
2. MATERIALS AND METHODS
2.1. Chemicals
All chemical reagents were obtained from Sigma‐Aldrich (St. Louis, MO, USA) except as otherwise stated. PSA (MW = 14.0 kDa) was supplied by Zhejiang Changxing Pharmaceutical Co., Ltd, Changxing, China. ODA was purchased from Fluka Chemical Co., Ronkonkoma, NY, USA. 1‐Ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide (EDC) and N‐hydroxysuccinimide (NHS) were supplied by Shanghai Medpep Co., Ltd, Shanghai, China. D2O and CDCl3 were sourced from J&K Scientific Co., Ltd, Beijing, China. DY‐9836 (DY) was gifted by Institute of Materia Medica, Zhejiang University.
2.2. Experimental animals
Ten‐ to 12‐week‐old male C57BL/6 mice weighing 22‐25 g were divided into 6 groups and housed under standard conditions with a 12/12 h light/dark cycle. Animals were acclimated to their environment for at least 1 week before initiating the experimental protocols. All animal use procedures were approved by the Committees at Zhejiang University and Leeds University for the Care and Use of Laboratory Animals. The animals were kept in cages for 45 days with food and water ad libitum.
2.3. Drug administration and BCAS mice model
Bilateral carotid artery stenosis in mice was prepared with minor modifications as described previously.21 Mice were anesthetized with chloral hydrate (400 mg/kg); both common carotid arteries were exposed through a midline cervical incision. A micro coil (Sawane Spring Co., Hamamatsu, Japan) with a 0.20 mm diameter was applied to the bilateral common carotid artery. The rectal temperature was monitored throughout the surgery, and the body temperature was maintained at 37 ± 0.5°C with a heating blanket. The sham‐operated mice involved bilateral exposure of the common carotid arteries but no stenosis procedure. Octadecylamine‐modified polysialic acid (PSA‐ODA) was synthesized as we reported previously.25 DY‐9836 was administered orally (p.o.) at 1 or 0.5 mg/kg, and PSA was administered intraperitoneally (i. p.) at a dose of 7 mg/kg, and PSA‐ODA/DY‐9836 was administered at a combination of PSA (7 mg/kg) and DY‐9836 (0.25 mg/kg), based on the protocol.25
2.4. Morris water maze test
The Morris water maze was performed as previously reported.24 It was carried out from day 36 to day 45 after the BCAS surgery. Place navigation test was carried out 4 consecutive days from day 37 to day 40 and consisted of 4 trials per day. Probe test was carried out on day 41, the platform was removed from the pool, and mice were tested on a 90‐seconds spatial probe trial. Working memory test was performed for 3 consecutive days and consisted of 5 trials per day. The data were processed and analyzed for each acquisition trial or probe trial as the escape latency (in seconds), the times of platform crossing (in numbers), and the time in the target quadrant (in seconds).
2.5. Immunohistochemical staining and analysis
After behavior task, animals were intracardially perfused with PBS followed by 4% PFA as previously described.26 Briefly, the brain sections were cut and incubated at room temperature in PBS with 0.01% Triton X‐100 for 30 minutes and followed by blocking with 3% bovine serum albumin for 1 hour. For immunolabeling, brain slices were probed with primary antibody overnight at 4°C. Antibodies included phospho‐CaMKII (Thr286/287; 1:200)27 and MAP‐2 (1:500; Millipore Corp, billerica, MA, USA), caspase‐1 (14F486, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and IL‐1β (H‐153, 1:50; Santa Cruz Biotechnology, USA) and nitrotyrosine (1:50; Millipore Corp). Nuclei were stained with DAPI (4, 6‐diamidino‐2‐phenylindole; Sigma‐Aldrich). After washing, the sections were incubated with Alexa Fluor 488‐conjugated anti‐rabbit IgG and Alexa Fluor 594‐conjugated anti‐mouse IgG (Invitrogen, Carlsbad, CA, USA). Signals were visualized using a Zeiss LSM 510 confocal microscope. The relative fluorescence intensity of immunostaining was quantified using Image J software (NIH, Bethesda, MD, USA).
2.6. Western blot analysis
After decapitation of mice at the end of behavior study, the brain was removed, and tissues from the hippocampal part were homogenized as previously reported.11 Briefly, the equal amounts of protein were applied to 10%‐13.5% acrylamide denaturing gels and shifted to the PVDF membrane (IPVH00010; Millipore Corp) for 1 hour at 250 mA. Membranes were blocked in 20 mmol/L Tris‐HCl (pH 7.4), 0.1% Tween 20 (TBS‐T), and 150 mmol/L NaCl containing 5% fat‐free milk for 1 hour and probed with specific antibodies against spectrin (Millipore Corp), NLRP3 (R&D Systems Inc., Minneapolis, MN, USA), calcineurin (Santa Cruz Biotechnology), and β‐actin (Sigma, St. Louis, MO, USA). The bands were visualized by enhanced chemiluminescence kit (1520301; Amersham Life Science, Beijing, China). Radiographic films were scanned for densitometry analysis and quantified using the NIH Image J program.
2.7. Statistical analysis
The data are presented as the mean ± SEM Statistical significance was determined using one‐way analysis of variance (ANOVA) followed by Tukey's test for multigroup comparisons. P < 0.05 indicated statistically significant differences.
3. RESULTS
3.1. DY‐9836 restores spatial learning and memory in BCAS mice
The bilateral common carotid artery stenosis mouse model is considered to some aspects of vascular cognitive impairment model.28 In this study, we first set out to investigate the pharmacological effect of DY‐9836 and its PSA‐ODA micelles on hypoperfusion‐induced VaD (Figure 1A). The behavioral memory paradigms were designed as reported previously.29, 30, 31 The changes in escape latency were probed to find the hidden platform produced during training trials in each group of mice (Figure 1B).
Figure 1.
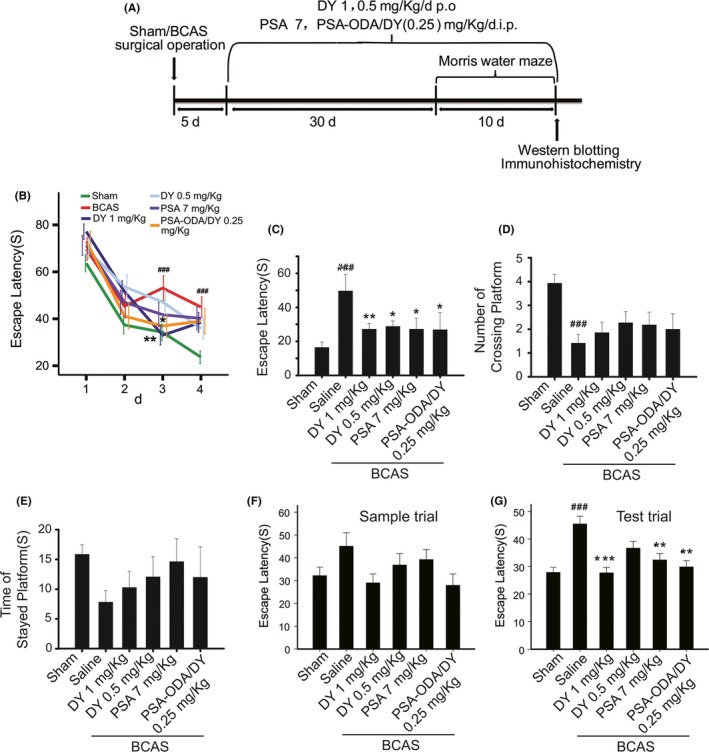
DY‐9836 enhances the hippocampal‐based spatial learning and memory in mice model of vascular dementia. (A) A schematic illustration of experimental plan. (B) Effects of DY‐9836 on changes in escape latency to find the hidden platform. (C‐E) Spatial probe trial and test to evaluate the effect of DY‐9836. DY‐9836, PSA and PSA‐ODA/DY significantly attenuated the escape latency. The number of crossing platform decreased in bilateral carotid artery stenosis (BCAS) mice model group, whereas time of staying at platform was significantly enhanced in all treated groups. (F, G) Short‐term memory test was performed, and the escape latency in sample trial and in the test trials was noted. Data represent mean ± SEM (n = 10‐12 per group). ### P< 0.001 vs sham‐operated group and *P < 0.05; **P < 0.01; ***P < 0.01 vs vehicle‐treated group. PSA, polysialic acid; ODA, octadecylamine
We found that performance of escape latency in the vehicle‐treated BCAS mice group was significantly impaired as compared to sham‐operated mice (P < 0.001). Notably, DY‐9836 attenuated this learning impairment was observed in mice after repeating daily administration at the doses of 1 mg/kg (Figure 1B). Moreover, the similar improvement in escape latency observed in DY‐9836 (0.25 mg/kg)‐encapsulated PSA‐ODA micelle group (Figure 1B).
Further, we carried out the 90‐seconds spatial probe trial at day 5 following the training trial (Figure 1C‐E). The vehicle‐treated mice searched the target platform for comparatively more time than the Sham‐operated group (P < 0.001). In the DY‐9836 (1 or 0.5 mg/kg), PSA (7 mg/kg), or PSA‐ODA/DY‐treated mice, a significant improvement in cognitive dysfunction observed, as revealed by significantly reduced escape latency compared with vehicle‐treated mice (Figure 1C). Here, the number of platform crossing on the probe trial day was significantly reduced in vehicle‐treated mice compared with sham‐operated controls and was not affected by chronic treatment with the drugs (Figure 1D).
There were no significant differences among groups during spatial probe trial (Figure 1E). The escape latency in the first trial (sample trial) and in the second to fifth trials (test trials) of the short‐term memory test is shown in Figure 1F,G. There was no difference in the escape latency in the sample trial (Figure 1F). DY‐9836 (1 mg/kg), PSA (7 mg/kg), or PSA‐ODA/DY treatment almost completely normalized the learning profile in BCAS mice (Figure 1G).
3.2. DY‐9836 prevents the dephosphorylation of CaMKII in BCAS mice
Ca2+/CaM‐dependent protein kinase II is localized subcellular to the dendrites and to the postsynaptic densities of excitatory synapses and considered as an important mediator of learning and memory.32 Here, immunofluorescence staining was performed to further confirm the effect of DY‐9836 on phospho‐CaMKII (Thr286) expression in the hippocampal region in BCAS mice. As shown in Figure 2, there was a significant decrease in the intensity of fluorescence for phospho‐CaMKII (Thr286) in CA1 pyramidal neurons of the hippocampus (Figure 2A and 2B). Similar decline in phospho‐CaMKII (Thr286) expression was also observed in hippocampal CA3 regions (Fig. S1). By contrast, DY‐9836 (1 mg/kg) restored the decrease in immunostaining for phospho‐CaMKII (Thr286) in hippocampal pyramidal neurons (Figure 2). Similar data obtained in the protocol of chronic administration of the DY‐9836 (1 mg/kg), administration of PSA‐ODA/DY to BCAS mice produced the same effect on phospho‐CaMKII (Thr286/287) (Figure 2). However, PSA (7 mg/kg) chronic treatment did not significantly affect the dephosphorylation of CaMKII in BCAS mice (Figure 2). In this study, we further tested the hypothesis that hypoperfusion induces calpain and calcineurin activation. Unexpectedly, the data demonstrated that no breakdown products of spectrin (150 or 145 kDa) or calcineurin (48 or 45 kDa) observed (Fig. S2).
Figure 2.

DY‐9836 prevents the dephosphorylation of CaMKII in bilateral carotid artery stenosis (BCAS) mice model. Immunohistochemical localization of phospho‐CaMKII and MAP‐2 was examined in the hippocampal CA1 (A). Markedly decreased phospho‐CaMKII (green) and MAP‐2 (red) staining were observed by confocal microscopy in the hippocampus of BCAS mice model. (B) Quantification of phospho‐CaMKII immunofluorescence expressed as mean intensity. DY‐9836, PSA, and PSA‐ODA/DY treatment restored the decrease in immunostaining for phospho‐CaMKII (Thr286/287) in hippocampal pyramidal neurons of BCAS mice. DAPI counterstaining indicates nuclear localization (blue). Data are representative of 3 independent experiments. *P < 0.05, vs sham operated mice. # P < 0.05, ## P < 0.01, ### P < 0.001, vs saline‐treated BCAS operated mice. Scale bar = 50 μm. PSA, polysialic acid; ODA, octadecylamine
3.3. Effect of DY‐9836 on NLRP3 inflammasome signaling in BCAS mice
We therefore analyzed some representative biochemical and molecular markers with the aim to support the above‐reported behavioral observations. Accumulating evidence showed that activation of NLRP3 regulates by affecting inflammatory cell infiltration in various diseases.33 Here, a dramatic increase in NLRP3 in the hippocampus of BCAS mice model was observed, whereas DY and PSA‐ODA/DY (0.25 mg/kg) treatment ameliorated BCAS‐induced neuronal inflammation, as demonstrated by the decrease in the NLRP3 expressions (Figure 3). These results showed that DY‐9836 administration mitigates the hippocampal NLRP3 activation in response to chronic cerebral hypoperfusion mediated by BCAS.
Figure 3.
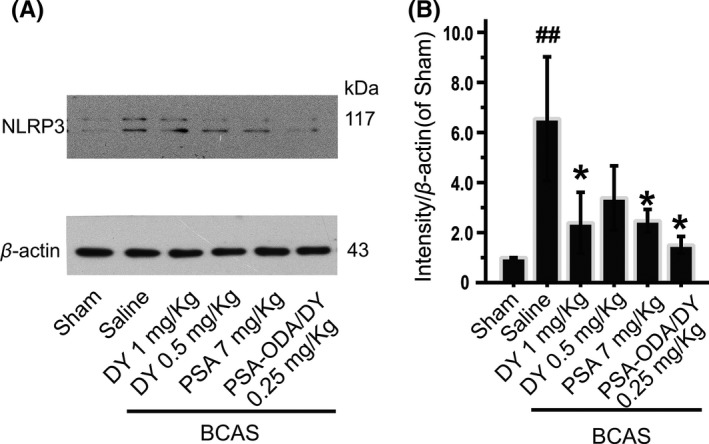
DY‐9836 inhibits the NLRP3 inflammasome activation in mice model of bilateral carotid artery stenosis (BCAS). (A) Temporal changes in expression of hippocampal NLRP3 in mice model of BCAS. Representative Western blots of NLRP3 in the hippocampal region in BCAS mice. Levels of β‐actin were used as the loading control. (B) Quantitative analysis of relative NLRP3 in BCAS mice was performed by intensity of expression with respect to sham‐operated group. Data are expressed as values of control animals (mean ± SEM, n = 5). ## P < 0.01 vs sham‐operated group and *P < 0.05 vs vehicle‐treated group
Caspase‐1 is an enzyme that proteolytically cleaves other proteins and involved in inflammasome formation and activation.34 To extend our observations to NLRP3 inflammasome signaling, we tested activity of caspase‐1 in BCAS mice with or without pharmacological intervention. Immunofluorescence of caspase‐1 was absent in hippocampal neurons of the sham‐operated group, whereas the significant elevation of caspase‐1 in BCAS mice was observed (Figure 4A,B). DY‐9836 (1 mg/kg) and PSA‐ODA/DY (0.25 mg/kg) treatment blocked the BCAS‐induced caspase‐1 immunoreactivity in the hippocampal CA1 regions in the brain (Figure 4A,B). A similar result was also observed in hippocampal CA3 regions (Fig. S3).
Figure 4.

DY‐9836 attenuates the caspase‐1 activation in bilateral carotid artery stenosis (BCAS) mice model. Representative image of hippocampal CA1 (A) pyramidal neurons stained with caspase‐1. (B) Quantification of caspase‐1 immunofluorescence expressed as mean intensity. Increased immunostaining of caspase‐1 was observed by confocal microscopy in the hippocampus of BCAS mice that was reversed by treatment of DY‐9836 (1 mg/kg) and PSA‐ODA/DY (DY, 0.25 mg/kg). DAPI counterstaining indicates nuclear localization (blue). Data are representative of 3 independent experiments. **P < 0.01, vs sham operated mice. ## P < 0.01, vs saline‐treated BCAS operated mice. Scale bar = 20 μm. PSA, polysialic acid; ODA, octadecylamine
3.4. DY‐9836 inhibits the nitrosative stress and IL‐1β activation in mice model of BCAS
We further tested the pathological role of protein tyrosine nitration on activation of NLRP3 inflammasome signaling. Interleukin‐1 contributes to the brain inflammatory disease.35 Unlike a sham‐operated group, our immunofluorescence data showed that nitrotyrosine expressed in the hippocampus neurons of vehicle group was significantly increased. In parallel with the observed nitrotyrosine formation, the expression of IL‐1β was obviously enhanced in the hippocampus of vehicle group compared with sham group. Furthermore, DY9836 (1 mg/kg) and PSA‐ODA/DY (0.25 mg/kg) treatment attenuated BCAS‐induced nitrotyrosine and IL‐1β immunoreactivity in the hippocampus (Figure 5A‐C). Therefore, we conclude that BCAS‐mediated nitrosative stress induces protein nitrotyrosine and likely contributes to proinflammatory cytokine IL‐1β activation that was corrected by DY‐9836 treatment. Overall, this set of data adds further support to the neurovascular protective effect of DY‐9836 observed during behavioral studies.
Figure 5.
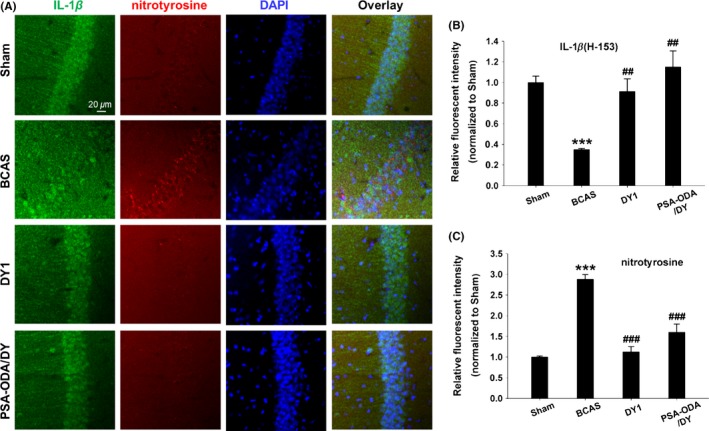
Nitrosative stress and proinflammatory IL‐1β activation in bilateral carotid artery stenosis (BCAS) mice are inhibited by DY‐9836 treatment. (A) Fluorescent immunohistochemical staining of IL‐1β (H‐153) and nitrotyrosine in the hippocampal CA1 region of BCAS mice. Antinitrotyrosine and IL‐1β (H‐153) staining were performed after BCAS. IL‐1β expression was disturbed with increased in nitrotyrosine‐positive staining in hippocampus of BCAS mice model. BCAS‐induced nitrotyrosine formation and IL‐1β expression colocalized in the CA1 region of hippocampus. Quantification of IL‐1β (H‐153) (B) and nitrotyrosine (C) immunofluorescence expressed as mean intensity. DY‐9836 (1 mg/kg) and PSA‐ODA/DY (DY, 0.25 mg/kg) significantly reduced nitrotyrosine immune reactivity and IL‐1β activation as compared to vehicle‐treated mice group. DAPI counterstaining indicates nuclear localization (blue). Data are representative of 3 independent experiments. ***P< 0.001, vs sham operated mice. ## P < 0.01, ### P < 0.001 vs saline‐treated BCAS operated mice. Scale bar = 20 μm. PSA, polysialic acid; ODA, octadecylamine
4. DISCUSSION
Understanding the mechanisms of cerebral lesions induced by hypoperfusion should result in appropriate preventative and therapeutic prospects. Brain hypoperfusion consequently results in loss of learning and memory.28, 36 In the present study, we demonstrated that DY‐9836 and its nanocarrier system elicit therapeutic effect to improve cognitive dysfunction in BCAS mouse model. The inhibitory effect on nitrosative stress and NLRP3 inflammasome signaling is a previously unreported neurovascular protective mechanism of DY‐9836 during pathological process of VaD.
Cognitive dysfunction with a decline in working memory correlates to the hippocampal damage was observed in mice after BCAS.23, 28 Here, DY‐9836 and its nanocarrier system effectively enhance learning and working memory in BCAS mice model as well as improve cognition. We recently reported that PSA‐ODA micelle could encapsulate hydrophobic drug DY‐9836 effectively and achieve a sustained release of the drug. Moreover, PSA‐ODA micelle exhibited excellent penetrability to BBB in vitro as well as in vivo.25 Consistently, we here confirmed that DY‐9836 reversed the autophosphorylation of CaMKII at threonine 286 in CA1 and CA3 regions of the hippocampus. Importantly, DY‐9836 (0.25 mg/kg)‐encapsulated PSA‐ODA micelle is resulted in decreased neurological dysfunction after BCAS, with the reduced dosages vs regular DY‐9836 (1 mg/kg).
Although we cannot rule out the precise mechanism of PSA on restoring the cognitive function in BCAS mice, it has been found to be associated with the cell migration, neural regeneration, and maintenance.36, 37 Consistently, we recently reported that PSA‐ODA micelle delivery system partially inhibited the dephosphorylation of CaMKII and synapsin I and increased the number of mature neurons in the hippocampus, which correlated with significant improvements in cognitive dysfunction in VaD mice.25 Based on this comparison, we speculate that DY‐9836 and PSA work synergistically to attenuate the BCAS‐induced cognitive dysfunction.
The role of inflammasomes in neurodegenerative diseases has been recently described.38, 39, 40 In the present study, the increased expression of NLRP3 in the hippocampus suggests that upregulation of NLRP3 associated with chronic hypoperfusion‐mediated inflammation. Here, DY‐9836 and PSA‐ODA/DY attenuated the expressions of NLRP3. Note that a significant activation of NLRP3 inflammasome, which initiates the proteolytic activity of caspase‐1, required for the dispensation and secretion of the inflammatory cytokines IL‐1β.41, 42 We showed that activation of IL‐1β caused by NLRP3/caspase‐1 mediating cell injury occurred in tyrosine nitration‐positive pyramidal neurons following BCAS. Consistently, DY‐9836 preferentially inhibits protein tyrosine nitration and caspase‐1/IL‐1β signaling. Indeed, it was reported that peroxynitrite play a critical role in triggering NLRP3 inflammasome activation.43, 44 Together with our observation, dual‐target modulators (low dose DY‐9836 + PSA) might particularly potent in inhibiting protein tyrosine nitration and subsequently lead to decreased NLRP3/caspase‐1/IL‐1β signaling, eventually attenuate the hypoperfusion‐induced CaMKII dephosphorylation and cognitive dysfunction.
With respect to the role of nitric oxide (NO) in synaptic plasticity during brain injury, it is produced by 3 isoforms of NOS which can also bind to the Ca2+/CaM.45, 46 Nevertheless, the further approach to determine the relative roles of nitrosative stress on activation of inflammasome signaling needs to be explored in future. The disturbance expression of NOSs in hippocampus might contribute the peroxynitrite formation and subsequently induce NLRP3/caspase‐1/IL‐1β signaling in the present context, whereas DY‐9836 and PSA‐ODA/DY can inhibit these detrimental cascades.
5. CONCLUSIONS
We hypothesize that BCAS‐mediated chronic cerebral hypoperfusion induces Ca2+/CaM‐dependent nitrosative stress, and therefore, the causes of protein nitrotyrosine and NLRP3/caspase‐1/IL‐1β activation ultimately result in progression of VaD, whereas DY‐9836 in novel nanodrug carrier system improves the cognitive deficit by inhibiting the nitrosative stress and inflammasome activation (Figure 6). In particular, PSA/ODA micelles encapsulated reduced dosage of candidate drugs, which is expected to achieve a breakthrough to therapy neurovascular diseases.
Figure 6.
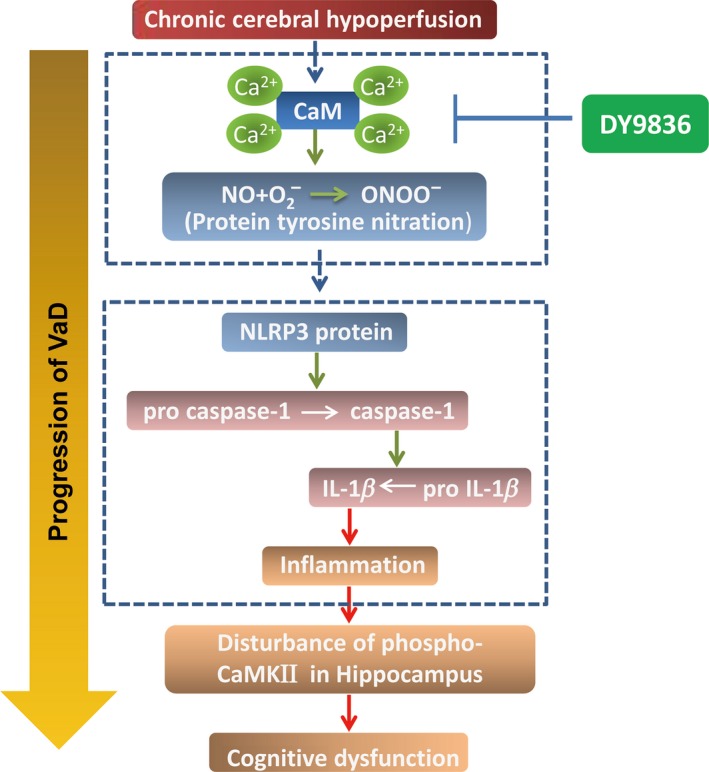
Schematic illustration of the mechanisms by which DY‐9836 inhibits NLRP3 activation and cognitive dysfunction in bilateral carotid artery stenosis mice model
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundations of China (81573411, 81260483), Zhejiang Provincial Natural Science Foundation of China (Z16H310003), and Talents Program of Ningxia Medical University (XT2015009).
Wang R, Yin Y‐X, Mahmood Q, et al. Calmodulin inhibitor ameliorates cognitive dysfunction via inhibiting nitrosative stress and NLRP3 signaling in mice with bilateral carotid artery stenosis. CNS Neurosci Ther. 2017;23:818–826. 10.1111/cns.12726
The first two authors contributed equally to this work.
REFERENCES
- 1. O'Brien JT, Erkinjuntti T, Reisberg B, et al. Vascular cognitive impairment. Lancet Neurol. 2003;2:89‐98. [DOI] [PubMed] [Google Scholar]
- 2. Hachinski V, Iadecola C, Petersen RC, et al. National Institute of Neurological Disorders and Stroke‐Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220‐2241. [DOI] [PubMed] [Google Scholar]
- 3. Moorhouse P, Rockwood K. Vascular cognitive impairment: current concepts and clinical developments. Lancet Neurol. 2008;7:246‐255. [DOI] [PubMed] [Google Scholar]
- 4. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population‐based cohorts. Neurologic diseases in the Elderly Research Group. Neurology. 2000;54:S4‐S9. [PubMed] [Google Scholar]
- 5. Erkinjuntti T, Gauthier S. The concept of vascular cognitive impairment. Front Neurol Neurosci. 2009;24:79‐85. [DOI] [PubMed] [Google Scholar]
- 6. Hill J, Fillit H, Shah SN, Del VM, Futterman R. Patterns of healthcare utilization and costs for vascular dementia in a community‐dwelling population. J Alzheimers Dis. 2005;8:43‐50. [DOI] [PubMed] [Google Scholar]
- 7. Nys GM, van Zandvoort MJ, van der Worp HB, et al. Early cognitive impairment predicts long‐term depressive symptoms and quality of life after stroke. J Neurol Sci. 2006;247:149‐156. [DOI] [PubMed] [Google Scholar]
- 8. Elgersma Y, Fedorov NB, Ikonen S, et al. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493‐505. [DOI] [PubMed] [Google Scholar]
- 9. Sun LH, Ban T, Liu CD, et al. Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA‐195 down‐regulation. J Neurochem. 2015;134:1139‐1151. [DOI] [PubMed] [Google Scholar]
- 10. Shioda N, Han F, Moriguchi S, Fukunaga K. Constitutively active calcineurin mediates delayed neuronal death through Fas‐ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem. 2007;102:1506‐1517. [DOI] [PubMed] [Google Scholar]
- 11. Tao RR, Wang H, Hong LJ, et al. Nitrosative stress induces peroxiredoxin 1 ubiquitination during ischemic insult via E6AP activation in endothelial cells both in vitro and in vivo. Antioxid Redox Signal. 2014;21:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU. CaMKII in cerebral ischemia. Acta Pharmacol Sin. 2011;32:861‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moriguchi S, Han F, Shioda N, et al. Nefiracetam activation of CaM kinase II and protein kinase C mediated by NMDA and metabotropic glutamate receptors in olfactory bulbectomized mice. J Neurochem. 2009;110:170‐181. [DOI] [PubMed] [Google Scholar]
- 14. Fukunaga K, Miyamoto E. A working model of CaM kinase II activity in hippocampal long‐term potentiation and memory. Neurosci Res. 2000;38:3‐17. [DOI] [PubMed] [Google Scholar]
- 15. Yamamoto Y, Shioda N, Han F, et al. Nobiletin improves brain ischemia‐induced learning and memory deficits through stimulation of CaMKII and CREB phosphorylation. Brain Res. 2009;1295:218‐229. [DOI] [PubMed] [Google Scholar]
- 16. Min D, Mao X, Wu K, et al. Donepezil attenuates hippocampal neuronal damage and cognitive deficits after global cerebral ischemia in gerbils. Neurosci Lett. 2012;510:29‐33. [DOI] [PubMed] [Google Scholar]
- 17. Lu YM, Han F, Shioda N, et al. Phenylephrine‐induced cardiomyocyte injury is triggered by superoxide generation through uncoupled endothelial nitric‐oxide synthase and ameliorated by 3‐[2‐[4‐(3‐chloro‐2‐methylphenyl)‐1‐piperazinyl]ethyl]‐5,6‐dimethoxyindazole (DY‐9836), a novel calmodulin antagonist. Mol Pharmacol. 2009;75:101‐112. [DOI] [PubMed] [Google Scholar]
- 18. Han F, Shirasaki Y, Fukunaga K. Microsphere embolism‐induced endothelial nitric oxide synthase expression mediates disruption of the blood‐brain barrier in rat brain. J Neurochem. 2006;99:97‐106. [DOI] [PubMed] [Google Scholar]
- 19. Bader RA, Silvers AL, Zhang N. Polysialic acid‐based micelles for encapsulation of hydrophobic drugs. Biomacromol. 2011;12:314‐320. [DOI] [PubMed] [Google Scholar]
- 20. Aliabadi HM, Elhasi S, Mahmud A, Gulamhusein R, Mahdipoor P, Lavasanifar A. Encapsulation of hydrophobic drugs in polymeric micelles through co‐solvent evaporation: the effect of solvent composition on micellar properties and drug loading. Int J Pharm. 2007;329:158‐165. [DOI] [PubMed] [Google Scholar]
- 21. Nishio K, Ihara M, Yamasaki N, et al. A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke. 2010;41:1278‐1284. [DOI] [PubMed] [Google Scholar]
- 22. Yamada K, Tanaka T, Mamiya T, Shiotani T, Kameyama T, Nabeshima T. Improvement by nefiracetam of beta‐amyloid‐(1‐42)‐induced learning and memory impairments in rats. Br J Pharmacol. 1999;126:235‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Popa‐Wagner A, Buga AM, Popescu B, Muresanu D. Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm (Vienna). 2015;122(Suppl 1):S47‐S54. [DOI] [PubMed] [Google Scholar]
- 24. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598‐2603. [DOI] [PubMed] [Google Scholar]
- 25. Wang XJ, Gao YP, Lu NN, et al. Endogenous polysialic acid based micelles for calmodulin antagonist delivery against vascular dementia. ACS Appl Mater Interfaces. 2016;8:35045‐35058. [DOI] [PubMed] [Google Scholar]
- 26. Wang H, Hong LJ, Huang JY, et al. P2RX7 sensitizes Mac‐1/ICAM‐1‐dependent leukocyte‐endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015;25:674‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tian Y, Yabuki Y, Moriguchi S, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res. 2014;56:1‐11. [DOI] [PubMed] [Google Scholar]
- 28. Shibata M, Yamasaki N, Miyakawa T, et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38:2826‐2832. [DOI] [PubMed] [Google Scholar]
- 29. D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60‐90. [DOI] [PubMed] [Google Scholar]
- 30. Koopmans G, Blokland A, van Nieuwenhuijzen P, Prickaerts J. Assessment of spatial learning abilities of mice in a new circular maze. Physiol Behav. 2003;79:683‐693. [DOI] [PubMed] [Google Scholar]
- 31. Paul CM, Magda G, Abel S. Spatial memory: theoretical basis and comparative review on experimental methods in rodents. Behav Brain Res. 2009;203:151‐164. [DOI] [PubMed] [Google Scholar]
- 32. Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567‐578. [DOI] [PubMed] [Google Scholar]
- 33. Mizushina Y, Shirasuna K, Usui F, et al. NLRP3 protein deficiency exacerbates hyperoxia‐induced lethality through Stat3 protein signaling independent of interleukin‐1beta. J Biol Chem. 2015;290:5065‐5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferreira I, Liberal J, Martins J, Silva A, Neves BM, Cruz MT. Inflammasome in dendritic cells immunobiology: implications to diseases and therapeutic strategies. Curr Drug Targets. 2017;18:1003‐1018. [DOI] [PubMed] [Google Scholar]
- 35. Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin‐1 production and inflammasome activation. Immunity. 2011;34:213‐223. [DOI] [PubMed] [Google Scholar]
- 36. El Maarouf A, Petridis AK, Rutishauser U. Use of polysialic acid in repair of the central nervous system. Proc Natl Acad Sci U S A. 2006;103:16989‐16994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colley KJ, Kitajima K, Sato C. Polysialic acid: biosynthesis, novel functions and applications. Crit Rev Biochem Mol Biol. 2014;49:498‐532. [DOI] [PubMed] [Google Scholar]
- 38. Du RH, Tan J, Yan N, et al. Kir6.2 knockout aggravates lipopolysaccharide‐induced mouse liver injury via enhancing NLRP3 inflammasome activation. J Gastroenterol. 2014;49:727‐736. [DOI] [PubMed] [Google Scholar]
- 39. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao AP, Dong YF, Liu W, et al. Nicorandil inhibits inflammasome activation and Toll‐like receptor‐4 signal transduction to protect against oxygen‐glucose deprivation‐induced inflammation in BV‐2 cells. CNS Neurosci Ther. 2014;20:147‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109:11282‐11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Franchi L, Eigenbrod T, Munoz‐Planillo R, Nunez G. The inflammasome: a caspase‐1‐activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feng L, Chen Y, Ding R, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015;12:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bellezza I, Grottelli S, Costanzi E, et al. Peroxynitrite activates the NLRP3 inflammasome cascade in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2017. [Epub ahead of print]. 10.1007/s12035-017-0502-x [DOI] [PubMed] [Google Scholar]
- 45. Pozo D, Reiter RJ, Calvo JR, Guerrero JM. Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J Cell Biochem. 1997;65:430‐442. [DOI] [PubMed] [Google Scholar]
- 46. Han F, Tao RR, Zhang GS, et al. Melatonin ameliorates ischemic‐like injury‐evoked nitrosative stress: involvement of HtrA2/PED pathways in endothelial cells. J Pineal Res. 2011;50:281‐291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials