

Summary
Aims
Antihistaminergic drugs have traditionally been used to treat vestibular disorders in the clinic. As a potential central target for antihistaminergic drugs, the inferior vestibular nucleus (IVN) is the largest subnucleus of the central vestibular nuclear complex and is considered responsible for vestibular‐autonomic responses and integration of vestibular, cerebellar, and multisensory signals. However, the role of histamine on the IVN, particularly the underlying mechanisms, is still not clear.
Methods
Using whole‐cell patch‐clamp recordings on rat brain slices, histamine‐induced effect on IVN neurons and the underlying receptor and ionic mechanisms were investigated.
Results
We found that histamine remarkably depolarized both spontaneous firing neurons and silent neurons in IVN via both histamine H1 and histamine H2 receptors. Furthermore, Na+–Ca2+ exchangers (NCXs) and background leak K+ channels linked to H1 receptors and hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channels coupled to H2 receptors comediate the histamine‐induced depolarization on IVN neurons.
Conclusion
These results demonstrate the multiple ionic mechanisms underlying the excitatory modulation of histamine/central histaminergic system on IVN neurons and the related vestibular reflexes and functions. The findings also suggest potential targets for the treatment of vestibular disorders in the clinic, at the level of ionic channels in central vestibular nuclei.
Keywords: H1 receptors, H2 receptors, Histamine, Inferior vestibular nucleus, Ionic mechanisms
Introduction
In the central nervous system, histamine, which synthetized solely in the tuberomammillary nucleus of the hypothalamus, acts as a general modulator for whole brain activity 1, 2, 3, 4, 5. The central histaminergic system extensively innervates almost the whole brain and plays an important role in many basic physiological functions, including arousal, endocrine functions, learning and memory, cognition, as well as motor control 1, 2, 3, 4, 5. Therefore, histamine is attracting more and more attention for its potential therapeutic value 6, 7, 8. In fact, antihistaminergic drugs have been used for almost a century in the clinical therapy of vestibular‐related diseases and their associated symptoms, such as imbalance, vertigo, motion sickness, nausea, and nystagmus 1, 7, 9, 10, 11. The therapeutic targets of antihistaminergic drugs include not only the peripheral vestibular organs, including the labyrinth in the inner ear 12, 13, but also the central vestibular nuclear complex, which holds a key position in the regulation of body balance and vestibular‐autonomic reflexes 14, 15, 16, 17, 18.
The inferior vestibular nucleus (IVN), also called the descending or spinal vestibular nucleus, is the largest nucleus of the vestibular nuclear complex. It receives primarily signals from the otolith, semicircular canals, as well as vermis of the cerebellum and sends direct projections to the vestibular nuclei, cerebellum, reticular formation, and spinal cord for regulating vestibular motor reflexes. In particular, neurons within the IVN possess axons that descend bilaterally in a position just off the midline near the dorsal surface of the pons and medulla through the medial vestibulospinal tract. These descending axons course caudally and enter the spinal cord, where they lie within the medial part of the ventral funiculus. This pathway projects to and modulates cervical and upper thoracic spinal motor neurons that innervate neck and head musculature 19, 20, 21, 22, 23. On the other hand, the outputs of IVN modulate the nucleus of the solitary tract, caudal part of parabrachial nucleus, nucleus ambiguous, and dorsal motor nucleus of the vagus to induce vestibular‐related autonomic responses. Lesion studies have revealed that during otolith stimulation in cats, the IVN is critical for vestibulo‐sympathetic reflex 24, 25. Moreover, the amount of Fos labeling neurons in the rat IVN was increased when animals performed Ferris‐wheel‐like rotations, which causes behavioral symptoms associated with motion sickness 26. Therefore, IVN may be a potential target for clinical treatment of vestibular symptoms and disorders.
Interestingly, it has been revealed that there is a moderately dense level of histaminergic fibers 8, 27, 28, 29, as well as histamine receptors 30, distributed throughout the IVN. Correspondingly, our previous extracellular recording study showed that histamine increased the firing rate of spontaneous firing IVN neurons by direct activation of histamine H1 and H2 receptors 30. However, the detailed ionic mechanisms underlying the action of histamine on IVN neurons remain largely unknown. On the other hand, studies on action and mechanisms of histamine in the central vestibular nuclei mostly concentrate on the medial vestibular nucleus (MVN) 9, 15, 17, 29, 31, 32 but neglect the IVN. In fact, unlike the MVN and other central vestibular subnuclei, IVN is a key station for integrating peripheral vestibular and cerebellar afferent inputs and holds an important integration role in vestibular motor and autonomic reflexes. Thus, in this study, the ionic mechanisms underlying the effect of histamine on IVN neurons were investigated. The results show that Na+–Ca2+ exchangers (NCXs) and background leak K+ channels coupled to H1 receptors and hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channels linked to H2 receptors comediate the histamine‐induced depolarization of IVN neurons.
Materials and Methods
Animals and Brain Slice Preparations
Brainstem slices were prepared from Sprague Dawley rats aged 12–14 days of either sex, in compliance with US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication 80‐23, revised 1996). All efforts were made to minimize the number of animals used and their suffering. After decapitation, according to the rat brain atlas 33, 300‐μM coronal brainstem slices containing the IVN were cut 30, 34, 35 and incubated in artificial cerebrospinal fluid (ACSF, composition in mM: 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 d‐glucose; pH = 7.4), equilibrated with 95% O2 and 5% CO2 at 35 ± 0.5°C for at least 1 h. Then, the slices were maintained at room temperature for about 20 min before recordings. For Na+ replacement experiments, the Na+‐free ACSF was as follows: 124 mM Tris–Cl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1.3 mM MgSO4, 26 mM NaHCO3, 2 mM CaCl2, and 10 mM d‐glucose (pH = 7.4).
Whole‐Cell Patch‐Clamp Recordings
Whole‐cell patch‐clamp recordings were performed as previously described 16, 17, 35. Briefly, the recording pipettes (3–5 MΩ) were filled with an internal solution (in nM): 140 K‐methylsulfate, 7 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 4 Na2‐ATP, 0.4 GTP‐Tris, adjusted to pH 7.25 with 1 M KOH. Patch‐clamp recordings were acquired with an Axopatch 700B amplifier (Axon Instruments, Foster City, CA, USA), captured through a Digidata‐1550 interface (Axon Instruments, Foster City, CA, USA), and analyzed by pClamp 10.4 (Axon Instruments). Under voltage‐clamp mode, the recording neurons were held at the potential of −60 mV. In slow‐ramp experiment, the voltage command ranged from −60 to −120 mV with dV/dt = −10 mV/s 17, 36, 37. Furthermore, to record depolarizing voltage sag, which is triggered by the activation of HCN channels, hyperpolarizing current steps (70–150 pA, 1 s) was employed under current‐clamp mode. The amplitude of voltage sag was calculated by subtracting the peak voltage amplitude from the steady‐state voltage.
Drugs
Drugs used in this study were as follows: histamine (10–100 μM), 2‐pyridylethylamine (2‐PyEA; 300 μM), BaCl2 (1 mM), and CsCl (2 mM) from Sigma (St. Louis, MO, USA); dimaprit (300 μM), mepyramine (3 μM), ranitidine (3 μM), ZD7288 (50 μM), KB‐R7943 (50 μM), and SN‐6 (10 μM) from Tocris (Bristol, UK); apamin (100 nM) from Abcam (Cambridge, UK); and TTX (0.3 μM) from Alomone Labs (Jerusalem, Israel). KB‐R7943 and SN‐6 were dissolved in DMSO and diluted to working concentrations in ACSF (DMSO <0.01%, final), while other drugs were prepared freshly in ACSF. All the drugs were applied by bath application as described previously 16, 17, 35.
Data Analysis
All data were analyzed with Origin 10 (MicroCal Software, Northampton, MA, USA) and statistical analysis results presented as means ± SEM. After having checked the normality of data (normality test, with a 5% confidence interval), Student's t‐test was employed for statistical comparison, and P‐values of <0.05 were considered to be significant.
Results
Histamine Depolarized IVN Neurons by the Activation of Both H1 and H2 Receptors
A total of 86 IVN neurons with the input resistance higher than 300 MΩ and whole‐cell membrane capacitance of 151 ± 6.4 pF were recorded in this study. Among the 86 recorded IVN neurons, 58 had spontaneous firing (mean firing rate = 8.7 ± 0.6 spikes/s, input resistance = 589.7 ± 96.1 MΩ, membrane capacitance = 148 ± 9.7 pF) and the remaining 28 were silent at rest (input resistance = 568.7 ± 120.6 MΩ, membrane capacitance = 155 ± 11.8 pF). These results are consistent with our and other previous reports 35, 38, 39 and reveal that neurons in the vestibular nuclear complex (including the IVN) have two different types of firing behavior. However, the electrophysiological properties, including input resistance and membrane capacitance, of these two populations seem to have no significant difference.
Of the 58 IVN neurons having spontaneous firing, histamine induced a significant increase in firing rate of 55 (55/58, 94.8%) neurons. As shown in Figure 1A, in current‐clamp recordings, brief (1 min) bath application of histamine (30 μM) increased the firing rate of the spontaneous firing IVN neurons from 8.1 ± 1.2 spikes/s to 15.8 ± 1.9 spikes/s (n = 5, P < 0.01). On the other hand, on 27 of the 28 (27/28, 96.4%) silent IVN neurons, 30 μM histamine evoked a strong depolarization, which increased neuronal firing (Figure 1B). The results indicate that histamine depolarizes a majority of spontaneous firing and silent neurons in the IVN. In addition, TTX was used to determine whether the histamine‐induced depolarization on IVN neurons was a direct postsynaptic effect. As shown in Figure 1C and D, although neuronal firing was impeded by 0.3 μM TTX, 10, 30, and 100 μM histamine still induced a significant depolarization of 5.0 ± 0.6, 10.1 ± 0.6, and 19.2 ± 1.4 mV on the IVN neurons (n = 6) in a concentration‐dependent manner, indicating that histamine‐induced excitation on IVN neurons was evoked by directly depolarizing the postsynaptic membrane, presumably through the activation of postsynaptic histamine receptors.
Figure 1.
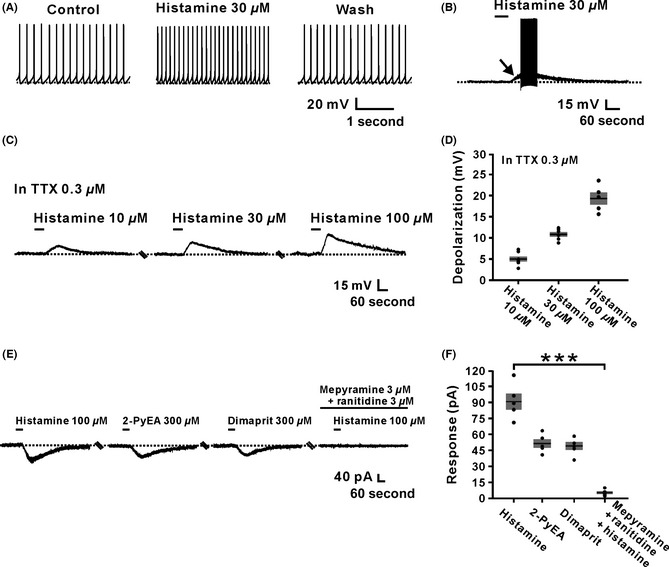
Histamine excited inferior vestibular nucleus (IVN) neurons by the activation of both H1 and H2 receptors. (A) Histamine (30 μM) increased the firing rate of an IVN spontaneous firing neuron. (B) Histamine excited an IVN silent neuron. The arrow indicates a strong depolarization induced by histamine. (C) Histamine induced a depolarization in dose‐dependent manner (10–100 μM) on one IVN neuron in the presence of 0.3 μM TTX. (D) Group data of tested IVN neurons (n = 6). (E) The histamine‐induced inward currents were mimicked by 2‐PyEA (300 μM) and dimaprit (300 μM), highly selective agonist for H1 and H2 receptors, respectively, and totally blocked by combined application of mepyramine and ranitidine, highly selective antagonists for H1 and H2 receptors, respectively. (F) Group data of the tested IVN neurons (n = 5, P = 0.0009). Data shown are means ± SEM. ***P < 0.001, significantly different from control. In this and the following figures, the short horizontal bars above the data indicated the 1‐min period of application of histamine or histamine receptor agonist, and the long horizontal bars denoted the exposure of the slice to histamine receptor and ionic channel antagonists.
Histamine exerts its postsynaptic action via three distinct receptor subtypes, namely H1, H2, and H4 receptors 1, 2, 3, 4, 5. However, in the IVN neurons, our previous immunohistochemical and extracellular electrophysiological studies have revealed that H1 and H2 instead of H4 receptors mediate the histamine‐induced increase in neuronal firing rates. In the present whole‐cell patch‐clamp recordings, this comediation of H1 and H2 receptors underlying the excitation of IVN neurons induced by histamine was further confirmed. As shown in Figure 1E and F, both 2‐PyEA and dimaprit, selective agonists for H1 and H2 receptors, respectively, mimicked the inward currents (52.4 ± 4.0 pA and 49.2 ± 3.6 pA, respectively) induced by histamine (−92.4 ± 7.5 pA) on IVN neurons (n = 5). Combined application of mepyramine and ranitidine, selective antagonists for H1 and H2 receptors, respectively, totally blocked the histamine‐evoked excitation (5.2 ± 1.4 pA, n = 5, P < 0.001). These results suggest that histamine excites IVN neurons by activating both H1 and H2 receptors, which not only confirmed our previous observations with extracellular recordings 30 but also provided an important clue for determining the downstream ionic mechanisms linked to these two histamine receptors.
Na+–Ca2+ Exchangers and Background Leak K+ Channels Comediate the Excitation Induced by the Activation of H1 Receptors on IVN Neurons
To evaluate the ionic basis underlying the excitation of IVN neurons induced by histamine, we employed 2‐PyEA and dimaprit to separate the current, respectively, mediated by H1 or H2 receptors. Firstly, we examined the change in input resistance when 2‐PyEA induced neuronal depolarization. As shown in Figure 2A, a step current (ranging from 0 to −12 pA in 3 pA steps) was run to acquire a series of corresponding changes in membrane potential, and then, an I–V curve was fitted by linear regression. Comparing the I–V curves before and after the application of 300 μM 2‐PyEA, we found a slight increase in membrane resistance accompanying the 2‐PyEA‐evoked depolarization (from 590.8 ± 68.1 MΩ to 663.8 ± 71.6 MΩ, n = 5; P < 0.05; Figure 2B and C), suggesting that 2‐PyEA induced a closure of ion channels on the postsynaptic membrane.
Figure 2.
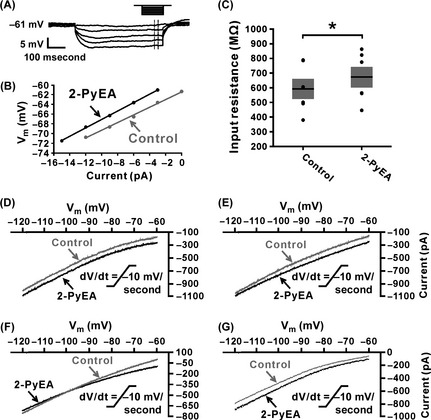
More than one ionic basis is involved in histamine H1 receptor‐mediated inward current. (A) The diagram showed how to acquire a series of voltage changes through applying a 500‐ms step current stimulation. (B) I–V curves were plotted using the data obtained from the protocol showed in (A) before (gray) and after (black) the application of 2‐PyEA (300 μM), a highly selective H1 receptor agonist. (C) Group data of changes in membrane resistance induced by 2‐PyEA (n = 5, P = 0.0386). Data shown are means ± SEM; *P < 0.05. (D–G): Four types of 2‐PyEA‐induced changes of I–V curves on inferior vestibular nucleus (IVN) neurons (n = 7, 4, 3, and 4, respectively) responding to a slow‐ramp command (dV/dt = −10 mV/s). The diversity of the 2‐PyEA‐induced changes in I–V relationships suggests that more than one ionic basis is involved in histamine H1 receptor‐mediated inward current on IVN neurons. Note that 2‐PyEA elicited an inward current that reversed near the calculated E k of −100 mV on 16.7% (3/18) of neurons (F).
Next, we applied slow‐ramp command tests to assess the dynamic features of current induced by the activation of H1 receptors. As shown in Figure 2D–G, there are four types of the I–V curves observed, indicating that more than one ionic mechanism are involved in the H1 receptor‐mediated depolarization on the IVN neurons. Notably, in 16.7% (3/18) of the recorded neurons, the two I–V curves intersect at −100 mV (Figure 2F), which means an inward current elicited by 2‐PyEA reverses near the calculated E k of −100 mV. The result, together with the 2‐PyEA‐induced increase in input resistance (Figure 2B and C), strongly suggests an involvement of K+ channels.
Furthermore, we used Ba2+, a broad spectrum blocker of potassium ion channels 40, to examine the dynamic properties of the 2‐PyEA‐induced current excluding K+ component. As shown in Figure 3A, after blocking K+ current, we only observed one change in the I–V curves induced by a slow‐ramp command in the absence and presence of 2‐PyEA. Subtracting the control from the current recorded during 2‐PyEA application yielded a difference current representing the 2‐PyEA‐induced current excluding the component of K+ (the right panel in Figure 3A). The difference current showed a trend of reversal at a potential more depolarized than −60 mV, which is highly consistent with the feature of NCX current. Considering that it is the Na+ influx contributing to the inward current produced by electrogenic NCX activation (3 Na+ ions entering in exchange for 1 Ca2+ ion extruded from the cell), we replaced the external Na+ with equimolar concentrations of Tris+, a relatively large organic cation that does not permeate Na+ channels and should not support Na+‐dependent current 17, 36. As shown in Figure 3B and C, perfusing the slice with BaCl2 (1 mM) partly blocked the 2‐PyEA‐elicited inward current (45.0% ± 10.5% of the control, n = 8, P < 0.01), and combined application of BaCl2 and Na+‐free ASCF totally blocked the current (n = 5, P < 0.001), suggesting a Na+‐dependent characteristic of the rest component of 2‐PyEA‐elicited inward current excluding the K+ current. It is well known that electrogenic NCXs are coupled to histamine H1 receptors in various brain structures/region 1, 2, 3, 4, 5, 41, including the MVN 16. To further confirm the involvement of electrogenic NCXs, we applied KB‐R7943 (50 μM), a potent and selective inhibitor for NCXs, and observed a partial inhibition of the 2‐PyEA‐induced inward current (64.9% ± 16.9% of the control, n = 5, P < 0.05, Figure 3D and E). Moreover, SN‐6 (10 μM), another specific blocker for NCXs, also partly blocked the 2‐PyEA‐elicited inward current (48.6% ± 6.6% of the control, n = 5, P < 0.05, Figure 3F and G). All of these results strongly suggest that NCXs participate in the mediation of excitation of IVN neurons induced by the activation of H1 receptors.
Figure 3.
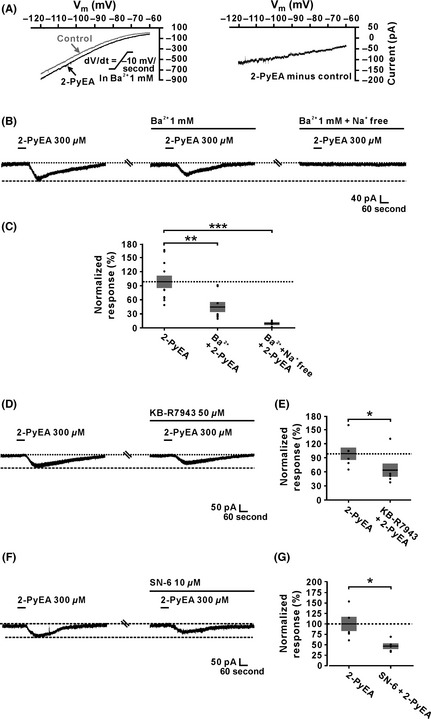
Activation of Na+–Ca2+ exchangers (NCXs) is involved in the histamine H1 receptor‐induced inward current on inferior vestibular nucleus neurons. (A) In the ACSF containing Ba2+, a blocker of K+ channels, the 2‐PyEA‐induced changes of I–V curves (the left panel) and current excluding K+ component (the right panel) in slow‐ramp command tests (dV/dt = −10 mV/s). (B and C) The 2‐PyEA‐induced inward current was partly blocked by BaCl2 (1 mM) (n = 8, P = 0.0087) and totally blocked by combined application of BaCl2 and Na+‐free ASCF (n = 5, P = 0.0006). (D and E) KB‐R7943 (50 μM), a blocker of NCXs, partly blocked the 2‐PyEA‐induced inward currents (n = 5, P = 0.0402). (F and G) The inward current induced by 2‐PyEA was partly blocked by SN‐6 (10 μM), another selective antagonist of NCXs (n = 5, P = 0.0316). Data shown are means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Additionally, we analyzed the characteristics of the 2‐PyEA‐induced K+ current component. Under the condition of blockage of NCXs by KB‐R7943, we used slow‐ramp command tests to obtain the I–V curves in the absence and presence of 2‐PyEA (Figure 4A1). The current induced by 2‐PyEA excluding the component of NCXs (n = 5, Figure 4A2 and A3) exhibited two significant features: a reversal potential near the calculated E k of −100 mV and a good linearity (R 2 = 0.985) over the voltage range tested. Considering 2‐PyEA increased membrane resistance (Figure 2C), the two features of 2‐PyEA‐induced K+ current indicate that the K+ channels blocked by histamine are the voltage‐insensitive background leak K+ channels 40. As there is still no selective and specific blocker for the voltage‐insensitive background leak K+ channel, and considering the background leak K+ channel and SK channel are the two types of K+ channels coupled to H1 receptors 1, 2, 41, we applied apamin, a selective antagonist of SK channels, and found that apamin has no effect on the excitatory response induced by 2‐PyEA (from 100.1% ± 13.0% to 95.4% ± 8.3%, n = 4, P = 0.538; Figure 4B and C). The results strongly indicate that the background leak K+ channels are the other ionic channels coupled to H1 receptors in the IVN neurons. Furthermore, as shown in Figure 4D–G, combined application of Ba2+ and KB‐R7943, or Ba2+ and SN‐6, respectively, nearly totally blocked the 2‐PyEA‐elicited inward current (in the test of Ba2+ and KB‐R7943, n = 9, P < 0.001; in the test of Ba2+ and SN‐6, n = 5, P < 0.001) on IVN neurons, suggesting a dual ionic mechanism, including the activation of NCXs and the closure of background leak K+ channels, underlying the activation of H1 receptors on IVN neurons.
Figure 4.
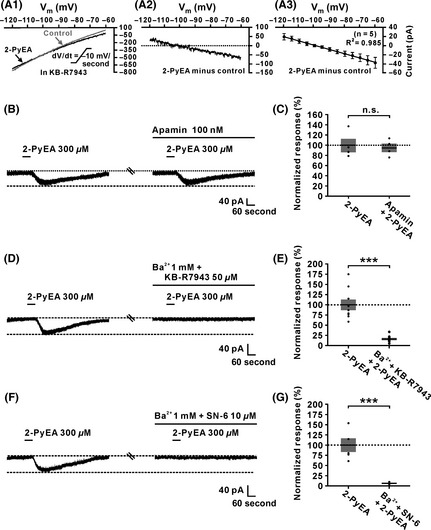
Closure of background leak K+ and activation of Na+–Ca2+ exchangers (NCXs) and channels comediated the histamine H2‐receptor‐induced inward current on inferior vestibular nucleus (IVN) neurons. (A1) In the ACSF containing KB‐R7943 (50 μM), a blocker for NCXs, 2‐PyEA‐induced changes of I–V curves of an IVN neuron in slow‐ramp command tests (dV/dt = −10 mV/s). (A2) The 2‐PyEA‐induced current excluding the component of NCXs on the same neuron. (A3) Group data on 5 tested IVN neurons. Note that in the presence of KB‐R7943, the 2‐PyEA‐induced current showed a good linearity (R 2 = 0.985) over the voltage range tested and reversed at the potential near the calculated E k of −100 mV, indicating histamine closed the background leak K+ channels (n = 5). (B and C) Apamin, the blocker of small‐conductance K+ channel (SK channel), had no influence on the inward current induced by 2‐PyEA (n = 4, P = 0.6437). (D) Combined application of KB‐R7943 and Ba2+ totally blocked 2‐PyEA‐induced inward current on an IVN neuron. (E) Group data of the tested IVN neurons (n = 5, P = 0.0003). (F) The inward current induced by 2‐PyEA was totally blocked by combined application of Ba2+ and SN‐6. (G) Group data of the tested IVN neurons (n = 5, P = 0.0002). Data shown are means ± SEM; n.s. indicates nonsignificant, ***P < 0.001.
Hyperpolarization‐Activated Cyclic Nucleotide‐Gated Channels were Coupled to H2 Receptors on IVN Neurons
The ionic mechanism underlying the current induced by the activation of H2 receptors was determined using dimaprit. As shown in Figure 5A, after the application of dimaprit (300 μM), membrane resistance decreased from 554.2 ± 76.9 MΩ to 476.4 ± 182.1 MΩ (n = 5, P < 0.01), suggesting that dimaprit caused an opening of ion channels on the postsynaptic membrane. Furthermore, in slow‐ramp command tests, the difference current representing the dimaprit‐induced current (Figure 5B) from the 5 IVN neurons exhibited a significant feature of hyperpolarization activation, which is consistent with the characteristics of current of HCN channels. As the activation of HCN channel also evokes a significant depolarizing voltage sag 42, the effect of dimaprit on voltage sag induced by hyperpolarizing current steps stimulation on IVN neurons was observed. The result showed that the sag was significantly increased by dimaprit (from 9.6 ± 0.2 mV to 12.1 ± 0.4 mV, n = 5, P < 0.01) (Figure 5C and D). Moreover, after blockade of HCN channels with ZD7288 (50 μM), a selective antagonist of HCN channels, the sag induced by the activation of HCN channels vanished and the enhancement of dimaprit on voltage sag was totally blocked (Figure 5C and D). Furthermore, we found that ZD7288 (50 μM) nearly totally blocked the dimaprit‐induced inward current (Figure 5E and F). In addition, CsCl (2 mM), another blocker for HCN channels, also fully abolished the dimaprit‐induced current (Figure 5G and H). All these results suggest that HCN channels contributed to the excitatory effect induced by the activation of H2 receptors on IVN neurons.
Figure 5.
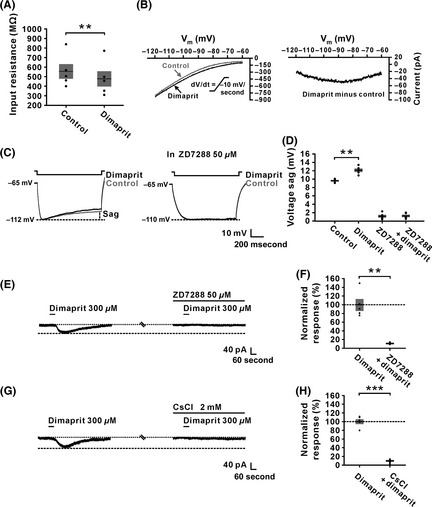
Hyperpolarization‐activated cyclic nucleotide‐gated channels were responsible for the inward current induced by the activation of H2 receptors on inferior vestibular nucleus (IVN) neurons. (A) Application of dimaprit (300 μM), a highly selective H2 receptor agonist, decreased the membrane resistance of recorded IVN neurons (n = 5, P = 0.0089). (B) The dimaprit‐induced changes of I–V curves (the left panel) and the difference current representing the current induced by the activation of H2 receptors (the right panel) in slow‐ramp command tests (dV/dt = −10 mV/s) showing a significant hyperpolarization activation feature of the current of HCN channels. (C) Inward rectification (sag) triggered by hyperpolarizing current steps on an IVN neuron was increased by dimaprit (300 μM) (the left panel). ZD7288 (50 μM), a highly selective HCN channel antagonist, totally blocked the increase in the sag induced by dimaprit (the right panel). (D) Group data of the tested IVN neurons (n = 5, P = 0.0062). (E and F) ZD7288 totally blocked the dimaprit‐induced inward current (n = 5, P = 0.0013). (G and H) CsCl, another antagonist of HCN channels, also totally blocked dimaprit‐induced inward current (n = 5, P = 0.0002). Data shown are means ± SEM; **P < 0.01, ***P < 0.001.
Moreover, selective histamine receptor antagonists were employed to confirm the above‐mentioned results observed using histamine receptor agonists. As shown in Figure 6A and B, the inward current induced by histamine was partly blocked by ranitidine, a selective antagonist for H2 receptor, and the residual current (i.e., histamine H1 receptor‐mediated current) was totally blocked by combined application of Ba2+ (1 mM) and KB‐R7943 (50 μM). The result confirmed that background leak K+ channels and NCXs comediated the excitatory effect of activation of H1 receptor on IVN neurons. On the other hand, the histamine‐induced inward current in mepyramine, a selective antagonist for H1 receptor, was totally blocked by ZD7288 (50 μM), confirming the result that HCN channels contributed to the excitatory effect induced by the activation of H2 receptors on IVN neurons (Figure 6C and D). Finally, to confirm NCXs, background leak K+ channels, and HCN channels are responsible for the histamine‐induced excitation on IVN neurons, ZD7288, KB‐R7943, and Ba2+ were applied together. As shown in Figure 6E and F, the histamine‐induced inward current was totally blocked by combined application of ZD7288, KB‐R7943, and Ba2+. Thus, all these results strongly suggested that NCXs and background leak K+ channels coupled to H1 receptors, as well as HCN channels linked to H2 receptors, comediated the depolarization of histamine on IVN neurons.
Figure 6.
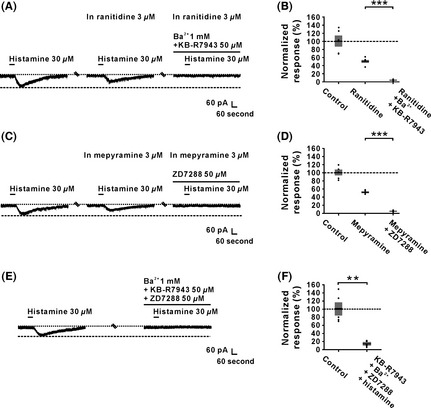
Na+–Ca2+ exchanger and leak K+ channel coupled to H1 receptor and HCN channels coupled to H2 receptor comediate the histamine‐induced excitation on inferior vestibular nucleus neurons. (A and B) The inward currents induced by histamine (30 μM) were partly blocked by ranitidine (3 μM), a highly selective H2 receptor antagonist, and the residual currents were totally blocked by combined application of Ba2+ and KB‐R7943 (n = 5, P = 0.0004). (C and D) The inward currents induced by histamine (30 μM) were partly blocked by mepyramine (3 μM), a highly selective H1 receptor antagonist, and the residual currents were totally blocked by ZD7288 (n = 5, P = 0.0006). (E and F) Combined application of ZD7288, KB‐R7943, and Ba2+ totally blocked the postsynaptic inward current induced by histamine (n = 5, P = 0.0023). Data shown are means ± SEM; **P < 0.01, ***P < 0.001.
Discussion
The vestibular nuclear complex in the brainstem is the most important node in the central processing of vestibular sensory information 9, 43. As a sensorimotor complex, central vestibular nuclei integrate multiple vestibular, motor, and visual signals to adjust posture and compensate head and eye movements. On the other hand, dysfunction of vestibular nuclear circuits results in vestibular disorders with characteristic symptoms, including vertigo, disorientations, postural imbalances, nausea, and vomiting 11, 44, 45, 46. Thus, the central vestibular nuclei are considered to be critical central targets for antihistaminergic drugs, which have been traditionally used to treat vestibular disorders in clinic. In fact, histamine extensively excites neurons in all the four vestibular nuclei 15, 16, 17 and actively modulates the vestibular‐related reflexes and vestibular compensation 9, 10, 46. Moreover, histamine can increase the release of acetylcholine 47, 48, 49, which has been implicated in the vestibular‐related symptoms of motion sickness in humans 50, 51. It has been suggested that histamine may also change the degree of activation on glycinergic and GABAergic neurotransmissions before and after unilateral labyrinthectomy to participate in the regulation of vestibular compensation 31. However, these direct homogeneous excitatory effect and indirect actions of histamine on different subnuclear neurons in the vestibular nuclei are mediated by various histamine receptors. Here, we further provide electrophysiological evidence, for the first time, that NCXs and background leak K+ channels coupled to H1 receptors as well as HCN channels linked to H2 receptors mediate the direct depolarization induced by histamine on the IVN neurons, which is different from the ionic mechanisms underlying the excitation of histamine on MVN neurons 17.
There are four major subnuclei in the central vestibular nuclei: the lateral (LVN), medial, superior, and inferior vestibular nucleus. Whole‐cell patch‐clamp recordings have revealed that histamine depolarizes and excites spontaneous firing and silent neurons in the LVN 16, type‐A and type‐B neurons in the MVN 17, as well as neurons in the IVN (the present study). Interestingly, in the LVN, only histamine H2 receptors are involved in the excitatory effect of histamine, whereas both H1 and H2 receptors comediate the histamine‐induced postsynaptic excitation in all the other three subnuclei, including IVN [30 and the present study]. However, the ionic mechanisms underlying the histamine‐induced depolarization on MVN and IVN neurons are not in complete accord.
Numerous types of ion channels have been reported to modulate the excitability of central vestibular nuclear neurons 52, 53, 54. Here, we found two types of ion channels/exchangers are involved in the histamine‐induced excitation on both IVN and MVN neurons. In comparison with MVN, both NCXs coupled to H1 receptors and HCN channels linked to H2 receptors contribute to the histamine‐induced excitation on MVN and IVN neurons. These same receptors and ionic mechanisms make histamine to modulate the activity of neurons in different vestibular subnucleus in a similar way to maintaining an appropriate level of excitability of whole vestibular nuclei. NCXs, which have a highly positive reversal potential 36, 37, guarantee a powerful driving force for depolarizing membrane potential. On the other hand, HCN channels, a kind of pacemaker channels activated during hyperpolarization, help accelerate membrane depolarization and the generation of neuronal activity 42. Therefore, we speculate that both NCXs and HCN channels endow histamine/histaminergic afferent inputs with an ability to rapidly modulate the neuronal activity of IVN and MVN. Through the activation of NCXs and HCN channels linked to H1 and H2 receptors, respectively, histamine/histaminergic inputs may effectively depolarize the IVN and MVN neurons and quickly increase their firing rates, and subsequently play a critical modulatory role on vestibular reflexes and functions.
Unlike the ion mechanisms underlying the histamine‐evoked depolarization on MVN neurons, histamine also induces the closure of background leak K+ channels coupled to H1 receptors on IVN neurons. The well‐known role of background leak K+ currents is to stabilize resting membrane potential and counterbalance depolarization 40, 55. Moreover, the inactivation of background leak K+ channels will consequently increase the neuronal membrane input resistance, which produces an effective amplification of the presynaptic input signals. Considering that the IVN plays a critical role in the integration of vestibular, cerebellar, and multisensory signals in vestibular nuclei, we suggest that an additional involvement of background leak K+ channels in the downstream mechanisms of IVN neurons’ excitation induced by histamine may help the neurons to hold an appropriate excitability and responsiveness through its modulation of neuronal membrane properties, so as to guarantee that the neurons produce an accurate integration of multiple impacting signals. Therefore, through differential ionic mechanisms, histamine/histaminergic inputs may finely modulate the activity and sensitivity of IVN and MVN neurons to meet their different functions in vestibular reflexes.
In the clinic, many histaminergic compounds have been used in preventing and treating Ménière's disease, vertigo, and motion sickness for a long time, such as betahistine, diphenhydramine, meclizine, cyclizine, and promethazine 56, 57, 58. However, all these clinical medications focus on histamine receptors but neglect the downstream ionic exchangers and channels. Interestingly, ion channel agents, such as calcium channel blockers, that is, nimodipine, cinnarizine, and flunarizine, may be useful in the treatment of vestibular‐related diseases 56. The mechanisms by which calcium channel blockers might prevent vertigo are still unclear. It is hypothesized that calcium channel blockers might act on calcium channels expressed in periphery vestibular hair cells and/or central vestibular nuclear neurons. From this perspective, the ion channels and exchangers linked to histamine receptors in the central vestibular nuclei, including the NCXs, background leak K+ channels, and HCN channels in the IVN demonstrated in the present study, may provide potential targets for treating vestibular disorders 14.
In summary, the present study reveals that histamine directly depolarizes IVN neurons by the mediation of NCXs and background leak K+ channels coupled to H1 receptors and HCN channels linked to H2 receptors. Through switching the functional status of ion channels and exchangers, histamine may effectively bias the excitability of the IVN neurons. Thus, in this way, the histaminergic afferent inputs may actively participate in the regulation of vestibular reflexes and vestibular disorders through the modulation of IVN neurons. From this perspective, the role of these ion channels and exchangers coupled to histamine receptors in the central vestibular neurons and their related vestibular functions and dysfunctions needs to be assessed further to look for more specific and effective targets for the treatment of vestibular disorders.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We thank Mr. Hong‐Zhao Li for technical assistance. The work was supported by grants 31171050, 31330033, 91332124, 31471112, J1103512, J1210026, and NSFC/RGC Joint Research Scheme 31461163001 from the National Natural Science Foundation of China; SRFDP/RGC ERG grant 20130091140003, NCET Program, and Fundamental Research Funds for the Central Universities 020814380004 and 20620140565 from the State Educational Ministry of China; grant BK2011014 from the Natural Science Foundation of Jiangsu Province, China; grant 2014M550283 from the China Postdoctoral Science Foundation; and grants 1202004C and 1302006C from the Jiangsu Planned Projects for Postdoctoral Research Funds.
The first two authors contributed equally to this work.
References
- 1. Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev 2008;88:1183–1241. [DOI] [PubMed] [Google Scholar]
- 2. Haas HL, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci 2003;4:121–130. [DOI] [PubMed] [Google Scholar]
- 3. Lin JS, Anaclet C, Sergeeva OA, Haas HL. The waking brain: an update. Cell Mol Life Sci 2011;68:2499–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haas HL, Lin JS. Waking with the hypothalamus. Pflugers Arch 2012;463:31–42. [DOI] [PubMed] [Google Scholar]
- 5. Li B, Zhu JN, Wang JJ. Histaminergic afferent system in the cerebellum: structure and function. Cerebellum Ataxias 2014;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin JS, Sergeeva OA, Haas HL. Histamine H3 receptors and sleep‐wake regulation. J Pharmacol Exp Ther 2011;336:17–23. [DOI] [PubMed] [Google Scholar]
- 7. Tiligada E, Kyriakidis K, Chazot PL, Passani MB. Histamine pharmacology and new CNS drug targets. CNS Neurosci Ther 2011;17:620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lacour M, Tighilet B. Vestibular compensation in the cat: the role of the histaminergic system. Acta Otolaryngol 2000;544(Suppl):15–18. [DOI] [PubMed] [Google Scholar]
- 9. Bergquist F, Dutia MB. Central histaminergic modulation of vestibular function – a review. Sheng Li Xue Bao 2006;58:293–304. [PubMed] [Google Scholar]
- 10. Lacour M, Sterkers O. Histamine and betahistine in the treatment of vertigo: elucidation of mechanisms of action. CNS Drugs 2001;15:853–870. [DOI] [PubMed] [Google Scholar]
- 11. Balaban CD. Vestibular autonomic regulation (including motion sickness and the mechanism of vomiting). Curr Opin Neurol 1999;12:29–33. [DOI] [PubMed] [Google Scholar]
- 12. Chávez H, Vega R, Soto E. Histamine (H3) receptors modulate the excitatory amino acid receptor response of the vestibular afferents. Brain Res 2005;1064:1–9. [DOI] [PubMed] [Google Scholar]
- 13. Soto E, Chávez H, Valli P, Benvenuti C, Vega R. Betahistine produces post‐synaptic inhibition of the excitability of the primary afferent neurons in the vestibular endorgans. Acta Otolaryngol 2001;545(Suppl):19–24. [DOI] [PubMed] [Google Scholar]
- 14. Lacour M, van de Heyning PH, Novotny M, Tighilet B. Betahistine in the treatment of Ménière's disease. Neurops ychiatr Dis Treat 2007;3:429–440. [PMC free article] [PubMed] [Google Scholar]
- 15. Wang JJ, Dutia MB. Effects of histamine and betahistine on rat medial vestibular nucleus neurones: possible mechanism of action of anti‐histaminergic drugs in vertigo and motion sickness. Exp Brain Res 1995;105:18–24. [DOI] [PubMed] [Google Scholar]
- 16. Zhang J, Han XH, Li HZ, Zhu JN, Wang JJ. Histamine excites rat lateral vestibular nuclear neurons through activation of post‐synaptic H2 receptors. Neurosci Lett 2008;448:15–19. [DOI] [PubMed] [Google Scholar]
- 17. Zhang XY, Yu L, Zhuang QX, Peng SY, Zhu JN, Wang JJ. Postsynaptic mechanisms underlying the excitatory action of histamine on medial vestibular nucleus neurons in rats. Br J Pharmacol 2013;170:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhuang QX, Wu YH, Wu GY, Zhu JN, Wang JJ. Histamine excites rat superior vestibular nuclear neurons via postsynaptic H1 and H2 receptors in vitro . Neurosignals 2013;21:174–183. [DOI] [PubMed] [Google Scholar]
- 19. Balaban CD, Porter JD. Neuroanatomic substrates for vestibulo‐autonomic interactions. J Vestib Res 1998;8:7–16. [PubMed] [Google Scholar]
- 20. Barmack NH. Central vestibular system: vestibular nuclei and posterior cerebellum. Brain Res Bull 2003;60:511–541. [DOI] [PubMed] [Google Scholar]
- 21. Shiba K, Siniaia MS, Miller AD. Role of ventral respiratory group bulbospinal expiratory neurons in vestibular‐respiratory reflexes. J Neurophysiol 1996;76:2271–2279. [DOI] [PubMed] [Google Scholar]
- 22. Uchino Y, Kudo N, Tsuda K, Iwamura Y. Vestibular inhibition of sympathetic nerve activities. Brain Res 1970;22:195–206. [DOI] [PubMed] [Google Scholar]
- 23. Xu F, Zhuang J, Zhou TR, Gibson T, Frazier DT. Activation of different vestibular subnuclei evokes differential respiratory and pressor responses in the rat. J Physiol 2002;544:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yates BJ, Miller AD. Properties of sympathetic reflexes elicited by natural vestibular stimulation: implications for cardiovascular control. J Neurophysiol 1994;71:2087–2092. [DOI] [PubMed] [Google Scholar]
- 25. Yates BJ, Siniaia MS, Miller AD. Descending pathways necessary for vestibular influences on sympathetic and inspiratory outflow. Am J Physiol 1995;268:R1381–R1385. [DOI] [PubMed] [Google Scholar]
- 26. Cai YL, Ma WL, Wang JQ, Li YQ, Li M. Excitatory pathways from the vestibular nuclei to the NTS and the PBN and indirect vestibulo‐cardiovascular pathway from the vestibular nuclei to the RVLM relayed by the NTS. Brain Res 2008;1240:96–104. [DOI] [PubMed] [Google Scholar]
- 27. Iwase M, Homma I, Shioda S, Nakai Y. Histamine immunoreactive neurons in the brain stem of the rabbit. Brain Res Bull 1993;32:267–272. [DOI] [PubMed] [Google Scholar]
- 28. Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiol Rev 1991;71:1–51. [DOI] [PubMed] [Google Scholar]
- 29. Tighilet B, Mourre C, Trottier S, Lacour M. Histaminergic ligands improve vestibular compensation in the cat: behavioural, neurochemical and molecular evidence. Eur J Pharmacol 2007;568:149–163. [DOI] [PubMed] [Google Scholar]
- 30. Peng SY, Zhuang QX, He YC, Zhu JN, Wang JJ. Histamine excites neurons of the inferior vestibular nucleus in rats by activation of H1 and H2 receptors. Neurosci Lett 2013;541:87–92. [DOI] [PubMed] [Google Scholar]
- 31. Bergquist F, Ruthven A, Ludwig M, Dutia MB. Histaminergic and glycinergic modulation of GABA release in the vestibular nuclei of normal and labyrinthectomised rats. J Physiol 2006;577:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tighilet B, Trottier S, Mourre C, Lacour M. Changes in the histaminergic system during vestibular compensation in the cat. J Physiol 2006;573:723–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates, 6th edn San Diego, CA: Academic Press, 2007. [Google Scholar]
- 34. Darlington CL, Gallagher JP, Smith PF. In vitro electrophysiological studies of the vestibular nucleus complex. Prog Neurobiol 1995;45:335–346. [DOI] [PubMed] [Google Scholar]
- 35. Yu L, Zhang XY, Chen ZP, Zhuang QX, Zhu JN, Wang JJ. Orexin excites rat inferior vestibular nuclear neurons via co‐activation of OX1 and OX2 receptors. J Neural Transm 2014;122:747–755. [DOI] [PubMed] [Google Scholar]
- 36. Wu M, Zaborszky L, Hajszan T, van den Pol AN, Alreja M. Hypocretin/orexin innervation and excitation of identified septohippocampal cholinergic neurons. J Neurosci 2004;24:3527–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J, Li B, Yu L, et al. A role for orexin in central vestibular motor control. Neuron 2011;69:793–804. [DOI] [PubMed] [Google Scholar]
- 38. Babalian A, Vibert N, Assie G, Serafin M, Mühlethaler M, Vidal PP. Central vestibular networks in the guinea‐pig: functional characterization in the isolated whole brain in vitro . Neuroscience 1997;81:405–426. [DOI] [PubMed] [Google Scholar]
- 39. Shao M, Popratiloff A, Hirsch JC, Peusner KD. Presynaptic and postsynaptic ion channel expression in vestibular nuclei neurons after unilateral vestibular deafferentation. J Vestib Res 2009;19:191–200. [DOI] [PubMed] [Google Scholar]
- 40. McCormick DA, Williamson A. Modulation of neuronal firing mode in cat and guinea pig LGNd by histamine: possible cellular mechanisms of histaminergic control of arousal. J Neurosci 1991;11:3188–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol 2001;63:637–672. [DOI] [PubMed] [Google Scholar]
- 42. Pape HC. Queer current and pacemaker: the hyperpolarization‐activated cation current in neurons. Annu Rev Physiol 1996;58:299–327. [DOI] [PubMed] [Google Scholar]
- 43. Cullen KE. The vestibular system: multimodal integration and encoding of self‐motion for motor control. Trends Neurosci 2012;35:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yabe T, de Waele C, Serafin M, et al. Medial vestibular nucleus in the guinea‐pig: histaminergic receptors. II. An in vivo study. Exp Brain Res 1993;93:249–258. [DOI] [PubMed] [Google Scholar]
- 45. Highstein SM, Holstein GR. The anatomy of the vestibular nuclei. Prog Brain Res 2006;151:157–203. [DOI] [PubMed] [Google Scholar]
- 46. Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol 2005;76:349–392. [DOI] [PubMed] [Google Scholar]
- 47. Horii A, Takeda N, Mochizuki T, Okakura‐Mochizuki K, Yamamoto Y, Yamatodani A. Effects of vestibular stimulation on acetylcholine release from rat hippocampus: an in vivo microdialysis study. J Neurophysiol 1994;72:605–611. [DOI] [PubMed] [Google Scholar]
- 48. Altinbas B, Yilmaz MS, Savci V, Jochem J, Yalcin M. Centrally injected histamine increases posterior hypothalamic acetylcholine release in hemorrhage‐hypotensive rats. Auton Neurosci 2015;187:63–69. [DOI] [PubMed] [Google Scholar]
- 49. Kraus MM, Prast H, Philippu A. Influence of the hippocampus on amino acid utilizing and cholinergic neurons within the nucleus accumbens is promoted by histamine via H₁ receptors. Br J Pharmacol 2013;170:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wood CD, Graybiel A. Theory of antimotion sickness drug mechanisms. Aerosp Med 1972;43:249–252. [PubMed] [Google Scholar]
- 51. Kohl RL, Homick JL. Motion sickness, a modulatory role for the central cholinergic nervous system. Neurosci Biobehav Rev 1983;7:73–85. [DOI] [PubMed] [Google Scholar]
- 52. Gittis AH, du Lac S. Firing properties of GABAergic versus non‐GABAergic vestibular nucleus neurons conferred by a differential balance of potassium currents. J Neurophysiol 2007;97:3986–3996. [DOI] [PubMed] [Google Scholar]
- 53. Gittis AH, Moghadam SH, du Lac S. Mechanisms of sustained high firing rates in two classes of vestibular nucleus neurons: differential contributions of resurgent Na, Kv3, and BK currents. J Neurophysiol 2010;104:1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Serafin M, Khateb A, de Waele C, Vidal PP, Mühlethaler M. Low threshold calcium spikes in medial vestibular nuclei neurons in vitro: a role in the generation of the vestibular nystagmus quick phase in vivo? Exp Brain Res 1990;82:187–190. [DOI] [PubMed] [Google Scholar]
- 55. Enyedi P, Czirják G. Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 2010;90:559–605. [DOI] [PubMed] [Google Scholar]
- 56. Hain TC, Uddin M. Pharmacological treatment of vertigo. CNS Drugs 2003;17:85–100. [DOI] [PubMed] [Google Scholar]
- 57. Wersinger E, Gaboyard‐Niay S, Travo C, et al. Symptomatic treatment of vestibular deficits: therapeutic potential of histamine H4 receptors. J Vestib Res 2013;23:153–159. [DOI] [PubMed] [Google Scholar]
- 58. Lacour M. Restoration of vestibular function: basic aspects and practical advances for rehabilitation. Curr Med Res Opin 2006;22:1651–1659. [DOI] [PubMed] [Google Scholar]