

Summary
Aims
Our previous studies showed that L‐3‐n‐butylphthalide (L‐NBP), an extract from seeds of Apium graveolens Linn (Chinese celery), improved cognitive ability in animal models of cerebral ischemia, vascular dementia, and Alzheimer's disease (AD). It is well known that cognitive deficit of AD is caused by synaptic dysfunction. In this study, we investigated the effect of L‐NBP on hippocampal synaptic function in APP/PS1 AD transgenic mice and related mechanisms.
Methods
Eighteen‐month‐old APP/PS1 transgenic (Tg) mice were administrated 15 mg/kg L‐NBP by oral gavage for 3 months. Synaptic morphology and the thickness of postsynaptic density (PSD) in hippocampal neurons were investigated by electron microscope. The dendritic spines, Aβ plaques, and glial activation were detected by staining. The expressions of synapse‐related proteins were observed by Western blotting.
Results
L‐NBP treatment significantly increased the number of synapses and apical dendritic thorns and the thickness of PSD, increased the expression levels of synapse‐associated proteins including PSD95, synaptophysin (SYN), β‐catenin, and GSK‐3β, and attenuated Aβ plaques and neuroinflammatory responses in aged APP/PS1 Tg mice.
Conclusion
L‐NBP may restore synaptic and spine function in aged APP Tg mice through inhibiting Aβ plaques deposition and neuroinflammatory response. Wnt/β‐catenin signaling pathway may be involved in L‐NBP‐related restoration of synaptic function.
Keywords: L‐3‐n‐butylphthalide, Synaptic plasticity, Mitochondrial function, Aβ, Golgi staining
Introduction
Alzheimer's disease (AD) is regarded as the major cause of dementia among elderly people. Multiple pathological factors have been found in the postmortem brains of patients with AD, such as Aβ plaques, tau‐protein tangles, neuroinflammation, neuron loss, and synapse loss. However, the etiology and pathogenesis of AD still remain unknown 1. At present, the therapeutic options for AD mainly include the acetylcholine esterase inhibitors and the NMDA receptor antagonist 2, which offer symptomatic improvement instead of effective cure or prevention. With the failure of clinical trials targeting Aβ, including semagacestat 3, 4, bapineuzumab 5, and solanezumab 6, it is important to critically re‐examine the design strategy of single‐target drug based on amyloid hypothesis. The researchers have become increasingly aware that a compound simultaneously targeting multiple risk factors of AD would be potentially useful to halt the progression of the multifactorial diseases 7.
L‐3‐n‐butylphthalide (L‐NBP) is a small molecule compound extracted from seeds of Apium graveolens Linn, Chinese celery, and its chemical structure is shown in Figure 1. Recently, racemic dl‐3‐n‐butylphthalide (dl‐NBP) has been approved as a drug for stroke treatment by China Food and Drug Administration. Interestingly, it has been reported that L‐NBP has more potent neuroprotective functions than dl‐NBP. L‐NBP could reverse cognitive impairment in AD animal models 8, 9, 10, 11 and reduce Aβ levels in APP transgenic mice 10 as well as in neuroblastoma SK‐N‐SH cells overexpressing wild‐type human APP695 12. L‐NBP also decreased neuronal loss induced by Aβ in primary cultured neurons and neuroblastoma SH‐SY5Y cells 13, 14, 15. Moreover, L‐NBP promoted hippocampal neurogenesis and improved behavioral recovery after cerebral ischemia in rats 16. These findings suggested that L‐NBP might be a potential multifunctional drug candidate for AD.
Figure 1.
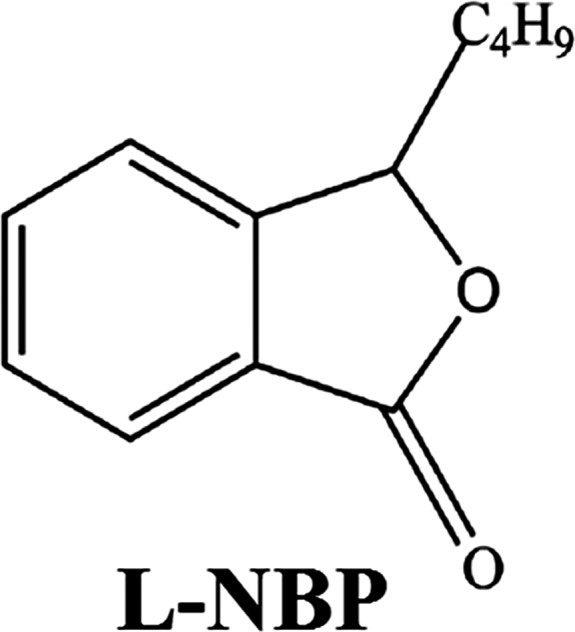
The chemical structure of L‐NBP.
Increasing evidence has shown a correlation between synaptic loss and cognitive decline in AD 17, 18, 19, 20, 21. As the hub of cognition 22, 23, the hippocampus is the principal focus of synaptic failure in AD. The loss of dendritic spines was apparent in the hippocampi of postmortem AD brains 24.
Although L‐NBP treatment showed promising effects at an early stage of AD, it is unclear whether L‐NBP could affect the synaptic plasticity in late stage of AD with well‐developed plaques. In this study, we investigated the effect of L‐NBP on hippocampal synaptic plasticity in aged APP/PS1 transgenic mice. L‐NBP improved synaptic dysfunction, reduced neuron loss, and restored spine density deficits in aged transgenic mice displaying severe Aβ pathology. In addition, there was a significant reduction on plaque burden and microgliosis in aged transgenic mice after L‐NBP treatment. Interestingly, L‐NBP upregulated the levels of PSD95, SYN, and β‐catenin. Overall, these results suggested that L‐NBP might be a potential therapeutic option for AD.
Materials and Methods
Animals and Treatment
L‐NBP (purity > 98.5%) was obtained from Institute of Materia Medica and diluted in vegetable oil at a concentration of 3 mg/mL. APP/PS1 double‐transgenic mice were obtained from Jackson Laboratory (strain name B6C3‐Tg(APPswe, PSEN1dE9) 85Dbo/J)25. Mice were housed in plastic cages at 23 ± 1°C with a 12‐h light/dark cycle and free access to water and food. All experiments were approved and performed in accordance with the institutional guidelines of the Experimental Animal Center of the Chinese Academy of Medical Science (Beijing, China).
Eighteen‐month‐old male APP/PS1 transgenic mice and age‐matched wild‐type mice were randomly divided into three groups: control‐treated wild‐type mice, control‐treated APP/PS1 mice, and L‐NBP‐treated APP/PS1 mice. L‐NBP‐treated group received L‐NBP by oral gavage once daily for 3 months at a dose of 15 mg/kg body weight. Control‐treated groups received vegetable oil alone. The general health of all the mice was monitored daily. The body weight of each mouse was recorded every week. Finally, 6 (WT control), 7 (Tg control), and 8 (Tg L‐NBP) mice were used to perform the experiments.
Electron Microscopy
The mice were anesthetized, and the brains were taken out. The hippocampus was dissected from left hemispheres. The right hemisphere was used for immunohistochemical staining. The CA1 and CA3 hippocampal sections were sliced into 1‐mm slices. They were postfixed with 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h, washed with 0.1 M PBS for three times (10 min each), and then exposed to 1% osmium tetroxide for 2 h. After several subsequent washes with water, the tissues were dehydrated with gradient alcohol (2 × 10 min 50%, 2 × 10 min 70%, 2 × 10 min 90%, 2 × 10 min 100%). Then, the sections were embedded in epon resin, and the randomly selected ultrathin sections were stained with uranyl acetate and lead citrate. To quantify the number of synapses and measure the thickness of PSD, three slides per animal and three fields within CA1 and CA3 area per slide were randomly chosen. Each field was imaged with 20,000× and 50,000× magnification using a transmission electron microscope (H‐7650; HITACHI, Tokyo, Japan). The number of synapses and the thickness of PSD in hippocampus CA1 and CA3 were analyzed by an experimenter blind to treatment and genotype using Image Pro Plus 6.0 software (Media Cybernetics Inc., Bethesda, MD, USA) from 15 photographs per mouse.
Golgi Staining for Dendritic Spines
Golgi staining was performed using the Rapid Golgi Staining Kit (FD Neuro Technologies, Columbia, NY, USA) according to the manufacturer's instructions. The tissue blocks were prepared with the mixture of solutions A and B equally at room temperature for 2 weeks in the dark, and then transferred to solution C at 4°C and kept for at least 2 days and up to 7 days. During this period, solutions AB and C were renewed within the first 24 h. The brains were rapidly frozen and sliced into sagittal sections at 200 μm thickness using a Leica microtome (Leica RM 2135; Wetzlar, Germany). Each section was mounted onto gelatin‐coated glass slides using solution C and allowed to dry at room temperature. The tissue sections were stained according to the manufacturer's protocol. The image of secondary and tertiary apical dendrites of hippocampal pyramidal neurons in CA1 and DG was viewed using bright field microscopy. The number of apical spines on hippocampal CA1 pyramidal neurons and DG neurons was counted by an experimenter blind to genotype and treatment group. Only neurons with a clear mushroom‐like body and a distinctive shaft, impregnated fully, and no obvious truncated dendrites were included. For each group, at least twelve randomly chosen neurons were analyzed. The second‐ or third‐order dendritic branches were selected for quantitative analysis. The number of spines was determined per micrometer of dendritic length from 10 photographs per mouse under 1000× magnification in the digitized images. The spine density was expressed as the number of thorns/10 μm of dendrite.
Immunofluorescence Staining
The OCT‐embedded blocks were cut serially on a Leica microtome into 40‐μm thick sagittal sections. For the staining of Aβ plaque and glial, the sections were incubated with 10% normal donkey serum to block nonspecific binding, followed by an overnight incubation with the primary antibodies, 6E10 (1:500, Covance, Princeton, NJ, USA) and anti‐Iba1 (1:200; Wako Pure Chemical Industries, Ltd., Chuo‐ku, Japan) at 4°C. After washing, the sections were incubated with secondary antibodies, including Alexa Fluor 488‐conjugated donkey anti‐mouse IgG (1:200; Thermo Fisher Scientific, Waltham, MA, USA) and Alexa Fluor 594‐conjugated donkey anti‐rat IgG (1:200; Thermo Fisher Scientific) for 2 h at room temperature. The sections were rinsed and transferred on slides, and the cover slipped in an antifading agent. The images were acquired using a Nikon camera mounted on a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan) and analyzed using Image Pro Plus 6.0 software. The threshold of detection was held constantly during analysis. The percentage of Aβ‐occupied area in the whole hippocampal area was calculated for 4–6 equidistant sections per mouse.
Western Blotting Analysis
Equal amounts of protein (60 μg) per lane were loaded and performed electrophoresis, and then transferred to polyvinylidene fluoride membranes. The membrane was blocked with 5% nonfat milk for 2 h, and then incubated with the following primary antibodies overnight at 4°C (anti‐SYN (1:2000; Abcam, Cambridge, UK), anti‐PSD95 (1:1000; Cell Signaling Technology, Danvers MA, USA), anti‐β‐catenin (1:1000; Cell Signaling Technology), and anti‐GSK‐3β (Ser9) (1:1000; Cell Signaling Technology)). The membranes were rinsed in Tris‐buffered saline (TBS) with 0.1% Tween‐20 (TBS‐T), incubated with horseradish peroxidase‐conjugated secondary antibodies at room temperature for 1 h, and detected using an enhanced chemiluminescence (ECL) kit. Densitometric evaluation was analyzed using the Quantity One image analysis software (Bio‐Rad, Hercules, CA, USA). Actin was used as a loading control.
Statistical Analysis
All data were expressed as mean ± the standard error of the means (SEM). One‐way analysis of variance (ANOVA) followed by post hoc LSD test was used for multiple comparisons (SPSS version 16.0, SPSS Inc., Chicago, IL, USA). Statistical significance was set to a value of P < 0.05.
Results
L‐NBP Increased the Number of Synapses and the Thickness of PSD in Hippocampal CA1 and CA3 Areas of APP/PS1 Mice
To explore the effect of L‐NBP on synapse function, we examined the number of synapses and the thickness of PSD using electron microscopy. Only synapses with clearly identifiable postsynaptic density, synaptic cleft, and presynaptic vesicles were counted. A single synapse was identified when a single presynaptic terminal was associated with multiple postsynaptic density on the same postsynaptic element. However, it was counted as more than one synapse, if a presynaptic terminal clearly formed synapses with more than one postsynaptic element 26. The results showed that the number of synapses in the Tg control group was significantly reduced compared with WT control group in CA1 (6.65 ± 0.43 vs. 16.68 ± 2.64, P < 0.01) and CA3 (9.94 ± 0.36 vs. 17.06 ± 0.99, P < 0.01) areas. L‐NBP markedly increased the number of synapses in CA1 by 94.59% and CA3 by 46.48% in APP/PS1 mice (Figure 2A–C). The thickness of PSD in the Tg control group showed a significant decrease compared with wild‐type mice, while L‐NBP administration increased the PSD thickness in APP/PS1 transgenic mice in CA1 area (WT: 59.57 ± 3.11, Tg: 38.51 ± 2.94, Tg L‐NBP: 59.95 ± 3.62) and CA3 area (WT: 68.34 ± 2.50, Tg: 34.44 ± 1.47, Tg L‐NBP: 55.39 ± 2.37) (Figure 2A, D–E). The results indicated that the chronic administration of L‐NBP could significantly increase the synaptic density and the thickness of PSD in the hippocampus region.
Figure 2.
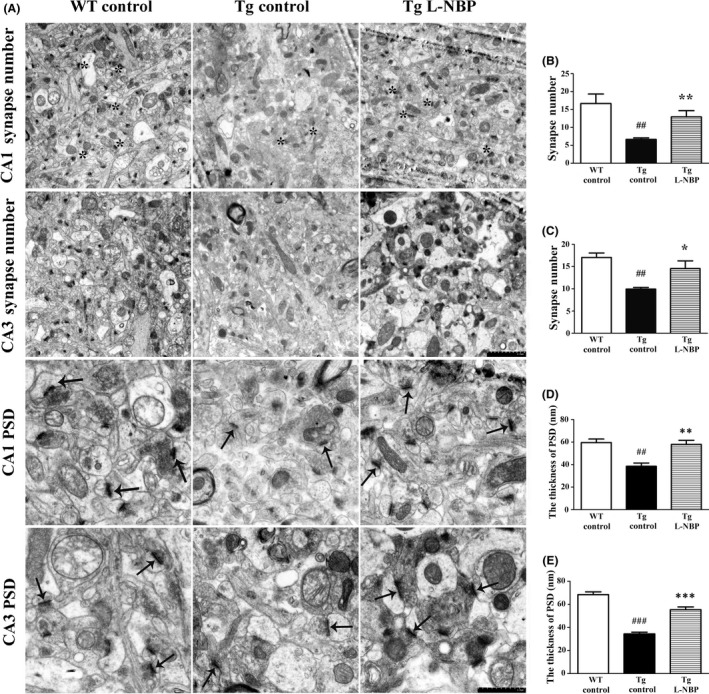
Effect of L‐NBP on synapse number and the thickness of PSD in the hippocampus CA1 and CA3 areas. (A) Electron micrographs showing synapse number (Magnification: ×20,000) and the thickness of PSD (Magnification: ×50,000) in one visual field of hippocampus CA1 and CA3 area. Synapses were indicated by asterisk (*). The thicknesses of PSD are indicated by arrowheads. (B–E) Quantification of synapse number (B–C) and the thicknesses of PSD (D–E). Data were expressed as the mean ± SEM. n = 3 mice per group. ## P < 0.01, ### P < 0.001, versus control‐treated wild‐type group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control‐treated APP/PS1 group.
L‐NBP Reversed Neuron Reduction, Mitochondria, and Golgi Apparatus Damage in Hippocampal CA1 and CA3 Areas of APP/PS1 Mice
The neurodegeneration and synaptic alterations are considered as the principal factors of cognitive decline in AD 27 and associated with alterations of neuron, mitochondria, and Golgi apparatus 28, 29, 30 in hippocampus. In this study, we attempted to reveal the morphological alterations of the neuron, mitochondria, and Golgi apparatus in APP/PS1 mice directly. In WT mice, plenty of neurons showed normal morphology, large and round body with clear membrane structure and well‐distributed chromatin, as well as mitochondria showing regular shapes with dense ridge. However, in APP/PS1 transgenic mice, abundant neurons showed degenerating characteristics with distinctive chromatin condensations, intact nuclear, disrupted cell membrane, and cytoplasmic blebbings. Moreover, a considerable number of mitochondria with disruption of the cristae were also observed, and the large number of cisternae of Golgi apparatus appeared to be fragmented in APP/PS1 transgenic mice. L‐NBP treatment could rescue these damages (Figure 3).
Figure 3.
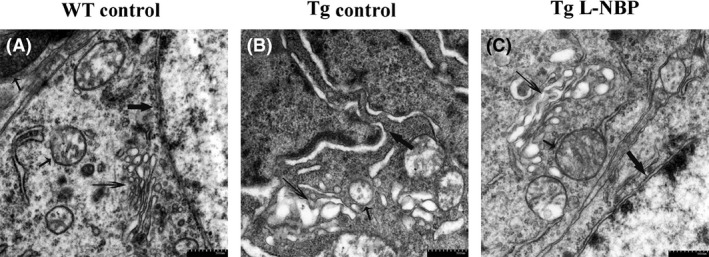
L‐NBP treatment rescued the hippocampal neurons, mitochondria, and Golgi apparatus damage. (A–C) Electron micrographs of hippocampal neurons of WT control (A), Tg control (B), Tg L‐NBP (C). Neuron of WT control animals (A) displayed smooth nuclear membrane and occasional condensations of nuclear chromatin (thick arrow) and had oval mitochondria with occasional lost cristae (short thin arrow) and regular cisternae of Golgi apparatus (long thick arrow). But in neuron of Tg control animals (B), nuclear membrane showed enfolding and disruption, nuclear shrinkages and chromatin clumping, mitochondria with fewer cristae appeared dilated. Golgi apparatus cisternae appeared to be fragmented. (C) L‐NBP treatment could rescue the damage. n = 3 mice per group.
L‐NBP Rescued the Reduction of Spine Density in APP/PS1 Mice
Another aspect of synaptic integrity is the number of spine numbers, which is considered to be correlate with excitatory synapses 31. Spine density of secondary and tertiary apical dendritic segments in hippocampal CA1 and DG regions was assessed using Golgi staining (Figure 4). In the sagittal section, neurons impregnated with the Golgi–Cox solution randomly display neurons that could reflect neural cells number (Figure 4A). The dendritic spines in dendritic compartments of both CA1 and DG neurons in the WT control group were regular and intense (10.72 ± 0.41 and 13.06 ± 0.14), but APP/PS1 mice showed a significant reduction in the number of dendritic spines (6.94 ± 0.28 and 7.59 ± 0.75). L‐NBP significantly increased the spine density in apical compartments of CA1 and DG segments in APP/PS1 mice (11.71 ± 0.37 and 13.93 ± 0.33) (Figure 4A, C). Together, these data indicated that L‐NBP ameliorated both structural and functional synaptic impairments of aged AD mice.
Figure 4.
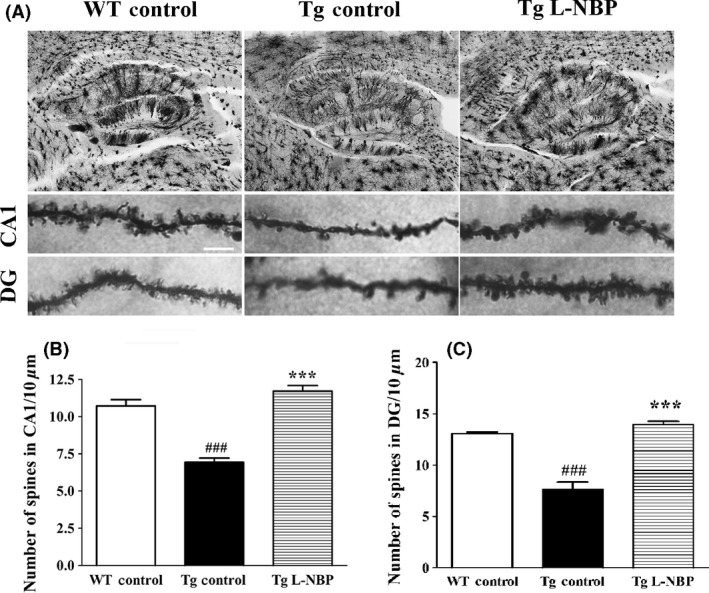
L‐NBP treatment improved spine density in the hippocampus of APP/PS1 mice. (A) Golgi staining of the hippocampal area (top), L‐NBP‐mediated preservation of the number of spines per given dendritic length in the hippocampus CA1 (middle) and DG (bottom of APP/PS1 mice. Scale bar=5 μm. (B–C) Quantitative analysis of spine density in CA1 (B) and DG (C). The spine number was decreased in the APP/PS1 mice treated with vehicle. L‐NBP rescued the impaired spine density both in CA1 and DG area. Data were expressed as the mean ± SEM. n = 7–12 dendrites per animal, 3–5 animals per group. ### P < 0.001 versus control‐treated wild‐type group. ***P < 0.001 versus control‐treated APP/PS1 group.
L‐NBP Attenuated Aβ Plaque Deposition in APP/PS1 Mice
Aβ accumulation and plaque deposition may induce synaptic failure, dendritic and axonal atrophy, and neuronal death 32, 33. L‐NBP seemed promote the structural synaptic integrity, so we wondered if this effect could accompany with ameliorating Aβ deposition in aged APP/PS1 mice. Our previous study has demonstrated that L‐NBP could lower Aβ levels and increase αAPPs levels in transgenic AD mice by regulating APP processing toward the nonamyloidogenic pathway 10. In this study, L‐NBP treatment reduced the levels of cerebral Aβ deposition by 66.60% (P < 0.01) compared with the control‐treated group in the APP/PS1 mice (Figure 5A–B). Furthermore, the fibrous structure of Aβ was surrounded by a large number of terminal neurite damage of degenerated neurons under the electron microscope (Figure 5D).
Figure 5.
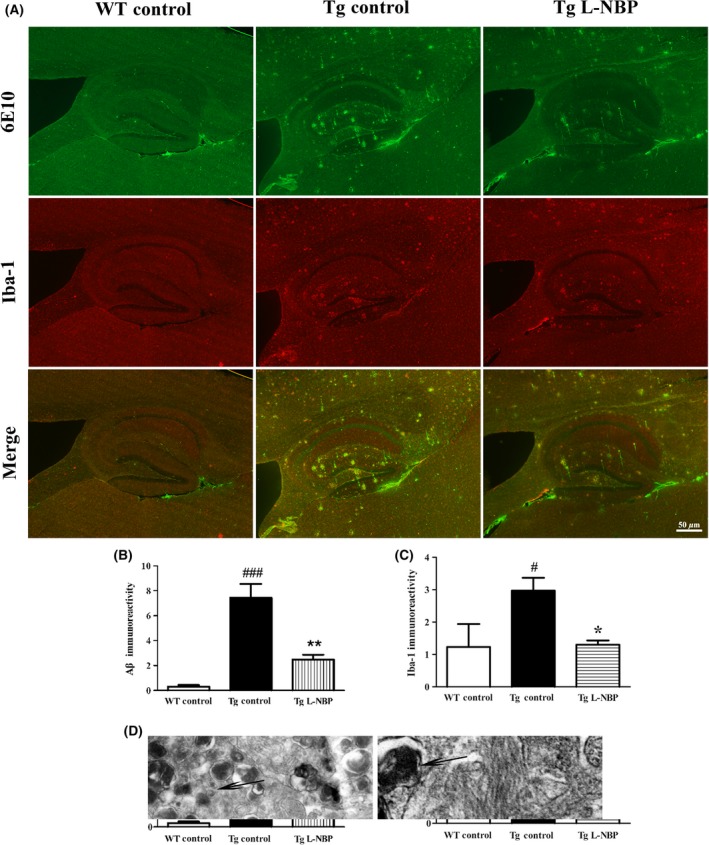
L‐NBP treatment reduced Aβ aggregation and microglia activation in the hippocampi of APP/PS1 mice. (A) Representative image of total Aβ plaque staining by 6E10 (top), activated microglia staining by Iba1 (middle) and the merged image (bottom) in hippocampus in control‐treated wild‐type mice (left), control‐treated APP/PS1 mice (middle), and L‐NBP‐treated APP/PS1 mice (right). (B–C) Quantitative image analysis was performed for 6E10 immunoreactivity (B) and Iba1 (C). (D) The fibrous structure of Aβ (short thick arrow) under the electron microscope was surrounded by a large number of degenerated neurons, which was reflected by the abundant of terminal neurite damage (long thick arrow). Data were expressed as the mean ± SEM. n = 3 mice per group. # P < 0.05, ### P < 0.001 versus control‐treated wild‐type group. ∗ P < 0.05, ∗∗ P < 0.01 versus control‐treated APP/PS1 group.
L‐NBP Inhibited Microglia Activation in APP/PS1 Mice
Neuroinflammation is considered as an essential player in the etiology of AD 34. Activated microglia and astrocytes are regarded as main neuroinflammatory cell types 35. Aβ deposition was always surrounded by extensive microgliosis in patients with AD and mouse models 36. We investigated the effect of L‐NBP in amyloid‐dependent gliosis by immunostaining using Iba‐1, 6E10, and GFAP. Activated microglia were detected in close proximity to Aβ plaques in APP/PS1 mice brain (Figure 5), in accordance with the previous reports. L‐NBP treatment significantly decreased the population of activated microglia surrounding amyloid plaques by 56.15% (P < 0.05) (Figure 5A and C). But no change was detected in astrocyte activation (data not shown).
L‐NBP Upregulated the Expressions of Synapse‐Associated Proteins
To further explore the mechanism of L‐NBP on protecting synapse in AD mouse model, we detected the expressions of synapse‐associated proteins in the hippocampus of APP/PS1 mice, including PSD95, SYN, β‐catenin, and phosphorylaed‐GSK‐3β (Ser9). The expressions of PSD and SYN were significantly decreased in APP/PS1 mice by 26.36% (P < 0.05) and 37.86% (P < 0.05), respectively, compared to WT control mice (Figure 6A–C). The expressions of β‐catenin and phosphorylaed‐GSK‐3β (Ser9)/total GSK‐3β were decreased in APP/PS1 mice by 12.12% and 35.04% compared to WT control mice, but there were no significant differences (Figure 6A, D–E). L‐NBP treatment significantly upregulated the levels of PSD95 by 46.05% and β‐catenin by 27.59% in APP/PS1 mice (Figure 6A, B, and D). However, L‐NBP did not change the expressions of SYN and phosphorylaed‐GSK‐3β (Ser9)/total GSK‐3β (Figure 6A, C, and E).
Figure 6.
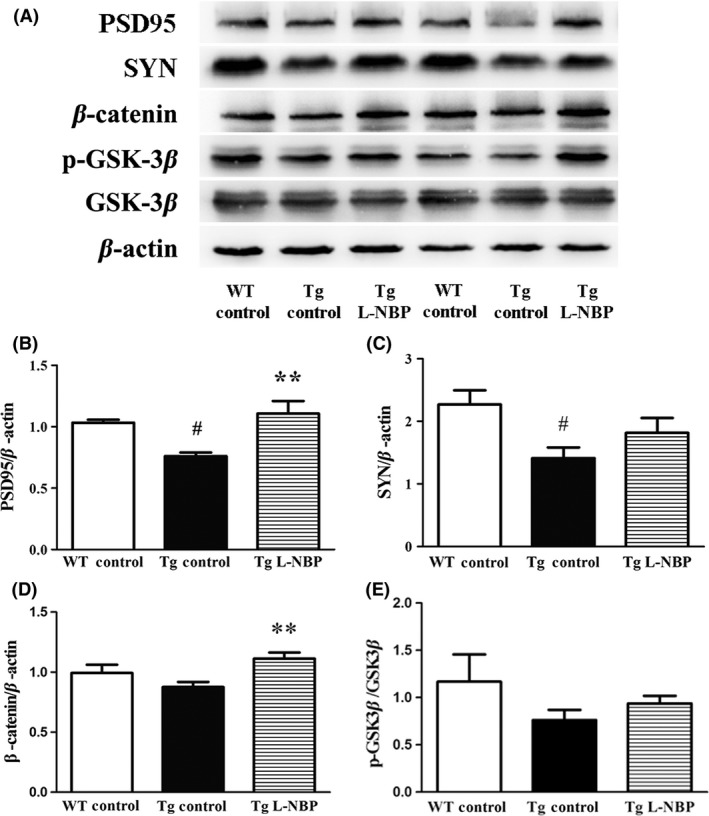
Effect of L‐NBP on the levels of postsynaptic density protein‐95 (PSD95), synaptophysin (SYN), β‐catenin, and phospho‐glycogen synthase kinase‐3β (GSK‐3β) in the mouse hippocampus. (A) Representative image of Western blots. (B–E) PSD95 (B), SYN(C), β‐catenin (D), and p‐GSK‐3β/total GSK‐3β (E) for quantitative analysis, normalized to the β‐actin band. Data were expressed as the mean ± SEM. n = 3–5 mice per group. # P < 0.05, versus control‐treated wild‐type group. **P < 0.01 versus control‐treated APP/PS1 group.
Discussion
AD is the most prevalent neurodegenerative disorder in aging populations worldwide, but the pathogenesis is still incompletely understood. Synaptic dysfunction and subsequent loss of neuronal cells happen at the early stage in neurodegenerative disorder 37. Recently, the concept of synaptic repair has been brought forward to tackle or ameliorate pathophysiology for AD 38. L‐NBP has been reported to inhibit oxidative injury, neuronal apoptosis, and glial activation, reduce Aβ plaque, and improve cognitive defects in a number of AD animal models. But, it is unknown whether L‐NBP can affect synaptic plasticity. In this study, we demonstrated the therapeutic potential of L‐NBP on modifying synaptic plasticity after the onset of amyloid pathology. We chose 18‐month‐old APP/PS1 mice because they displayed abundant amyloid plaques and might mimic the late pathological state in patients with AD. We found that L‐NBP improved the cognitive ability in both 6‐month and 12‐month APP/PS1 mice previously. In this study, we focused on the effect of L‐NBP on hippocampal synaptic plasticity and neuropathology. The dose and duration of treatment of L‐NBP were similar to the previous reports 11.
The number of synapses and spine density are related to synaptic plasticity 39. The PSD, a cytoskeletal specialization within the postsynaptic membrane, can organize neurotransmitter receptors and determine the physiological strength or weight of a synapse 40. In this study, we found that the number of synapses and the thickness of PSD were significantly reduced in APP/PS1 mice, and L‐NBP treatment reversed these changes. Dendritic spines receive most of the excitatory synaptic input and contribute to synaptic connections 41, 42. Here, we found that L‐NBP rescued the spine density reduction in APP/PS1 mice. Consistent with this, our previous studies showed that potassium 2‐(1‐hydroxypentyl)‐benzoate (PHPB), a derivative of L‐NBP, enhanced long‐term potentiation (LTP) in APP/PS1 mice 43. We did not detect whether L‐NBP affected the synaptic plasticity in this study due to the limitation of animals. However, the recent findings showed that L‐NBP had no effect on the number of synapses and the thickness of PSD of hippocampus in 6‐month‐old WT mice (data not shown). In addition, PHPB did not show any effect on LTP in WT mice 43. Thus, we deduced that L‐NBP might not regulate synapse functions in WT mice.
Aβ extracted from AD brains potently impaired synaptic structure and function in the hippocampus 44, and Aβ might be involved in aggravating the synaptic dysfunction of AD 33. Our previous study showed that L‐NBP treatment significantly lowered total Aβ plaque deposition in triple transgenic AD mice 10. In the present study, we also found that L‐NBP significantly decreased Aβ plaque load, and the population of reactive microglia surrounding amyloid plaques in the aged AD mice. Aβ accumulation and plaque deposition resulted in synaptic failure, dendritic and axonal atrophy, and neuronal damage 32, 33. Coincidentally, we observed large number of degenerated neurons surrounding the amyloid fibrils, indicating that neuronal death may be closely related to Aβ plaque.
Oxidative stress is one of the key factors in the pathophysiology of AD 34, 45, 46, 47, and mitochondrial dysfunction may play a pivotal role in oxidative stress. Mitochondria accumulate at the presynaptic and postsynaptic sites. The functional synapses require mitochondria to supply ATP for neurotransmission 48, and impaired synaptic plasticity is likely due to increased oxidative stress in APP/PS1 mouse brain 49. L‐NBP has been reported to significantly inhibit mitochondrial membrane potential reduction and reactive oxygen species production 13. In this study, L‐NBP was found to rescue mitochondria damage in neurons of aged APP/PS1 mice. It suggested that L‐NBP might improve synaptic impairment by regulating mitochondrial function.
In the present study, we randomly chose three mice per group to do immunohistochemical staining and electron microscopy. It is well known that a larger sample size is necessary to reach solid conclusions. Actually, we designed five samples per group for each experiment at the beginning of the study. However, some mice gradually died because of poor physical condition of aging. To obtain the most reliable data, we randomly took 15 photographs per mouse in electron microscopy detection and 10 photographs per mouse in Golgi staining and performed the statistically analysis. They showed a clear tendency of L‐NBP effects. In the future, a study with a larger sample size will be performed to confirm the results.
PSD95 is located preferentially in dendritic spines and plays a key role in regulating the formation, function, and plasticity of excitatory synapses 50. As a presynaptic marker, SYN is closely related to synaptogenesis 51. GSK‐3β and β‐catenin are involved in neurite outgrowth 51, 52. Stable β‐catenin is a critical mediator of dendritic morphology, and closely relevant to axonal length, dendritic processes, as well as the density of dendritic spines in hippocampal neurons 53, 54. β‐catenin also acts as a core factor in the canonical Wnt signal transduction pathway, and the intracellular content and phosphorylation status of β‐catenin determined the downstream cascade of Wnt pathway 55. Wnt signaling plays a key role in neuronal synapse formation and remodeling by recruiting presynaptic and postsynaptic components 56. Dysfunctional Wnt signaling is characterized by reduced β‐catenin level and constitutively active GSK‐3β. GSK‐3β is activated by the phosphorylation at Tyr216 and inhibited by phosphorylating Ser9. Activated GSK‐3β not only phosphorylates tau to facilitate tangle formation, but also promotes β‐catenin degradation. Inhibition of GSK‐3β results in neuroprotective effects in AD models. In this study, L‐NBP treatment significantly increased the expression of PSD95 and β‐catenin, suggesting that L‐NBP might reverse synaptic dysfunction via modulating Wnt/β‐catenin pathway.
Conclusion
In this study, we first demonstrated that L‐NBP improved the synaptic functions and reduced Aβ plaque load and microglia activation in aged APP/PS1 AD transgenic mice. Additionally, L‐NBP partly reversed the reduction of synapse‐associated proteins. Wnt/β‐catenin signaling pathway or cell adhesion correlated with β‐catenin might be involved in L‐NBP‐modulating synaptic plasticity. L‐NBP might be a potential therapeutic option for AD.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Xiao‐Liang Wang, Email: wangxl@imm.ac.cn.
Ying Peng, Email: ypeng@imm.ac.cn.
References
- 1. Peng D, Pan X, Cui J, Ren Y, Zhang J. Hyperphosphorylation of tau protein in hippocampus of central insulin‐resistant rats is associated with cognitive impairment. Cell Physiol Biochem 2013;32:1417–1425. [DOI] [PubMed] [Google Scholar]
- 2. Felsenstein KM, Candelario KM, Steindler DA, Borchelt DR. Regenerative medicine in Alzheimer's disease. Transl Res 2014;163:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cummings J. What can be inferred from the interruption of the semagacestat trial for treatment of Alzheimer's disease? Biol Psychiatry 2010;68:876–878. [DOI] [PubMed] [Google Scholar]
- 4. Extance A. Alzheimer's failure raises questions about disease‐modifying strategies. Nat Rev Drug Discov 2010;9:749–751. [DOI] [PubMed] [Google Scholar]
- 5. Blennow K, Zetterberg H, Rinne JO, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol 2012;69:1002–1010. [DOI] [PubMed] [Google Scholar]
- 6. Lannfelt L, Moller C, Basun H, et al. Perspectives on future Alzheimer therapies: amyloid‐beta protofibrils – a new target for immunotherapy with BAN2401 in Alzheimer's disease. Alzheimers Res Ther 2014;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carreiras MC, Mendes E, Perry MJ, Francisco AP, Marco‐Contelles J. The multifactorial nature of Alzheimer's disease for developing potential therapeutics. Curr Top Med Chem 2013;13:1745–1770. [DOI] [PubMed] [Google Scholar]
- 8. Peng Y, Xu S, Chen G, Wang L, Feng Y, Wang X. l‐3‐n‐Butylphthalide improves cognitive impairment induced by chronic cerebral hypoperfusion in rats. J Pharmacol Exp Ther 2007;321:902–910. [DOI] [PubMed] [Google Scholar]
- 9. Peng Y, Xing C, Xu S, et al. L‐3‐n‐butylphthalide improves cognitive impairment induced by intracerebroventricular infusion of amyloid‐beta peptide in rats. Eur J Pharmacol 2009;621:38–45. [DOI] [PubMed] [Google Scholar]
- 10. Peng Y, Sun J, Hon S, et al. L‐3‐n‐butylphthalide improves cognitive impairment and reduces amyloid‐beta in a transgenic model of Alzheimer's disease. J Neurosci 2010;30:8180–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng Y, Hu Y, Xu S, et al. L‐3‐n‐butylphthalide reduces tau phosphorylation and improves cognitive deficits in AbetaPP/PS1‐Alzheimer's transgenic mice. J Alzheimers Dis 2012;29:379–391. [DOI] [PubMed] [Google Scholar]
- 12. Peng Y, Hu Y, Xu S, Feng N, Wang L, Wang X. L‐3‐n‐butylphthalide regulates amyloid precursor protein processing by PKC and MAPK pathways in SK‐N‐SH cells over‐expressing wild type human APP695. Neurosci Lett 2011;487:211–216. [DOI] [PubMed] [Google Scholar]
- 13. Lei H, Zhao CY, Liu DM, et al. l‐3‐n‐Butylphthalide attenuates beta‐amyloid‐induced toxicity in neuroblastoma SH‐SY5Y cells through regulating mitochondrion‐mediated apoptosis and MAPK signaling. J Asian Nat Prod Res 2014;16:854–864. [DOI] [PubMed] [Google Scholar]
- 14. Peng Y, Hu Y, Feng N, Wang L, Wang X. L‐3‐n‐butyl‐phthalide alleviates hydrogen peroxide‐induced apoptosis by PKC pathway in human neuroblastoma SK‐N‐SH cells. Naunyn Schmiedebergs Arch Pharmacol 2011;383:91–99. [DOI] [PubMed] [Google Scholar]
- 15. Peng Y, Xing C, Lemere CA, et al. l‐3‐n‐Butylphthalide ameliorates beta‐amyloid‐induced neuronal toxicity in cultured neuronal cells. Neurosci Lett 2008;434:224–229. [DOI] [PubMed] [Google Scholar]
- 16. Yang LC, Li J, Xu SF, et al. L‐3‐n‐butylphthalide Promotes Neurogenesis and Neuroplasticity in Cerebral Ischemic Rats. CNS Neurosci Ther 2015;21:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572–580. [DOI] [PubMed] [Google Scholar]
- 18. Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer's disease and other dementias. Neurology 1989;39:355–361. [DOI] [PubMed] [Google Scholar]
- 19. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 20. Knafo S, Alonso‐Nanclares L, Gonzalez‐Soriano J, et al. Widespread changes in dendritic spines in a model of Alzheimer's disease. Cereb Cortex 2009;19:586–592. [DOI] [PubMed] [Google Scholar]
- 21. Baloyannis SJ, Mauroudis I, Manolides SL, Manolides LS. Synaptic alterations in the medial geniculate bodies and the inferior colliculi in Alzheimer's disease: a Golgi and electron microscope study. Acta Otolaryngol 2009;129:416–418. [DOI] [PubMed] [Google Scholar]
- 22. Dusek JA, Eichenbaum H. The hippocampus and memory for orderly stimulus relations. Proc Natl Acad Sci U S A 1997;94:7109–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 1992;99:195–231. [DOI] [PubMed] [Google Scholar]
- 24. Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol 2004;33:377–387. [DOI] [PubMed] [Google Scholar]
- 25. Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co‐expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 2001;17:157–165. [DOI] [PubMed] [Google Scholar]
- 26. Silva I, Mello LE, Freymuller E, Haidar MA, Baracat EC. Estrogen, progestogen and tamoxifen increase synaptic density of the hippocampus of ovariectomized rats. Neurosci Lett 2000;291:183–186. [DOI] [PubMed] [Google Scholar]
- 27. Bitner RS, Bunnelle WH, Anderson DJ, et al. Broad‐spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci 2007;27:10578–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baloyannis SJ, Costa V, Michmizos D. Mitochondrial alterations in Alzheimer's disease. Am J Alzheimers Dis Other Demen 2004;19:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baloyannis SJ. Mitochondria are related to synaptic pathology in Alzheimer's disease. Int J Alzheimers Dis 2011;2011:305395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baloyannis SJ, Manolidis SL, Manolidis LS. Synaptic alterations in the vestibulocerebellar system in Alzheimer's disease–a Golgi and electron microscope study. Acta Otolaryngol 2000;120:247–250. [DOI] [PubMed] [Google Scholar]
- 31. Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 2002;39:29–54. [DOI] [PubMed] [Google Scholar]
- 32. Fol R, Braudeau J, Ludewig S, et al. Viral gene transfer of APPsalpha rescues synaptic failure in an Alzheimer's disease mouse model. Acta Neuropathol 2016;131:247–266. [DOI] [PubMed] [Google Scholar]
- 33. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 34. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140:918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gerenu G, Liu K, Chojnacki JE, et al. Curcumin/melatonin hybrid 5‐(4‐hydroxy‐phenyl)‐3‐oxo‐pentanoic acid [2‐(5‐methoxy‐1H‐indol‐3‐yl)‐ethyl]‐amide ameliorates AD‐like pathology in the APP/PS1 mouse model. ACS Chem Neurosci 2015;6:1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meraz‐Rios MA, Toral‐Rios D, Franco‐Bocanegra D, Villeda‐Hernandez J, Campos‐Pena V. Inflammatory process in Alzheimer's Disease. Front Integr Neurosci 2013;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lacor PN, Buniel MC, Chang L, et al. Synaptic targeting by Alzheimer's‐related amyloid beta oligomers. J Neurosci 2004;24:10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF‐based synaptic repair as a disease‐modifying strategy for neurodegenerative diseases. Nat Rev Neurosci 2013;14:401–416. [DOI] [PubMed] [Google Scholar]
- 39. Bender RA, Zhou L, Wilkars W, et al. Roles of 17ss‐estradiol involve regulation of reelin expression and synaptogenesis in the dentate gyrus. Cereb Cortex 2010;20:2985–2995. [DOI] [PubMed] [Google Scholar]
- 40. Bressloff PC, Earnshaw BA. A dynamic corral model of receptor trafficking at a synapse. Biophys J 2009;96:1786–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rochefort NL, Konnerth A. Dendritic spines: from structure to in vivo function. EMBO Rep 2012;13:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure‐stability‐function relationships of dendritic spines. Trends Neurosci 2003;26:360–368. [DOI] [PubMed] [Google Scholar]
- 43. Li PP, Wang WP, Liu ZH, et al. Potassium 2‐(1‐hydroxypentyl)‐benzoate promotes long‐term potentiation in Abeta1‐42‐injected rats and APP/PS1 transgenic mice. Acta Pharmacol Sin 2014;35:869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shankar GM, Li S, Mehta TH, et al. Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamilton A, Holscher C. The effect of ageing on neurogenesis and oxidative stress in the APP(swe)/PS1(deltaE9) mouse model of Alzheimer's disease. Brain Res 2012;1449:83–93. [DOI] [PubMed] [Google Scholar]
- 46. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging 2000;21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hernandez‐Zimbron LF, Rivas‐Arancibia S. Oxidative stress caused by ozone exposure induces beta‐amyloid 1‐42 overproduction and mitochondrial accumulation by activating the amyloidogenic pathway. Neuroscience 2015;304:340–348. [DOI] [PubMed] [Google Scholar]
- 48. Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 2006;26:7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rui Y, Gu J, Yu K, Hartzell HC, Zheng JQ. Inhibition of AMPA receptor trafficking at hippocampal synapses by beta‐amyloid oligomers: the mitochondrial contribution. Mol Brain 2010;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han K, Kim E. Synaptic adhesion molecules and PSD‐95. Prog Neurobiol 2008;84:263–283. [DOI] [PubMed] [Google Scholar]
- 51. Seo MK, Lee CH, Cho HY, et al. Effects of antipsychotic drugs on the expression of synapse‐associated proteins in the frontal cortex of rats subjected to immobilization stress. Psychiatry Res 2015;229:968–974. [DOI] [PubMed] [Google Scholar]
- 52. Orme MH, Giannini AL, Vivanco MD, Kypta RM. Glycogen synthase kinase‐3 and Axin function in a beta‐catenin‐independent pathway that regulates neurite outgrowth in neuroblastoma cells. Mol Cell Neurosci 2003;24:673–686. [DOI] [PubMed] [Google Scholar]
- 53. Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 2008;31:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu X, Malenka RC. Beta‐catenin is critical for dendritic morphogenesis. Nat Neurosci 2003;6:1169–1177. [DOI] [PubMed] [Google Scholar]
- 55. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009;10:468–477. [DOI] [PubMed] [Google Scholar]
- 56. Wan W, Xia S, Kalionis B, Liu L, Li Y. The role of Wnt signaling in the development of Alzheimer's disease: a potential therapeutic target? Biomed Res Int 2014;2014:301575. [DOI] [PMC free article] [PubMed] [Google Scholar]