

Neurodegenerative diseases including AD (Alzheimer's disease) display several common neuropathological changes such as neuroinflammation, tau pathology, and neuron loss 1, 2. However, it remained unknown which types of pathology occur as early events during the progression of the disease. This is an important question to be investigated, as it is believed that prevention of early pathological changes is pivotal to developing effective therapies for neurodegenerative diseases. Presenilins, NCT (nicastrin), PEN2 (presenilin enhancer 2), and Aph1 (anterior pharynx defective 1) are four essential subunits of the γ‐secretase complex. It is well known that presenilin mutations are the major cause of familial AD 3, 4, 5. Recent evidence has strongly suggested that there is loss of presenilin function mechanism in the pathogenesis of AD 5, 6. Consistent with this notion, a number of recently published γ‐secretase subunit‐based mouse models were reported to display AD‐like neurodegeneration in an age‐dependent manner 6, 7, 8, 9, 10, 11. However, as neuron loss takes place in the above models at very young ages, for example, 2–3 months, it is difficult to dissect out early pathological changes.
Findings
In this study, we generated a new line of NCT cKO mice by crossing NCT f/f with the T29 CaMKIIα‐Cre transgenics (Tg), in which Cre recombinase starts to express in excitatory neurons of the forebrain at 1.5–2 months 12, 13. Our biochemical analysis confirmed significantly reduced NCT protein levels in NCT cKO mice across ages (Figure 1A. Ps < 0.01), for example, there was about 30% of reduction on NCT. Whereas levels of full‐length APP (APP‐FL) in NCT cKO mice were not different, as compared to controls (Figure 1A. Ps > 0.1), those for APP C‐terminal fragment (APP‐CTF) in NCT cKOs were massively increased (Figure 1A), confirming decreased γ‐secretase activity.
Figure 1.
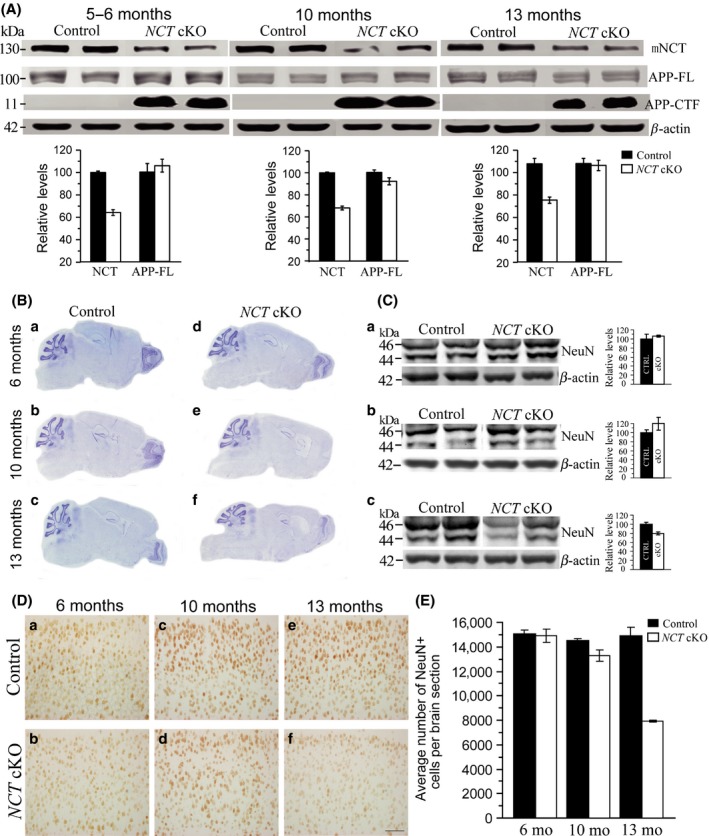
Age‐dependent loss of mature neurons in forebrain‐specific NCT cKO mice. (A) Biochemical analyses on NCT, APP‐FL, and APP‐CTF in NCT cKO mice across ages (n = 3–4/group). Western blotting confirmed significantly decreased levels of NCT in three cKO groups (5–6 months: control = 100 ± 1.2%, cKO = 64.3 ± 2.6%, P = 0.00009, two‐tailed Student t‐test; 10 months: control = 100 ± 0.6%, cKO = 68.0 ± 1.7%, P = 0.003; 13 months: control = 100 ± 4.4%, cKO = 69.9 ± 4.2%, P = 0.007). Western blotting on APP‐FL showed unchanged levels in NCT cKO groups at 5–6 (P = 0.56), 10 (P = 0.11), or 13 (P = 0.81) months of age. Western results for APP‐CTF indicated massive accumulation in NCT cKO mice aged at 5–6, 10, or 13 months. (B) Nissl staining for NCT cKO mice using brain sections aged at 6, 10, and 13 months. The cortex morphology and the cortex size were normal in NCT cKOs at 6 months (a,d). The cortex became smaller in NCT cKOs at 10 months (b,e). The cortex size was further reduced, and the lateral ventricle became bigger in NCT cKOs at 13 months (c,f). (C) Western analysis on NeuN. There were no significant differences on protein levels of NeuN in NCT cKO brains at 5–6 (a: P = 0.55) or 10 (b: P = 0.10) months of age. NeuN levels were significantly decreased in NCT cKO mice at 13 months, as compared to age‐matched littermate controls (c: P = 0.01). (D) NeuN immunostaining. There was no difference on immuno‐reactivity of NeuN in NCT cKO brains at 6 (a–b) and 10 (c–d) months. Immuno‐reactivity of NeuN in NCT cKO brains was decreased at 13 months (e–f). Scale bar=100 μm. (E) Quantification data on the average number of cortical NeuN+ cells per brain section. There was no significant difference between NCT cKOs and controls at 6 (P = 0.82) or 10 (P = 0.13) months. However, the average number of cortical NeuN+ cells was significantly decreased in NCT cKO mice at 13 months (P = 0.009).
Nissl staining showed no detectable change in brain morphology of NCT cKO mice at 6 months (Figure 1B–a,d). In contrast, the cortex size of NCT cKO mice became significantly smaller than that of age‐matched littermate controls at 10 (Figure 1B–b,e) and 13 (Figure 1B–c,f) months. To determine at which age mutant mice began to exhibit evident neuron loss, Western blotting and IHC (immunohistochemistry) using NeuN, a marker for mature neurons, were conducted. Relative protein levels of NeuN were significantly decreased in the cortex of NCT cKO mice at 13 (Figure 1C–c) months but not at 5–6 (Figure 1C–a) or 10 months (Figure 1C–b), suggesting a possibility that there was a loss of the total number of mature neurons. Consistent with biochemical results, immuno‐reactivity of NeuN in the cortex of NCT cKO mice was unchanged at 6 (Figure 1D–a,b) or 10 months (Figure 1D–c,d), but was reduced at 13 months (Figure 1D–e,f). Using a stereological method, we counted the average number of NeuN‐positive (+) cells across a number of brain sections. We found that the average number of cortical NeuN+ cells per brain section was significantly decreased in NCT cKO mice at 13 months but not at 5–6 or 10 months (Figure 1E).
Neuroinflammation is often associated with neurodegeneration. To determine at which age inflammatory responses would appear in NCT cKO mice, GFAP IHC was conducted. Highly increased immuno‐reactivity for GFAP was readily observed in the cortex (Figure 2A–a,d) and the hippocampus (data not shown) of NCT cKO mice at 6 months. There was massive elevation of GFAP immuno‐reactivity in NCT cKOs at 10 (Figure 2A–b,e) or 13 months (Figure 2A–c,f), suggesting severe astroglial activation. Western analysis confirmed increased levels of GFAP in NCT cKOs at each age tested (Figure 2B).
Figure 2.
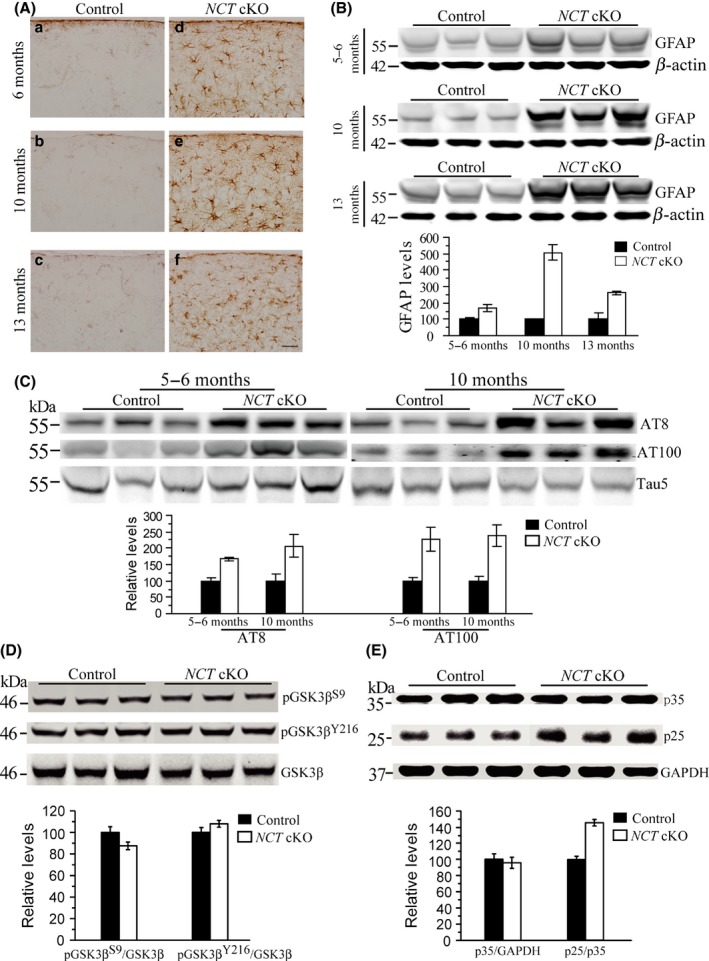
Astroglial activation and tau hyperphosphorylation in NCT cKO mice. (A) Immunostaining of GFAP. GFAP+ cells were intensively seen in the cortex of NCT cKO mice at 6 (a,d), 10 (b,e), and 13 (c,f) months of age, but were hardly detected in age‐matched controls. Scale bar = 40 μm. (B) Western analysis on GFAP. Protein levels of GFAP were significantly increased in NCT cKO mice at 5–6 (P = 0.008, two‐tailed Student t‐test), 10 (P = 0.00002), and 13 months (P = 0.00001), as compared to age‐matched littermate controls. (C) Western blotting on p‐tau using antibodies of AT8 and AT100. Levels of p‐tau were significantly increased in the cortex of NCT cKO mice at 5–6 (For AT8: P = 0.004; For AT100: P = 0.04) and 10 months (For AT8: P = 0.031; For AT100: P = 0.02). (D) Western analyses on GSK3β and p‐GSK3β. Relative levels of p‐GSK3β S9 (P = 0.7) and p‐GSK3β Y216 (P = 0.4) were not decreased in the cortex of NCT cKO mice. (E) Western analyses on p25 and p35. Levels of p25 but not p35 were increased in NCT cKO mice (P = 0.0006).
Tau hyperphosphorylation is also believed to be a driving force for neuro‐degeneration 2. To study at which age levels of phosphorylated tau (p‐tau) began to increase in NCT cKO mice, antibodies of AT8 (against p‐tau at epitopes of Ser202/Thr205) and AT100 (against p‐tau at epitopes of Thr212/ Ser214) were used. AT8 or AT100 Western blotting revealed significantly increased p‐tau levels in NCT cKO mice at ages such as 6 or 10 months (Figure 2C), suggesting that tau hyperphosphorylation takes place prior to neuron loss. To investigate which type of tau kinases was responsible for the change of p‐tau, we analyzed GSK3β and CDK5 14. Whereas relative levels of pGSK3β Y216 and pGSK3β S9 were not significantly reduced (Figure 2D), those of p25 were increased in NCT cKO mice (Figure 2E). In contrast, levels of total GSK3β and total p35 were unchanged in NCT cKOs as compared to age‐matched littermate controls, suggestive of enhanced CDK5 activity.
Discussion
Compared to 50% reduction on NCT protein levels in the Tabuchi line (2009) of NCT cKO, the inactivation efficiency of NCT was low in this line (e.g., ~30% reduction on NCT levels). This is likely due to the use of the T29 line of CaMKIIα‐Cre, which starts to express Cre recombinase at relatively late stage, as compared to the lines of CaMKIIα‐Cre reported previously 8, 9. Interestingly, this line of NCT cKO exhibited significantly reduced cortical neuron number at 13 months. In contrast, dramatic neuron loss was reported in other lines of NCT cKO mice at very young ages 8, 9. However, due to the late age by which evident cortical neuron loss takes place in this line of NCT cKO, this could allow us to dissect out sequential pathological events during the progression of neurodegeneration. Indeed, astroglial activation and p‐tau elevation were already detected in NCT cKO at as early as 3.5 months (data not shown), which is much earlier than the age when prominent neuron loss occurred. Overall, these findings strongly suggest that both tau hyperphosphorylation and neuroinflammation may be early pathological events in neurodegenerative diseases. We observed elevated p‐tau levels in NCT cKO mice and have demonstrated that changes on p‐tau were likely caused by enhanced activity of CDK5 but not GSK3β. Tau hyperphosphorylation in NCT cKO mice may act as a driving force for neurodegeneration. Although the exact role of neuroinflammation observed in NCT cKO mice remains to be investigated, it may also be a trigger to neuron death. Given that there is no effective treatment for neurodegenerative diseases, prevention of early neuropathology should be considered as potential strategies for the treatment of neurodegenerative diseases.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We would like to thank He Wang, Tingting Liu, Chaoli Huang, Xiaoyan Zou, and Huahong Yu for their contributions to this study. The work was supported by grants from the National Natural Science Foundation of China (31271123), the National Basic Research Program of Ministry of Science and Technology of China (2014CB942804), and the Natural Science Foundation of Jiangsu Province (BK20140018).
References
- 1. Stancu IC, Vasconcelos B, Terwel D, Dewachter I. Models of beta‐amyloid induced Tau‐pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener 2014;9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001;24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 3. Sun L, Zhao L, Yang G, et al. Structural basis of human γ‐secretase assembly. Proc Natl Acad Sci USA 2015;112:6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Y, Bohm C, Dodd R, et al. Structural biology of presenilin 1 complexes. Mol Neurodegener 2014;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shen J, Kelleher RJ 3rd. The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc Natl Acad Sci USA 2007;104:403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xia D, Watanabe H, Wu B, et al. Presenilin‐1 knockin mice reveal loss‐of‐function mechanism for familial Alzheimer's disease. Neuron 2015;85:967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee SH, Sharma M, Sudhof TC, Shen J. Synaptic function of nicastrin in hippocampal neurons. Proc Natl Acad Sci USA 2014;111:8973–8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sesele K, Thanopoulou K, Paouri E, et al. Conditional inactivation of nicastrin restricts amyloid deposition in an Alzheimer's disease mouse model. Aging Cell 2013;12:1032–1040. [DOI] [PubMed] [Google Scholar]
- 9. Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age‐related neurodegeneration. J Neurosci 2009;29:7290–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saura CA, Choi S‐Y, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron 2004;42:23–36. [DOI] [PubMed] [Google Scholar]
- 11. Feng R, Wang H, Wang J, et al. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin‐1 and presenilin‐2. Proc Natl Acad Sci USA 2004;101:8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng S, Zhang C, Xu C, et al. Age‐dependent neuron loss is associated with impaired adult neurogenesis in forebrain neuron‐specific Dicer conditional knockout mice. Int J Biochem Cell 2014;57:186–196. [DOI] [PubMed] [Google Scholar]
- 13. Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA 2003;100:4855–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang L, Cheng S, Yin Z, et al. Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol Neurodegener 2015;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]