

Summary
Aim
To treat neurodegenerative disorders such as Parkinson's disease (PD), drugs must be able to cross the blood–brain barrier (BBB). Patients with PD are deficient in dopamine (DA), a neurotransmitter that cannot pass through the BBB. Liposomes modified by adding polyethylene glycol (PEGylated liposomes (PLs)) can be conjugated with antibody to form DA–PEGylated immunoliposomes (DA‐PILs), and we tested their use as carriers of DA for treating PD.
Methods
PEGylated liposomes (PLs) were prepared by evaporation method, and [3H]dopamine was encapsulated within the dried lipid film using a freeze/thaw cycle to form DA‐PL. Thiolated OX26 MAb, an antitransferrin receptor monoclonal antibody, was then conjugated to 46‐nm PEGylated liposomes. Particle size, zeta potential, and stability were assessed, and in vivo effects were determined after the intravenous injection of DA, DA‐PL, and DA‐PIL by examining brain tissue in normal rats and rats that underwent transection of the medial forebrain bundle to induce PD.
Results
The uptake of DA‐PIL in the brains of this PD rat model increased about 8‐fold compared with that of DA alone and about 3‐fold compared with that of encapsulated DA–PEGylated liposomes (DA‐PL). The volume of distribution of DA‐PIL in the brain by the perfusion method was 4‐fold higher than that of DA‐PL, indicating that conjugation of OX26 MAb to the transferrin receptor of brain capillary endothelium mediated the effective delivery of DA to brain tissue.
Conclusions
Dopamine can be effectively delivered to the brain by means of a PIL‐based drug delivery system in PD rats.
Keywords: Blood–brain barrier, Dopamine, OX26 monoclonal antibody, Parkinson's disease, PEGylated immunoliposomes
Introduction
The major symptoms of Parkinson's disease (PD) are movement disorders such as akinesia/bradykinesia, rigidity, and tremor 1. Every year, more than 10 million people globally are diagnosed with neurodegenerative diseases such as Alzheimer's disease or PD 2. It has been proposed that the most significant characteristic of motor impairment in PD is the result of selective cell death of dopaminergic neurons in the substantia nigra pars compacta, with the consequent reduction in the dopamine content of the striatum 3, 4. Currently, several therapeutic agents are being used to relieve the symptoms of PD. The mainstay of therapy is levodopa (l‐DOPA), which replaces the lost dopamine in the striatum, and its effects in patients with early‐stage PD are excellent. However, as the disease progresses, the numbers of viable dopaminergic neurons that can convert l‐DOPA into dopamine decrease, and the effects of the drug are significantly reduced 3, 5. Dopamine agonists that act like dopamine, such as pramipexole, ropinirole, bromocriptine, and apomorphine, are used for all stages of PD and have proven efficacy. Compared with l‐DOPA, these drugs have longer plasma half‐lives and are associated with fewer instances of fluctuations in motor function and dyskinesia 6. Despite such advantages, they have several significant side effects such as sleepiness, drowsiness, confusion, hallucinations, or sedation that interfere with driving and other activities 7. Drug therapy in PD including these dopamine agonists has focused mainly on relieving symptoms by replacing the lost dopamine in the striatum.
The diffusion of an active agent across the blood–brain barrier (BBB) depends on its ability to cross the lipid membrane 8. However, many drugs do not possess properties such as a high lipid solubility, low molecular size, and positive charge 9. Many previous studies have used both intracerebral and intravenous administration of liposomes for the targeted delivery of drug to the brain. Caelyx that is doxorubicin encapsulated in PEGylated liposomes (PLs) has shown positive results in glioblastomas and metastatic tumors 10, 11 even though these liposomes are relatively inert and cannot pass across the BBB 9. PEGylated immunoliposomes (PILs), which are 100 nm in diameter and are conjugated to approximately 50 receptor‐specific monoclonal antibody (MAb) molecules, have been used to transport drugs across blood vessel walls 12, 13. An antibody against OX26 was able to facilitate the passing of liposomes across the BBB via the endogenous transferrin receptor (TfR) 14, 15.
In the present study, we formulated a dopamine‐loaded PEGylated immunoliposome (DA‐PIL) that exhibits significantly higher brain transport and a longer half‐life than either free dopamine (DA) or dopamine‐encapsulated PEGylated liposomes (DA‐PLs). We then injected DA, DA‐PL, and DA‐PIL in a PD rat model, measured in vivo brain uptake, and performed pharmacokinetic studies.
Materials and Methods
Materials
[3H]Dopamine (20.7 Ci/mmol) was purchased from PerkinElmer Inc. (Waltham, MA, USA). Distearoylphosphatidylcholine, 1‐palmitoyl‐2‐oleoyl‐sn‐glycerol‐3‐phosphatidylcholine (POPC), 1,2‐dioleoyl‐3‐trimethylammonium‐propane (DOTAP), 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(polyethylenleglycol‐2000) (DSPE‐PEG2000), and 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[maleimide(polyethyleneglycol)‐2000] (DSPE‐PEG2000‐maleimide) were all purchased from Avanti Polar Lipids (Alabaster, AL, USA). Chloroform (CHCl3) and 2‐iminothiolane (Traut's reagent) were purchased from Pierce Biotechnology, Inc. (Rockford, IL, USA). Sepharose G‐25 was purchased from Amersham Pharmacia Biotech (Barcelona, Spain), and sepharose CL‐4B was obtained from GE Healthcare (Buckinghamshire, UK). The monoclonal antibody against OX26 (OX26 MAb) was purchased from AbD Serotec (Oxford, UK). Centriprep‐30 was purchased from the Millipore Corporation (Bedford, MA, USA). All other reagents and solvents were of analytical grade or higher.
Preparation of PEGylated Liposomes (PLs)
The PL was prepared as previously reported 12, 16. Distearoylphosphatidylcholine (5.2 mM), cholesterol (4.5 mM), DSPE (0.3 mM), and a linker lipid (DSPE‐PEG2000‐maleimide) (0.015 mM) were dissolved in chloroform/methanol (2:1, vol:vol), followed by evaporation. The lipids were dispersed in 300 μL of 0.05 M Tris–HCl buffer (pH 7.4) and were sonicated for 20 min. [3H]Dopamine (0.01 nmol) (DA) was added to the lipids, and the liposome/[3H]DA dispersion was frozen on ethanol/dry ice for 5 min and then thawed at 40°C for 2 min. This freeze/thaw cycle was repeated 20 times to ensure encapsulation. Subsequently, the liposome dispersion was diluted to a lipid concentration of 10 mM, followed by extrusion with the push/pull extruder (Avestin, Ottawa, Canada) seven, five, and three times each through two stacks each of polycarbonate membranes with pore sizes of 400, 200, and 100 nm pore size. The non‐PL was removed by passing the mixture through sepharose CL‐4B, and the PL was stored at 4°C.
Preparation of PEGylated Immunoliposomes (PILs)
The PIL was prepared as previously reported 12, 16, 17, 18, 19, 20. OX26 MAb was iodinated using iodine‐125 (125I) and chloramine‐T according to a previous report 21. The radiolabeled protein was then purified by Sephadex G‐25 gel filtration chromatography and was more than 96% TCA precipitable. The [125I]OX26 antibody was thiolated using Traut's reagent (2‐iminothiolane), and OX26 MAb was dissolved in 0.05 M of sodium borate buffer/0.1 mM of EDTA (pH 8.5) 22. After incubation for 60 min at room temperature, the OX26 MAb solutions were concentrated and the buffer was exchanged with 0.1 M of sodium phosphate (pH 8.0) using a Centriprep‐30 concentrator. The thiolated OX26 MAb was conjugated with PL, and the nonthiolated PIL was removed by passing the mixture through Sephadex G25 1.5‐cm × 18‐cm filtration columns and were eluted with buffers (HEPES buffer—0.05 M of HEPES, pH 7.4) and phosphate‐buffered saline (PBS) buffer (0.01 M of NaCl, pH 7.4). The aliquot size of each fraction of the PIL sample (1 mL) was determined by scintillation counting in a beta‐radiation liquid scintillation counter (Model LS6500) (Beckman Instruments, Fullerton, CA, USA).
Characterization of PL
The average diameter and zeta potential of PL were measured three times per sample (n = 3) using a laser scattering technique (Nano ZS 90; Malvern, Worcestershire, UK) 12, 16, 17, 18. To avoid multiple scattering effects, the samples were diluted 1:10, 1:50, and 1:100 with acetate buffer (0.25 mol/L, pH 3.5 ± 0.1).
Stability of PIL
The stability of the PIL + plasma (1:1) mixture and the PIL + saline (1:1) mixture was investigated 12, 16, 17, 18 by incubating the samples for 0, 1, 12, 24, 48, and 72 h at both 37°C and 25°C. All measurements were performed in triplicate.
Animal Model of PD
Adult male Wistar rats (Charles River Laboratory, Wilmington, DE, USA) weighing 250–300 g were used for the PD model. All animal experiments were approved by the Committee for the Ethics of Animal Experiments at Sookmyung Women's University (SMWU‐IACUC‐1405‐009). The animals were housed at 25°C with a 12‐h dark/light cycle.
To simulate PD, the rats underwent transection of the medial forebrain bundle (MFB) as previously reported 23, 24. These rats were anesthetized intraperitoneally with ketamine (70 mg/kg) and xylazine (8 mg/kg). A small hole was made 3.0 mm caudal to the bregma and 2.8 mm lateral to the midline in the right brain area. A cannula, housing a retractable wire Scouten knife (David Kopf Instruments, Tujunga, CA, USA), was lowered into the hole to a depth of 9.0 mm from the dural surface and then extended 2.2 mm toward the midline, and was slowly raised 3.0 mm and subsequently lowered back to its original position to ensure complete excision of the whole MFB. After surgery, the animals were kept on a heating plate at 37°C until recovery was complete.
Pharmacokinetics and Brain Delivery of PIL
The pharmacokinetic study was performed in normal male Sprague Dawley rats (SD) and the PD model rats (PD) 12, 25. All the rats were anesthetized with 100 mg/kg of ketamine by intramuscular injection. The left femoral vein was cannulated with a polyethylene tube (PE50) (Natsume, Tokyo, Japan), and 0.2 mL of Ringer‐HEPES buffer (141 mM of NaCl, 4.0 mM of KCl, 2.8 mM of CaCl2, 10 mM of HEPES, pH 7.4) containing 5 μCi each of [3H]DA, [3H]DA‐PL, and [3H]DA‐PIL, was injected intravenously. After the heparinized PE50 tube was implanted in the left femoral artery (for anticoagulation), arterial blood samples (0.3 mL) were collected at 0.25, 1, 2, 5, 15, 30, and 60 min after the intravenous injection of the sample. The volume of blood collected was replaced with an equal volume of saline (containing 100 U of heparin/mL of saline). Centrifugation for 10 min at 560 g and 4°C was used to separate the plasma from the rest of the blood. At 60 min after the intravenous injection, the animals were decapitated and their brains were removed. The plasma and brain samples were solubilized with 2N of NaOH and analyzed for 3H radioactivity using a liquid scintillation counter (Model LS6500, Beckman Instruments Inc. Fullerton, CA, USA).
WinNonlin Professional software (Pharsight, Mountain View, CA, USA) was used for nonlinear regression analysis of the pharmacokinetic parameters. The radioactivity in the plasma (dpm/mL) was converted to a percentage of injected dose (ID) per milliliter (mL), and the %ID/mL value was fitted to the following bi‐exponential equation:
The pharmacokinetic parameters were computed using the intercepts A 1 and A 2 and the slopes k 1 and k 2. These pharmacokinetic parameters included the area under the plasma concentration curve (AUC) at 60 min, the steady‐state AUC (AUCss), and the systemic clearance (CL) 12, 25. The volume of distribution (V d) (μL/g of brain) is the ratio of brain radioactivity (dpm/g of brain) divided by the plasma radioactivity (dpm/μL) at 60 min. The brain clearance (μL/min/g), also called the permeability surface area product (PS), was determined as follows:
where C p(T) is the 60‐min plasma concentration (%ID/mL) and V 0 is the plasma volumes for the respective brain, as reported previously 25. The terminal brain uptake, expressed as %ID/g of brain, was calculated using the equation %ID/g = PS × AUC.
Internal Carotid Artery Perfusion (ICAP) Method
The internal carotid artery perfusion (ICAP) technique was carried out, as previously reported 20, 25. Rats were anesthetized by intramuscular injection of a combination of ketamine (100 mg/kg) and xylazine (2 mg/kg). After coagulation of the occipital, superior thyroid, and pterygopalatine arteries, the external carotid artery was catheterized using a polyethylene tube (PE‐10) (Natsume, Tokyo, Japan) to allow perfusion of the internal carotid artery. The perfusate with Krebs–Henseleit buffer at pH 7.4 containing 0.5 μCi/mL each of DA, DA‐PL, and DA‐PIL was oxygenated with O2:CO2 (95:5) and heated to 37°C. The infusion was begun using a syringe pump (Harvard Apparatus, South Natick, MA, USA) with a perfusion rate of 1.25 mL/min for 5 min, along with simultaneous ligation of the right common carotid artery to prevent mixing of the perfusate with the systemic circulation. After the rats were decapitated, the ipsilateral cerebral hemisphere was removed and weighed. This tissue was solubilized in Soluene‐350 at 60°C for 3 h, and the radioactivity was measured using a liquid scintillation counter (Beckman). The volume of distribution (V D) of the DA, DA‐PL, DA‐PIL, and [14C]sucrose as a blood volume marker in the brain was calculated as the dpm/g of brain divided by the dpm/μL of the perfusate.
Statistical Analysis
All data are expressed as means ± SEM. All plots were drawn using SigmaPlot version 7.0 (Systat Software, Point Richmond, CA). Repeated‐measures ANOVA analysis and two‐way ANOVA with Dunnett's post hoc test were used to compare mean values between the formulations. In all cases, P < 0.05 indicated differences that were statistically significant. Noncompartmental analyses of pharmacokinetic data were performed using WinNonlin 5.0.1 (Pharsight).
Results
Preparation of PL and Dopamine Encapsulation
The efficiency by which dopamine is encapsulated into the PEGylated liposomes (PLs) was evaluated at several concentration ratios of DSPE‐PEG2000 and DSPE‐PEG2000‐maleimide. The encapsulation yields were 35 ± 1% (P < 0.001), 27 ± 1% (P < 0.05), and 26 ± 1%, with total concentrations of DSPE‐PEG2000 and DSPE‐PEG2000‐maleimide of 0.4, 0.5, and 0.8 μmol, respectively. The concentration ratio of DSPE‐PEG2000 to DSPE‐PEG2000‐maleimide was 3:1. Moreover, the encapsulation efficiency of dopamine in PL was significantly increased by about 26.4 ± 0.02% using a glass tube in 0.3 μmol of DSPE‐PEG2000 and 0.1 μmol of DSPE‐PEG2000‐maleimide, as compared with using a polypropylene tube (18%). Thus, the amounts of DSPE‐PEG2000 and DSPE‐PEG2000‐maleimide were fixed at 0.3 μmol (1.5 mol % of lipid) and 0.1 μmol (0.5 mol % of lipid). Finally, we used approximately 46 ± 4 nm as the mean diameter of DA‐PL, which was made using an extruder, and exhibited a zeta potential of 6 ± 1 mV, and its encapsulation efficiency was 35 ± 1%.
Preparation of PIL
In order to prepare immunoliposomes, thiolated antibody was linked to the distal end of lipid‐conjugated maleimide‐PEG by means of a thioether bond. The degree of coupling of the thiolated MAb OX26 (OX26‐SH), with stabilized liposomes containing DSPE‐PEG2000 and DSPE‐PEG2000‐maleimide, was evaluated by Sepharose CL4B gel filtration chromatography. The coupling of the OX26 MAb with PL that included 0.4 μmol of DSPE‐PEG2000 and 0.1 μmol of DSPE‐PEG2000‐maleimide was approximately 58% higher than the 52% for the degree of coupling of OX26 MAb and PL that included 0.8 μmol of DSPE‐PEG2000 and 0.1 μmol DSPE‐PEG2000‐maleimide, and the PIL ranged in size from 42 to 50 nm.
Physical Stability of PIL
The in vitro stability of DA‐PIL was demonstrated in terms of the percentage of DA retained in the liposomes, as shown in Figure 1. When PIL was cultured for 72 h in normal saline at room temperature, its levels of stability were 87 ± 2% at 24 h and 78 ± 1% at 72 h. In addition, the stability of DA‐PIL in rat plasma was approximately 67 ± 1% at 24 h and 51 ± 4% at 72 h after incubation.
Figure 1.
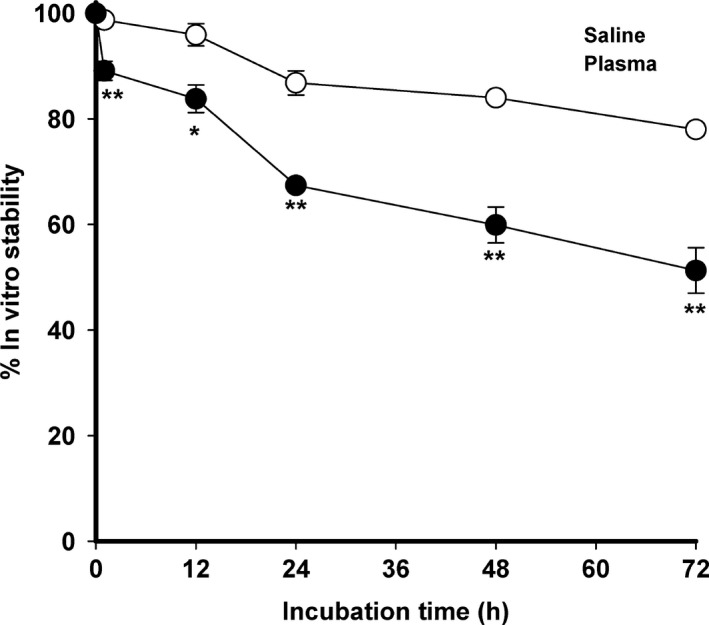
In vitro stability of DA‐PIL at different time points following incubation in normal saline at room temperature, or rat plasma at 37°C. DA‐PILs, encapsulated dopamine PEGylated immunoliposomes. Data are means ± SEM (n = 3). * P < 0.05, ** P < 0.01, significantly different from normal saline.
Pharmacokinetics and Brain Distribution of DA, DA‐PL, and DA‐PIL
DA‐PL and DA‐PIL were used immediately after the liposomes had been prepared. The disappearance of free DA, DA‐PL, and DA‐PIL from the plasma compartment in the PD rats is graphically displayed in Figure 2 in a bi‐exponential manner. Figure 2 shows the results of the plasma–time profile data comparing the pharmacokinetic parameters of DA, DA‐PL, and DA‐PIL in the PD rats. Free DA disappeared rapidly from the circulation with a clearance of 2.31 ± 0.33 mL/min/kg, as compared with DA‐PL (0.18± 0.03 mL/min/kg, P < 0.001) and DA‐PIL (0.18 ± 0. 05 mL/min/kg, P < 0.001) (Table 1). The 10‐fold difference in disappearance from the circulation between DA and DA‐PL or between DA and DA‐PIL is indicative of appropriate retention time of the dopamine encapsulated in the liposomes or immunoliposomes. In the PD rats, DA‐PL and DA‐PIL exhibited moderately prolonged circulation, with a half‐life (t 1/2) in plasma of 116 ± 24 min and 107 ± 35 min, respectively. This t 1/2 level was somewhat higher than that for free DA (t 1/2 = 45.6 ± 13.4 min), possibly owing to the faster clearance of DA in plasma. The total area under the plasma concentration curve (AUCt) of DA‐PL and DA‐PIL increased approximately 14‐fold (609 ± 102%ID min/mL, P < 0.05) and 16‐fold (703 ± 187%ID min/mL, P < 0.05) as compared to the AUCt of free DA (45.0 ± 6.0%ID min/mL) in the PD rats. The mean residence times (MRT) of DA‐PL and DA‐PIL after intravenous administration were approximately 27% and 23% longer than the MRT of free DA, indicating a slow absorption process associated with PL or PIL. The distribution volumes at steady state (V dss) of DA‐PL and DA‐PIL in the PD rats were 27.0 ± 1.5 mL/kg and 21.7 ± 1.6 mL/kg, respectively. These were 81% (P < 0.01) and 85% (P < 0.01) less than the V dss of free DA (143 ± 29 mL/kg). On analysis of the brain tissue, the BBB permeability surface area (PS) product of DA was 1.73 ± 0.12 μL/min/g of brain. The AUC of DA at 60 min was 24.4 ± 2.4%ID min/mL, smaller than the AUC of DA‐PL (175 ± 12%ID min/mL) and the AUC of DA‐PIL (220 ± 18%ID min/mL), indicating that DA accumulated poorly in brain tissue (0.04 ± 0.01%ID/g). The PS of DA‐PL was decreased about 3‐fold compared with the PS of DA, whereas the AUC of DA‐PL at 60 min was 7‐fold higher than the AUC of DA. These results showed a 2‐fold increase in brain drug uptake (0.10 ± 0.02%ID/g in DA‐PL, P < 0.05) as compared with the value of DA (Figure 3). After the administration of DA‐PIL, the BBB PS value was 2.92 ± 0.50 μL/min/g that is about 2‐fold higher than that of DA (P < 0.05) and about 5‐fold higher than that of DA‐PL (P < 0.05), with the elevation of AUC at 60 min, and it induced to the highest brain delivery (%ID/g value). In other words, DA‐PIL administration showed the highest level of brain uptake (0.32 ± 0.06%ID/g, P < 0.01), increasing about 8‐fold, as compared with the level of brain uptake of DA (Figure 3).
Figure 2.
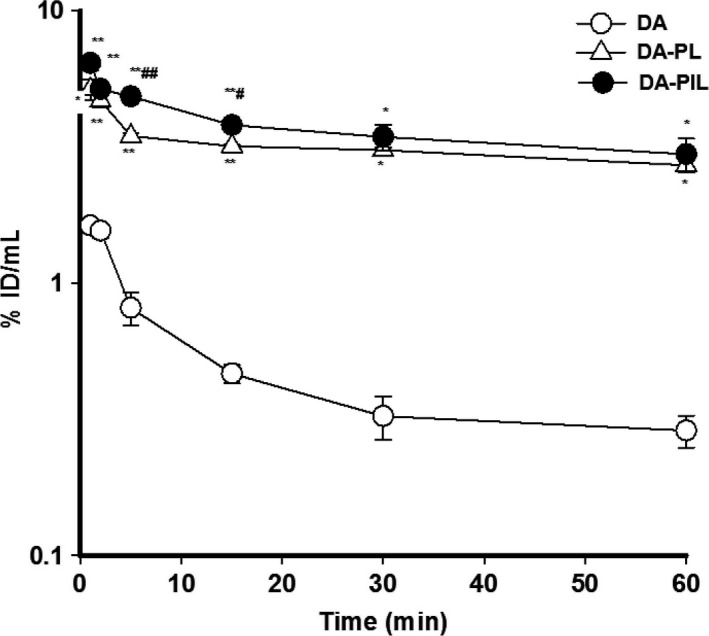
The percentage of injected dose per milliliter of the plasma–time profiles of DA (○), encapsulated DA‐PL (▵), and DA‐PIL (●) in PD rats up to 60 min after intravenous injection. DA, free dopamine; DA‐PLs, encapsulated dopamine PEGylated liposomes; DA‐PILs, encapsulated dopamine PEGylated immunoliposomes. Data are means ± SEM (n = 3 or 4). * P < 0.01; **P < 0.001 significantly different from DA. # P < 0.05, ## P < 0.01, significantly different from DA‐PL.
Table 1.
Pharmacokinetic parameters of DA, DA‐PL, and DA‐PIL after intravenous injection in PD rats
| Parameter | DA | DA‐PL | DA‐PIL |
|---|---|---|---|
| t 1/2 (min) | 45.6 ± 13.4 | 116 ± 24 | 107 ± 35 |
| AUC (%ID min/mL) | 45.0 ± 6.0 | 609 ± 102* | 703 ± 187* |
| CL (mL/min/kg) | 2.30 ± 0.30 | 0.18 ± 0.03*** | 0.18 ± 0.10*** |
| MRT (min) | 21.5 ± 1.2 | 27.2 ± 0.4** | 26.5 ± 0.8** |
| V dss (mL/kg) | 143 ± 29 | 27.0 ± 1.5** | 21.7 ± 1.6** |
t 1/2, half‐life; AUC, area under the curve; ID, injected dose; CL, clearance; MRT, mean residence time; V dss, volume of distribution.Parameters were computed from the serum radioactivity profile in Figure 2. Each value represents means ± SEM (n = 3–4). *P < 0.05, **P < 0.01, ***P < 0.001, significantly different when compared with dopamine (DA).
Figure 3.
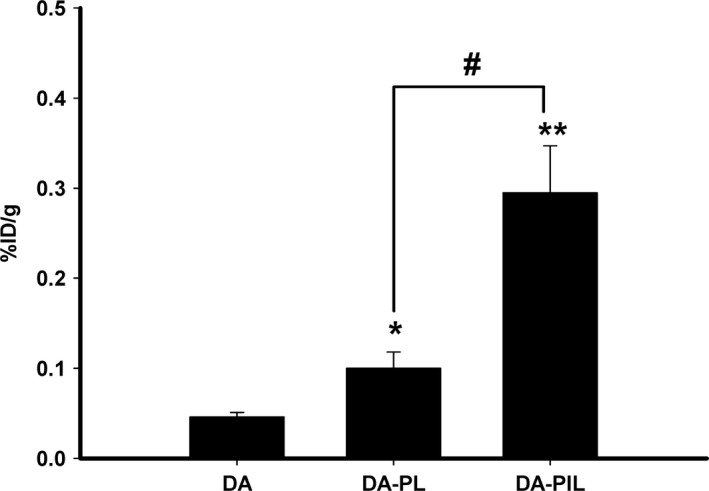
The brain uptake (%ID/g) for DA, DA‐PL, and DA‐PIL at 60 min after intravenous injection. DA, free dopamine; DA‐PLs, encapsulated dopamine PEGylated liposomes, DA‐PILs, encapsulated dopamine PEGylated immunoliposomes. %ID/g, percentage of the injected dose per gram of brain tissue. Data are means ± SEM (n = 3 or 4). * P < 0.05, ** P < 0.01, significantly different from DA, # P < 0.05, significantly different from DA‐PL.
The direct brain uptake of DA, DA‐PL, and DA‐PIL in PD rats was studied using the internal carotid artery perfusion (ICAP) method. In Figure 4, the V D of DA‐PL (13.5 ± 0.6 μL/g) was higher than that of DA (7.4 ± 1.4 μL/g), and the V D of DA‐PIL (46.0 ± 5.7 μL/g) was significantly higher (P < 0.01) than the DA‐PL in the control rats. The brain V D of DA‐PIL significantly increased about 4‐fold more than that of DA‐PL (44.0 ± 3.0 μL/g vs. 12.0 ± 0.7 μL/g) (P < 0.001). Thus, brain uptake in PD rats was increased with the use of OX26 MAb (Figure 4).
Figure 4.
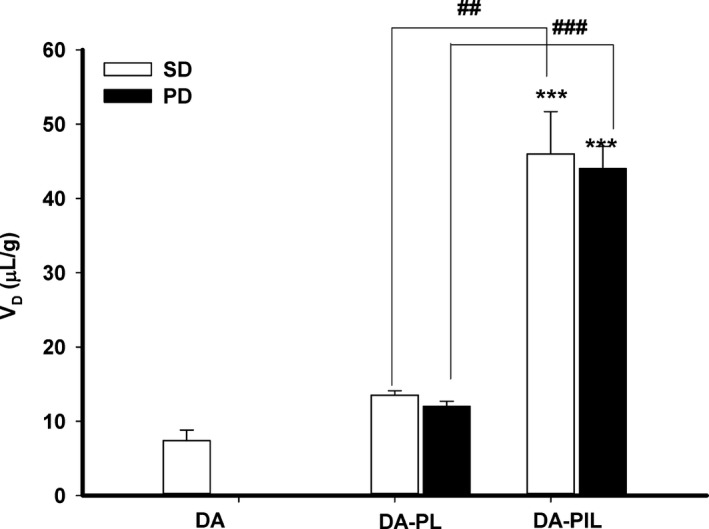
The brain volume of distribution of DA, DA‐PL, and DA‐PIL in the brain parenchyma measured by internal carotid artery perfusion. DA, free dopamine; DA‐PLs, encapsulated dopamine PEGylated liposomes; DA‐PILs, encapsulated dopamine PEGylated immunoliposomes; SD, control Sprague Dawley rat group; PD, experimental group of rat model of Parkinson's disease (PD). Data are means ± SEM (n = 3,4). ***P < 0.001, significantly different from DA, ## P < 0.01, ### P < 0.001, significantly different from DA‐PL.
Discussion
PD, an incurable and progressive neurodegenerative disorder, is characterized by a decrease in dopamine levels as a result of the death of dopaminergic neurons in the substantia nigra pars compacta, a region of the brain that regulates motor activity through projecting dopaminergic axons to the striatum 26, 27, 28. Therapeutic approaches have focused on replenishing dopamine in order to relieve the symptoms of PD. However, it is difficult for dopamine to affect the central nervous system (CNS) because it does not cross the BBB owing to its hydrophilicity, low level of permeability, and rapid plasma clearance 6. Although l‐DOPA replacement therapy, a precursor of dopamine administration, has been used to reduce the clinical symptoms of PD, its therapeutic effects diminish, during the latter stages of the disease 3, 5, 7. Therefore, the ability to produce dopamine in the brain or transport it to the brain in high doses is an important goal.
To maintain CNS homeostasis, the BBB plays a central role in regulating the exchange of molecules into and out of the brain. The BBB consists of endothelial cells (ECs), pericytes, and astrocytes, and creates a neurovascular unit with the adjacent neurons 29. The development of therapeutic agents to treat neurodegenerative diseases is limited in that their entry into the brain is commonly restricted by the BBB 30. Therefore, bypassing the BBB is an important factor when considering therapeutic approaches to nervous system disorders 31.
The use of nanotechnology‐based approaches came into the spotlight as a way to deliver drugs with increased targeting efficiency 32. PEGylated liposomes (PLs) have been used to lengthen blood residence times of the drug and potentially increase bioavailability according to delivery across the BBB 20, 21 The reverse transcytosis of apo‐ and holo‐transferrin between the brain and the blood is mediated by the BBB transferrin receptor (TfR), since the TfR is expressed on the abluminal membrane of the capillary endothelium in the brain 33, 34. The conjugation of lipid nanocapsules to OX26 MAb directed against TfR has led to the high specificity and efficiency of drug delivery to target tissues 35.
Since the intact nanoparticles pass through the BBB without changing the properties of the BBB, it is necessary to control the avidity as well as their size and surface charge 12. In particular cases, nanoparticles of 100 nm or smaller have been shown to pass through brain tissue, such as nanoparticles that have near‐neutral zeta potentials and are coated with a dense layer of polyethylene glycol 36. Our results showed that dopamine is encapsulated into PEGylated liposomes (PLs) at a yield of 34%. The dopamine‐encapsulated PL (DA‐PL) showed a mean particle size of 46 ± 4 nm and a mean zeta potential of 6 ± 1 mV. Accordingly, DA‐PL can pass across the BBB if they are small enough in size and have electrical characteristics close to neutral. About 85 nm of PEGylated immunoliposomes (PILs) is able to enter brain cells that express TfR on the surface, using receptor‐mediated endocytosis 12, 37. The mean in vitro stabilities of DA‐PIL were 87 ± 2% at 24 h and 78 ± 1% at 72 h in normal saline at room temperature, and 67 ± 1% at 24 h and 51 ± 4% at 72 h in rat plasma at 37°C (Figure 1). When the liposomes are coated with PEG, the blood residence time is longer, the pharmacokinetic profile is improved, and the biodistribution is altered 38. Moreover, it is believed that the polymer coating of liposomes increases their biological half‐life owing to reduced interactions with plasma proteins or cell surface receptors 39, 40. We found that DA‐PIL, as compared with DA, showed a very significant augmented AUC, a lower volume of distribution, and a decreased clearance rate in PD rats. Also, DA‐PL showed similar PK parameter values as DA‐PIL. We observed very prolonged blood mean residence time of DA‐PIL and DA‐PL compared to DA (Table 1 and Figure 2).
Furthermore, DA‐PIL had the lowest plasma clearance at the same time point, up to 60 min following intravenous injection, and DA‐PIL appeared to slow down the decay process. And it was most stable in circulation in PD rats (Figure 2). The stability of a drug in circulation is important for successful drug delivery, and this can be confirmed by prolonged circulation and retention properties 41. The decreased plasma clearance of the OX26 PEGylated immunoliposomes is due to the targeting of the liposomes to tissues such as the liver and brain, which express high levels of the TfR in the microcirculation 33, 34. In comparison with DA, the brain uptake level of DA‐PL into the brain increased 3‐fold in PD rats (reflected by its %ID/g); moreover, the brain uptake of DA‐PIL from the blood showed a 7‐fold increase compared with DA‐PL in PD rats (Figure 3). The brain delivery level of DA‐PIL was 4‐fold higher than that of morphine (0.08%ID/g) 42. According to these results, a higher degree of brain delivery can be achieved with the administration of DA encapsulated in PEGylated liposomes (DA‐PLs), as compared with free DA administration, as well as DA‐PL coupled to the thiolated OX26 MAb (DA‐PIL), as compared with DA‐PL in the PD rats. The brain uptake of DA‐PL without the OX26 MAb attached is approximately 0.040 ± 0.002%ID/g, indicating little transport of the PL across the BBB in vivo. The brain uptake (%ID/g) of the OX26‐conjugated PIL, which is a function of both the plasma AUC and the PS, was approximately 3‐fold greater than brain uptake of the unconjugated version. This increase in the brain uptake of DA‐PIL in the PD rats was due mainly to the elevated plasma PS value. The BBB PS of DA‐PIL is similarly increased by approximately 6‐fold that of DA‐PL. Furthermore, in the internal carotid artery perfusion study, the brain tissue volume of distribution (V D) of DA‐PIL was significantly enhanced by either encapsulation of DA into PEGylated liposomes or the coupling of OX26 MAb to DA‐PL in both the SD and the PD rats (Figure 4). The V D of DA was similar to the level of sucrose such as in the intravascular space. The V D of DA‐PL was significantly increased, as compared with DA in the SD rat, and the V D of DA‐PIL was significantly increased about 4‐fold that of DA‐PL in the SD and PD rats (Figure 4). Therefore, the DA‐PIL passed through the BBB to achieve outstanding delivery of DA to the brain.
In terms of the limitations of this study, it is known that the MFB‐transected animal model of PD does not induce the selective loss of DA fibers and that the lesion induced is not likely to be restricted to the nigrostriatal pathway 24. Therefore, further studies will be needed, to validate our results by applying DA‐PIL to other animal models of PD that will induce the selective degeneration of DA neurons with neurotoxins, such as 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) 43, 44 or 6‐hydroxydopamine (6‐OHDA) 45.
In summary, a BBB drug delivery system using PL and OX26 MAb should be effective in neurodegenerative diseases such as PD, because it allows the successful transport of drug into the brain.
Conclusion
Based on our in vitro and in vivo results in this rat model of PD, the characteristics of a PEGylated OX‐26‐conjugated immunoliposomal (PIL) formulation of dopamine allowed the effective delivery of this drug across the BBB. Therefore, the efficient and specific delivery of dopamine into brain tissue by means of PIL implies that this strategy may be a promising approach for treating patients with Parkinson's disease. In the future, we plan to carry out efficacy studies of Parkinson's disease in clinically relevant models.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Appendix S1 ANOVA analysis result.
Acknowledgments
The authors thank Temdara Tun for technical assistance. This research was supported by the National Research Foundation of Korea (NRF) (NRF‐2010‐0008046), and an NRF grant funded by the Korean government (MSIP) (2011‐0030074).
References
- 1. Lindner MD, Emerich DF. Therapeutic potential of a polymer‐encapsulated L‐DOPA and dopamine‐producing cell line in rodent and primate models of Parkinson's disease. Cell Transplant 1998;7:165–174. [DOI] [PubMed] [Google Scholar]
- 2. Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest 2005;115:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron 2003;39:889–909. [DOI] [PubMed] [Google Scholar]
- 4. Villalba RM, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP‐treated monkeys. Exp Neurol 2009;215:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deleu D, Northway MG, Hanssens Y. Clinical pharmacokinetic and pharmacodynamic properties of drugs used in the treatment of Parkinson's disease. Clin Pharmacokinet 2002;41:261–309. [DOI] [PubMed] [Google Scholar]
- 6. Camargo SM, Vuille‐dit‐Bille RN, Mariotta L, et al. The molecular mechanism of intestinal levodopa absorption and its possible implications for the treatment of Parkinson's disease. J Pharmacol Exp Ther 2014;351:114–123. [DOI] [PubMed] [Google Scholar]
- 7. Brooks DJ. Dopamine agonists: their role in the treatment of Parkinson's disease. J Neurol Neurosurg Psychiatry 2000;68:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fong CW. Permeability of the blood‐brain barrier: molecular mechanism of transport of drugs and physiologically important compounds. J Membr Biol 2015;248:651–669. [DOI] [PubMed] [Google Scholar]
- 9. Garcia‐Garcia E, Andrieux K, Gil S, Couvreur P. Colloidal carriers and blood‐brain barrier (BBB) translocation: a way to deliver drugs to the brain? Int J Pharm 2005;298:274–292. [DOI] [PubMed] [Google Scholar]
- 10. Hau P, Fabel K, Baumgart U, et al. Pegylated liposomal doxorubicin‐efficacy in patients with recurrent high‐grade glioma. Cancer 2004;100:1199–1207. [DOI] [PubMed] [Google Scholar]
- 11. Koukourakis MI, Koukouraki S, Fezoulidis I, et al. High intratumoural accumulation of stealth liposomal doxorubicin (Caelyx) in glioblastomas and in metastatic brain tumours. Br J Cancer 2000;83:1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci USA 1996;93:14164–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci USA 2000;97:7567–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olivier JC, Huertas R, Lee HJ, et al. Synthesis of pegylated immunonanoparticles. Pharm Res 2002;19:1137–1143. [DOI] [PubMed] [Google Scholar]
- 15. Pardridge WM. Tyrosine hydroxylase replacement in experimental Parkinson's disease with transvascular gene therapy. NeuroRx 2005;2:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pardridge WM. Gene targeting in vivo with pegylated immunoliposomes. Methods Enzymol 2003;373:507–528. [DOI] [PubMed] [Google Scholar]
- 17. Pardridge WM, Kang YS, Yang J, et al. Enhanced cellular uptake and in vivo biodistribution of a monoclonal antibody following cationization. J Pharm Sci 1995;84:943–948. [DOI] [PubMed] [Google Scholar]
- 18. Kang YS, Pardridge WM. Use of neutral avidin improves pharmacokinetics and brain delivery of biotin bound to an avidin‐monoclonal antibody conjugate. J Pharmacol Exp Ther 1994;269:344–350. [PubMed] [Google Scholar]
- 19. Pardridge WM. Drug delivery to the brain. J Cereb Blood Flow Metab 1997;17:713–731. [DOI] [PubMed] [Google Scholar]
- 20. Yoshikawa T, Pardridge WM. Biotin delivery to brain with a covalent conjugate of avidin and a monoclonal antibody to the transferrin receptor. J Pharmacol Exp Ther 1992;263:897–903. [PubMed] [Google Scholar]
- 21. Triguero D, Buciak JB, Yang J, et al. Blood‐brain barrier transport of cationized immunoglobulin G: enhanced delivery compared to native protein. Proc Natl Acad Sci USA 1989;86:4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsh JW. Antibody‐mediated routing of diphtheria toxin in murine cells results in a highly efficacious immunotoxin. J Biol Chem 1988;263:15993–15999. [PubMed] [Google Scholar]
- 23. Song DY, Yang YC, Shin DH, et al. Axotomy‐induced dopaminergic neurodegeneration is accompanied with c‐Jun phosphorylation and activation transcription factor 3 expression. Exp Neurol 2008;209:268–278. [DOI] [PubMed] [Google Scholar]
- 24. Song DY, Yu HN, Park CR, et al. Down‐regulation of microglial activity attenuates axotomized nigral dopaminergic neuronal cell loss. BMC Neurosci 2013;14:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee NY, Lee KB, Kang YS. Pharmacokinetics, placenta, and brain uptake of paclitaxel in pregnant rats. Cancer Chemother Pharmacol 2014;73:1041–1045. [DOI] [PubMed] [Google Scholar]
- 26. Terao Y, Fukuda H, Ugawa Y, et al. New perspectives on the pathophysiology of Parkinson's disease as assessed by saccade performance: a clinical review. Clin Neurophysiol 2013;24:1491–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawlor PA, During MJ. Gene therapy for Parkinson's disease. Expert Rev Mol Med 2004;6:1–18. [DOI] [PubMed] [Google Scholar]
- 28. Kong P, Zhang B, Lei P, et al. Neuroprotection of MAO‐B inhibitor and dopamine agonist in Parkinson disease. Int J Clin Exp Med 2015;8:431–439. [PMC free article] [PubMed] [Google Scholar]
- 29. Tajes M, Ramos‐Fernández E, Weng‐Jiang X, et al. The blood‐brain barrier: structure, function and therapeutic approaches to cross it. Mol Membr Biol 2014;31:152–167. [DOI] [PubMed] [Google Scholar]
- 30. Vlieghe P, Khrestchatisky M. Medicinal chemistry based approaches and nanotechnology‐based systems to improve CNS drug targeting and delivery. Med Res Rev 2013;33:457–516. [DOI] [PubMed] [Google Scholar]
- 31. Kabanov AV, Gendelman HE. Nanomedicine in the diagnosis and therapy of neurodegenerative disorders. Prog Polym Sci 2007;32:1054–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pardridge WM. Blood‐brain barrier delivery of protein and non‐viral gene therapeutics with molecular Trojan horses. J Control Release 2007;122:345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Pardridge WM. Rapid transferrin efflux from brain to blood across the blood‐brain barrier. J Neurochem 2001;76:1597–1600. [DOI] [PubMed] [Google Scholar]
- 34. Skarlatos S, Yoshikawa T, Pardridge WM. Transport of [125I]transferrin through the rat blood‐brain barrier. Brain Res 1995;683:164–171. [DOI] [PubMed] [Google Scholar]
- 35. Béduneau A, Saulnier P, Hindré F, et al. Design of targeted lipid nanocapsules by conjugation of whole antibodies and antibody Fab' fragments. Biomaterials 2007;28:4978–4990. [DOI] [PubMed] [Google Scholar]
- 36. Song H, Zhang J, Han Z, et al. Pharmacokinetic and cytotoxic studies of pegylated liposomal daunorubicin. Cancer Chemother Pharmacol 2006;57:591–598. [DOI] [PubMed] [Google Scholar]
- 37. Ishida T, Kirchmeier MJ, Moase EH, et al. Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim Biophys Acta 2001;1515:144–158. [DOI] [PubMed] [Google Scholar]
- 38. Chekhonin VP, Zhirkov YA, Gurina OI, et al. PEGylated immunoliposomes directed against brain astrocytes. Drug Deliv 2005;12:1–6. [DOI] [PubMed] [Google Scholar]
- 39. Schnyder A, Huwyler J. Drug transport to brain with targeted liposomes. NeuroRx 2005;2:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci USA 1991;88:11460–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lasic DD, Papahadjopoulos D. Liposomes revisited. Science 1995;267:1275–1276. [DOI] [PubMed] [Google Scholar]
- 42. Wu D, Kang YS, Bickel U, Pardridge WM. Blood‐brain barrier permeability to morphine‐6‐glucuronide is markedly reduced compared with morphine. Drug Metab Dispos 1997;25:768–771. [PubMed] [Google Scholar]
- 43. Langston JW, Ballard P, Tetrud JW, et al. Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science 1983;219:979–980. [DOI] [PubMed] [Google Scholar]
- 44. Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP): seven cases. Neurology 1985;35:949–956. [DOI] [PubMed] [Google Scholar]
- 45. Ungerstedt U. Adipsia and aphagia after 6‐hydroxydopamine induced degeneration of the nigro‐striatal dopamine system. Acta Physiol Scand Suppl 1971;367:95–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 ANOVA analysis result.