

Summary
Aims
Astrocytic S100B and receptor for advanced glycation endproducts (RAGE) have been implicated in Parkinson׳s disease (PD) pathogenesis through yet unclear mechanisms. This study attempted to characterize S100B/mRAGE (signaling isoform) axis in a dying‐back dopaminergic (DAergic) axonopathy setting, which mimics an early event of PD pathology.
Methods
C57BL/6 mice were submitted to a chronic MPTP paradigm (20 mg/kg i.p., 2 i.d‐12 h apart, 5 days/week for 2 weeks) and euthanized 7 days posttreatment to assess mRAGE cellular distribution and S100B/mRAGE density in striatum, after probing their locomotor activity (pole test and rotarod). Dopaminergic status, oxidative stress, and gliosis were also measured (HPLC‐ED, WB, IHC).
Results
This MPTP regimen triggered increased oxidative stress (augmented HNE levels), gliosis (GS/Iba1‐reactive morphology), loss of DAergic fibers (decreased tyrosine hydroxylase levels), and severe hypodopaminergia. Biochemical deficits were not translated into motor abnormalities, mimicking a presymptomatic PD period. Remarkably, striatal neurotrophic S100B/mRAGE levels and major neuronal mRAGE localization coexist with compensatory responses (3‐fold increase in DA turnover), which are important to maintain normal motor function.
Conclusion
Our findings rule out the involvement of S100B/mRAGE axis in striatal reactive gliosis, DAergic axonopathy and warrant further exploration of its neurotrophic effects in a presymptomatic compensatory PD stage, which is a fundamental period for successful implementation of therapeutic strategies.
Keywords: MPTP, Presymptomatic, RAGE, S100B, Striatum
Introduction
RAGE (Receptor for advanced glycation endproducts) is a multiligand receptor of the immunoglobulin superfamily long implicated in the sustainment of glial activation, oxidative stress, and neurotoxicity 1, 2, 3, 4. RAGE is expressed in both full‐length membrane‐bound (mRAGE) and soluble (sRAGE) forms. Whereas mRAGE is the RAGE signaling isoform, sRAGE lacks the transmembrane domain and is a competitive inhibitor of mRAGE acting as a decoy for ligands 5. Therefore, it is vital to dissect different RAGE isoforms as it is likely that subtle shifts in ratio of RAGE variants can significantly impact intracellular signaling.
Brain deleterious events have been linked to RAGE ligands (e.g., S100 proteins, amyloid peptide, HMGB‐1) that get complex three‐dimensional structures in oxidative settings 6. RAGE activation following accumulation of such ligands perpetuates neurodegeneration by triggering its own upregulation via a positive feedback loop 7. Nonetheless, a range of adult cell types shows low levels of RAGE expression underlying RAGE physiological function. Consistently, physiological levels of RAGE ligands may modulate brain plasticity and damage repair 8. RAGE pleiotropic effects seem to be strongly dependent on the cell type and the context 9. For example, brain physiological levels of S100B tonically activate trophic RAGE signaling in neurons 10. In contrast, S100B overproduction causes excessive neuronal RAGE stimulation that culminates in overproduction of ROS and neurotoxicity in injured brain tissue 11. Accordingly, S100B is envisaged as a double‐edged sword because it could be a neurotrophin 12 or a damage‐associated molecular protein 13, depending on its levels.
Increased expression of both S100B 14 and RAGE 15 were reported in patients with Parkinson's disease (PD), suggesting that S100B/RAGE axis can be involved in PD neurodegenerative process. Moreover, RAGE was proposed to play a role in microglia‐mediated dopaminergic degeneration in ventral midbrain of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐treated mice 16, a gold standard PD animal model 17. Therefore, we hypothesized that continuous accumulation of S100B and subsequent RAGE upregulation would foster glial reactivity and DAergic axonopathy in striata. Hence, we attempted to characterize the neurochemical, immunohistochemical, and functional correlate of striatal S100B/mRAGE axis in a chronic MPTP mouse model of PD.
Materials and Methods
Animals and MPTP Protocol
Male adult C57BL/6J mice (3 months old; 24–28 g; Charles River Laboratories, Barcelona, Spain) were housed four per cage, under controlled environmental conditions [12‐h light/dark schedule at room temperature (RT) of 23 ± 1°C, with food and water supplied ad libitum]. All experiments were approved by the Institutional Animal Care and Use Committee from Faculty of Medicine, Coimbra University, and were performed following the European Community directive (2010/63/EU). The animal procedures were performed in strict accordance with the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Academy Press 1996).
Herein we applied a new chronic MPTP paradigm (Figure 1). We considered this regimen as RAGE ligands accumulation occurs preferentially in chronically stressed environments, which more closely mimic the progressive nature of PD 7. Briefly, animals were injected intraperitoneally (i.p.) with 20 mg/kg of MPTP hydrochloride in saline solution (Sigma‐Aldrich, St. Louis, MO, USA; MPTP, n = 8) or with saline solution (0.9% NaCl; SAL, n = 8), twice daily 12 h apart for 10 days, and a pause time of 2 consecutive days at the end of the first 5 days of administration. Technical recommendations of MPTP safety handling were strictly enforced 18. All animals survived this dosing regimen and none showed weight reductions. Sacrifice of both saline (n = 4)‐ and MPTP‐treated mice (n = 4) were performed 7 days after last administration by decapitation, following motor function assessment. Brains were rapidly dissected and immediately frozen in liquid nitrogen and stored at −80°C until protein levels assessment and dopamine (DA) and metabolites quantification. Remaining animals (SAL; n = 4 and MPTP; n = 4) were deeply anesthetized with pentobarbital and transcardially perfused with 0.1 M PBS followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. The brains were removed and postfixed for 24 h in 4% PFA and then dehydrated in 30% sucrose in 0.1 M PBS for 24 h for subsequent immunohistochemistry analysis. All chemicals (ultrapure and pro analysis quality) were purchased from Sigma‐Aldrich and Merck AG (Darmstadt, Germany).
Figure 1.
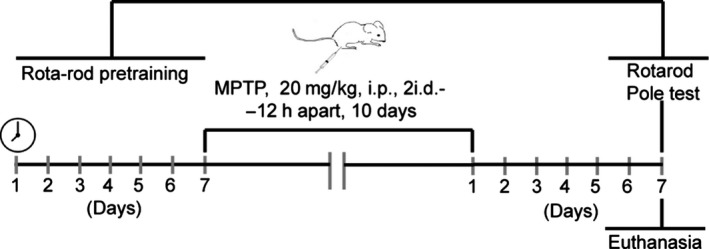
Experimental design including MPTP administration protocol, locomotor behavior assessment, and euthanasia.
Locomotor Assessment: Rotarod and Pole Tests
Animals [SAL (n = 8) and MPTP (n = 8)] were submitted to rotarod and pole tests at day 7 posttreatment. These tests have been extensive and successfully used to probe motor performance and coordination following DA system lesions in MPTP‐treated mice 19, 20. All tests were carried out between 9:00 and 17:00 h in a sound‐attenuated room under low‐intensity light (12 lx) and were scored by the same person in an observation room where the mice had been habituated for at least 1 h before the beginning of the tests.
Rotarod Test
Motor coordination and balance were evaluated using the rotarod test. The rotarod apparatus (Letica Scientific Instruments, LE 8200 model) consisted of a rotating spindle (3 cm of diameter distancing 15 cm of base) with 5 individual compartments distancing 3 cm so that 5 animals could be simultaneously tested. Animals were initially trained to maintain themselves on the rotating rod at 15 rotations per minute (RPM) for 120 seconds (habituation phase), prior to the trial. This assured that all the animals reached an analogous baseline. An increasing speed protocol was implemented herein, as the sensitivity to motor disability has been shown to improve when animals are forced to move at increasingly faster speeds 21. Briefly, animals were placed on the rotating rod and tested using an increasing rotating speed (from 10 to 22 rpm) for 240 sec. The time that mice remained on the rotarod was recorded over 3 trials with an intertrial interval of approximately 5 min. Data are presented as the mean time on the rotating bar over 3 test trials.
Pole Test
Pole test was performed to determine the degree of bradykinesia and slight modifications to a previous protocol were implemented 20. A metal pole (50 cm high and 8 mm diameter), doubly wrapped with cloth tape to prevent slipping and whose base was positioned in the home cages, was used for this test. Mice were placed head upward on the top of the pole. Thereafter, time taken to orientate the body completely downwards (Tturn) and to land on all four paws on the floor was recorded (TLA) with a cutoff of 60 seconds. The test consisted of 3 trials (20 min interval between each) and trial average was used for statistical analysis.
Quantification of Dopamine and Metabolites by HPLC‐ED
Right striata were sonicated in ice‐cold 0.2 M perchloric acid and centrifuged (15.500 × g, 7 min, 4°C), and the supernatants were used to determine the content of dopamine (DA) and metabolites 3,4‐dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) by high‐performance liquid chromatography (HPLC‐ED) with an electrochemical detector, as previously described 22, 23. The pellet was resuspended in 1 M NaOH and stored at −80°C for total protein quantification by the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, MA, Waltham, USA). Concentration of DA and its metabolites were determined by comparison with peak areas of standards and expressed in nanogram per mg of protein.
Western Blot Analysis
Left striata were used for total protein extracts as previously described 23 and total protein concentration was determined by the bicinchoninic acid (BCA) protein assay (Pierce Biotechnology, Rockford, IL, USA). Equal amounts of protein were separated using 8–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and electrophoretically transferred to a polyvinylidene difluoride membrane (Merck Millipore, Madrid, Spain) and blocked by 5% defatted milk for 2 h (Silva et al. 2014). Membranes were incubated overnight at 4°C with the following primary antibodies: mouse anti‐TH (1:1000; MAB318, Merck Millipore), rabbit anti‐HNE (1:500; 393207, Calbiochem, Merck Millipore), mouse anti‐GFAP (1:1000; IF03L, Calbiochem, Merck Millipore), rabbit anti‐S100β (1:500; JBC1771181, Merck Millipore), and rabbit anti‐RAGE raised against the C‐terminal domain recognizing specifically the full‐length membrane‐bound isoform (1:500; Ab3611, Abcam, Cambridge, UK). Membranes were then incubated with alkaline phosphatase‐conjugated IgG secondary antibodies (mouse or rabbit 1:10,000; GE Healthcare, Little Chalfont, UK). Finally, membranes were visualized on Typhoon FLA 9000 (GE Healthcare) and analyzed using Image Quant 5.0 software (Molecular Dynamics, Inc., Sunnyvale. CA, USA). Results were normalized against internal controls β‐actin (1:1000; A5316, Sigma‐Aldrich) or GAPDH (1:1000; MAB374, Merck Millipore) and then expressed as percentage of control.
Immunohistochemistry
Striatum anatomical limits were identified using a mouse brain atlas (AP, +1.32 to +0.5 mm). Striatal coronal sections of 40 μm thickness were collected from cryostat (Leica CM3050S, Nussloch, Germany) in 0.1 M PBS and used for free‐floating immunohistochemistry. Briefly, slices were washed twice with 0.1 M PBS, blocked with 0.25% Triton X‐100 and 5% normal fetal bovine serum (FBS) in 0.1 M PBS for 1 h at room temperature, and then incubated for 24 h at 4°C with the following primary antibodies: rabbit anti‐RAGE (1:200; Ab3160, Abcam), mouse anti‐GFAP (1:1000; IF03L, Calbiochem), mouse antiglutamine synthetase (GS; 1:500; MAB302, Millipore), goat antiionized calcium‐binding adapter molecule 1 (Iba1; 1:200, Ab5076, Abcam), mouse anti‐NeuN (1:500; MAB377, Millipore), or mouse anti‐TH (1:500; MAB318, Millipore).
Sections were then rinsed with 0.1 M PBS (3 × 10 min) and incubated with 4′,6‐diamidino‐2‐phenylindole (DAPI; 1:5000; D1306, Invitrogen, Carlsbad, CA, USA) for nuclear staining and the secondary fluorescent antibodies for 2 h at room. Finally, sections were rinsed with 0.1 M PBS (3 × 10 min) and mounted with glycergel (Dako mounting medium). Samples were imaged using a confocal laser scanning microscope (LSM 710 Meta, Carl Zeiss, Gottingen, Germany).
Statistical Analysis
Differences in locomotor and biochemical data were compared using unpaired Student's t‐test. The accepted level of significance for the tests was P < 0.05. Data are expressed as means ± SEM, and analyses were performed using GraphPad Prism 5.0 software for Windows.
Results
Mice Chronically Exposed to MPTP Maintain Normal Motor Function Regardless Severe Disruption of Dopaminergic Nerve Terminals
MPTP‐treated animals displayed motor performances similar to control animals in both tests (P > 0.05, Figure 2A,B). Nonetheless, MPTP imposed a severe disturbance of striatal dopaminergic system as demonstrated by a significant reduction in TH (~68% of control, P < 0.01), DA, and its metabolites levels (~11%, ~21%, and ~40% of control, respectively; P < 0.001) and a significant increase in DA turnover (DOPAC+HVA/DA ratio, ~300% of control; P < 0.05) 7 days after the toxic insult (Figure 2C–G).
Figure 2.
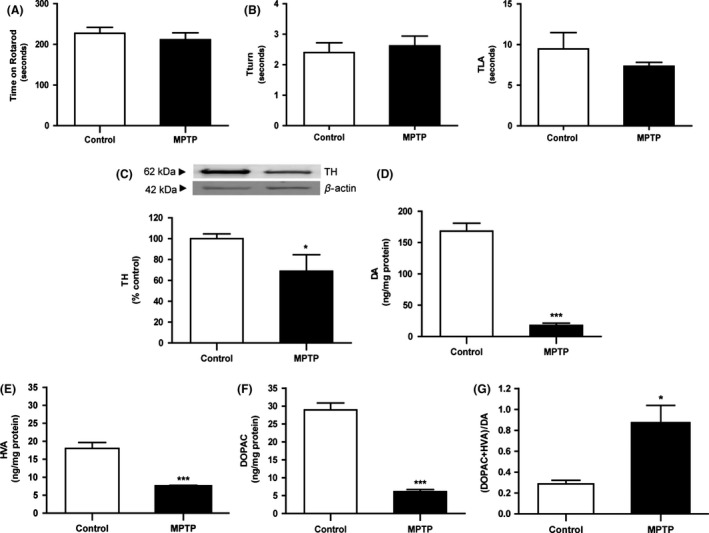
Effect of MPTP chronic treatment on motor skills and striatal dopamine status 7 days post‐MPTP. MPTP‐treated mice showed normal motor function, exhibiting similar performances of control animals in (A) time spent on rotarod and (B) time until they turned completely downward (Tturn) and landed floor (TLA) in pole test. Normal motor function occurred in the presence of striatal dopaminergic disruption MPTP driven, as shown by reduced (C) TH, (D) DA, (E) DOPAC, and (F) HVA tissue levels and a significant increase in (G) (DOPAC+HVA)/DA, which reflects dopamine turnover. TH protein levels were assessed by Western blot, normalized with β‐actin levels, and expressed as % of control. DA and metabolites were assessed by HPLC‐ED and expressed as ng/mg protein. Results *P < 0.05, ***P < 0.001 versus SAL‐treated animals, using an unpaired Student's t‐test.
Striata of Chronically MPTP‐Treated Mice Show an Oxidant and Gliotic Profile
Chronic MPTP treatment triggered an accumulation of phospholipid peroxidation 4‐hydroxy‐2‐nonenal (HNE) adducts (≈75 and 200 kDa) in striata when compared with control animals (122 and 125% of control, respectively; P < 0.05), 7 days postdosing (Figure 3A). This oxidant setting paralleled a robust gliotic profile. In fact, a significant increase in glial fibrillary acidic protein (GFAP) levels (175%, P < 0.05; Figure 3B) and profound astrocytic morphological changes comprising enlarged cell bodies and increased ramified processes were observed upon MPTP, as highlighted by GFAP and glutamine synthetase (GS) immunostaining (Figure 3C). Reactive profile of microglia cells, classically described to respond prior to astrogliosis in the acute phase of MPTP insult 24, is less robust than the astrocytic one. Nevertheless, actin‐cross‐linking ionized calcium‐binding adaptor molecule 1 (Iba1) immunostaining showed a reduced complexity of microglia shape with shortened and thicker processes branching off from soma in striata of MPTP‐treated mice when compared to control mice (Figure 3D).
Figure 3.
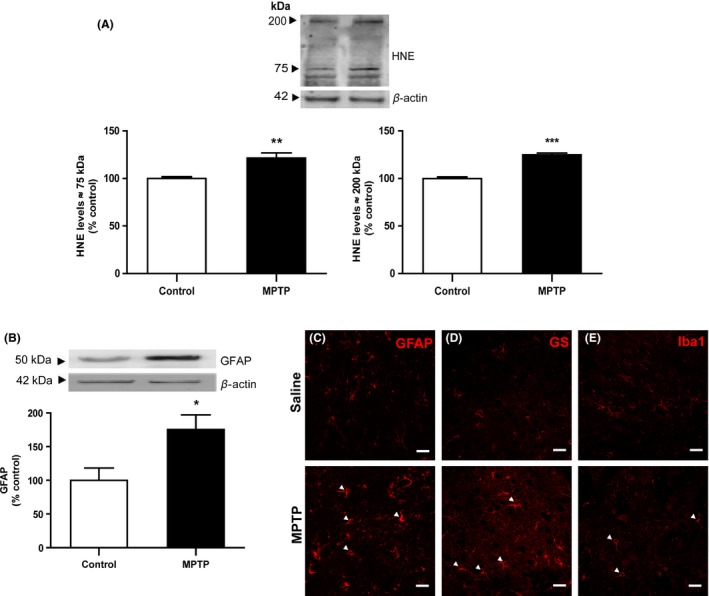
Effect of chronic MPTP treatment on striatal oxidative status and glial reactivity. MPTP triggered an oxidant setting confirmed by the accumulation of HNE protein adducts (75 and 200 kDa) in intoxicated animals (A). This oxidative status concurred with a gliotic profile, typified by increased GFAP levels (B) and enlarged astrocytic cell bodies with increased ramified processes (C,D—GFAP and GS immunofluorescence, respectively), and shortened and thicker microglia processes (E—Iba1 immunofluorescence), in MPTP‐treated animals when compared to control animals (arrows in MPTP conditions). HNE protein levels were assessed by Western blot, normalized with β‐actin levels, and expressed as % of control. Results are expressed as mean ± SEM of 4 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus SAL‐treated animals, using an unpaired Student's t‐test. Confocal images are representative of four animals per group. Scale bar: 20 μm.
Mice Chronically Exposed to MPTP Display Striatal Physiological S100B Levels and Basal Neuronal mRAGE Expression
Impact of chronic MPTP on S100B/mRAGE protein levels and mRAGE cellular localization was assessed by Western blot and double‐labeling immunohistochemistry. Both S100B and mRAGE remained at physiological levels (107 and 101% of control, respectively; P > 0.05; Figure 4A,B) regardless the oxidant and gliotic profile MPTP driven. mRAGE was predominantly found in striatal neurons from saline‐ and MPTP‐treated mice and showed a punctuated distribution mostly localized in nuclei and perikarya of nearly all neuron‐specific protein (NeuN)‐positive cells (Figure 4C). mRAGE immunofluorescence was further detected in somas of both resting and reactive astrocytes (Figure 4D). mRAGE immunofluorescence does not seem to be localized in microglia cells (Figure 4E) nor in DAergic axons (Figure 4F) in basal conditions, a distribution pattern that remained unchanged after MPTP treatment.
Figure 4.
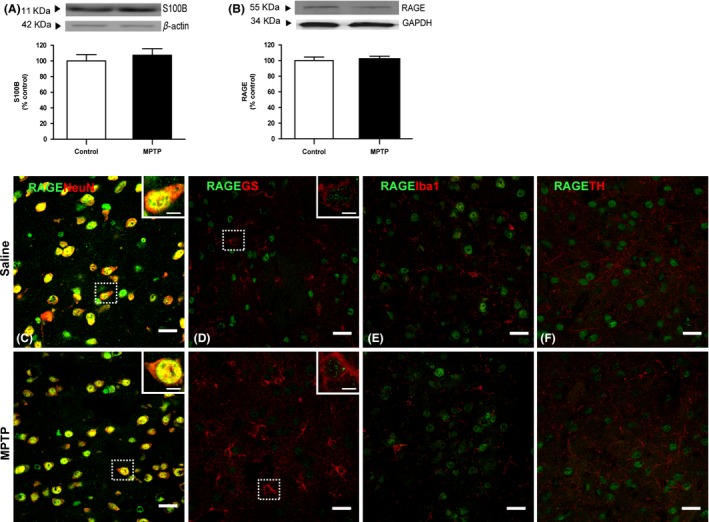
Effect of chronic MPTP treatment on S100B/RAGE axis. Physiological levels of both (A) S100B and (B) mRAGE proteins were observed in striatal total homogenates of MPTP‐treated animals. Double immunofluorescent staining for RAGE (green) and neuronal marker NeuN, astrocytic GS, microglial Iba1, or TH‐positive fibers (red) in striata of saline‐ and MPTP‐treated mice was performed. mRAGE was found to be highly expressed in striatal neurons, showing a punctate distribution mostly localized in nuclei and perikarya of NeuN‐positive cells (colabeling is in yellow) in both saline‐ and MPTP‐treated animals (C). RAGE immunostaining was also found in soma of GS‐positive cells in both saline‐ and MPTP‐treated mice (D), regardless of the astrocytic reactive status. RAGE does not seem to be localized in Iba1‐positive cells (E) nor in TH‐positive fibers from both saline‐ and MPTP‐injured striata (F). S100B and RAGE protein levels were assessed by Western blot, normalized with β‐actin/GAPDH levels, and expressed as % of control. Results are expressed as mean ± SEM of 4 animals per group. Confocal images are representative of four animals per group. Scale bar 20 μm and 5 μm for zoomed images.
Conclusion
Nearly all of the available treatments for PD progression are symptomatic in nature and do not appear to slow or reverse its natural course. Novel therapeutic strategies for PD are therefore critical. This prompted us to characterize striatal S100B/mRAGE axis in a chronic MPTP mouse model of PD. Chronic MPTP protocols provide a sustained oxidative setting that favor accumulation of RAGE ligands in striatum, a brain area comprising early events in PD‐related pathology 25. Our results clearly demonstrate that this MPTP model triggered a severe DAergic disruption as shown by depletion of striatal DA and its metabolites DOPAC and HVA. These findings are aligned with DAergic toxicity reported for other chronic and acute MPTP mice models 26. DA depletion surmounts the range of decrease in TH suggesting that DAergic dysfunction is more prominent than DAergic terminal degeneration in this MPTP regimen.
Most of these biochemical events occur over several years prior to diagnosis of typical PD motor symptoms 27. Multifactorial mechanisms occurring at corticobasal ganglia‐thalamo loop are suggested to display key compensatory roles for motor function until striatal DAergic deficits reach a critical threshold 28. MPTP experimental models revolve around a controversial correlation between motor deficits and striatal DA levels. In fact, motor disabilities are transient and identified shortly after treatment, sometimes when mice are still intoxicated by MPTP 29, 30. Not surprisingly, our chronic MPTP model displayed normal motor function 7 days posttreatment, in spite of profound striatal hypodopaminergia. Increased DA turnover is one of the striatal compensatory mechanisms engaged in maintaining the minimum concentration of DA required for normal motor function in PD 19, 31, 32. Consistently, we report a 3‐fold increase in DA to metabolite ratio in MPTP‐treated mice, suggesting that such striatal compensatory actions are recruited in this experimental setting and contribute to maintain motor skills. Overall, this MPTP model presents biochemical and locomotor similarities with presymptomatic PD stage, fundamental for successful implementation of strategies aimed at altering the progressive evolution of PD 27.
Evidences arising from both patients with PD 15 and experimental PD models 18 suggest oxidative stress as a key player in PD pathology. This was observed in our model by strong HNE‐immunoreactive bands in MPTP‐treated animals, probably reflecting distinct protein adducts arising from lipoxidative damage, as previously reported in early stages of human parkinsonism 15. Glial cells also contribute to PD progression with release of oxidative stress mediators involved in inflammatory processes 33, 34, 35. Consistently, our immunohistochemical data clearly showed a sustained gliotic profile in MPTP‐treated animals, lending additional credence to the protocol used herein. As environments laden with oxidative stress are prone to accumulation of RAGE ligands 6, we investigated the likely accumulation of ligands/mRAGE in MPTP chronically stressed striata. Remarkably, we found normal density of astrocytic ligand S100B and mRAGE in spite of an oxidative and gliotic setting.
Identity of RAGE‐bearing cells also dictates the outcome of RAGE biology 8. Our results clearly showed a prominent neuronal mRAGE localization as RAGE is present virtually in all NeuN‐positive cells in both saline‐ and MPTP‐treated striata. This finding is consistent with RAGE being distributed in 98% of striatal spiny projection neurons and in all interneurons both in WT and in Huntington's disease (HD) animal model 36. Importantly, a robust neuronal localization for RAGE was already described in nigral DAergic neurons following MPTP 16. Nevertheless, RAGE immunofluorescence did not extend to striatal TH‐positive axons, which was further confirmed by our findings. Regarding glial cells, evenly distribution of mRAGE in soma of astrocytes was unchanged following astrogliosis in striata from MPTP mice. Therefore, even though RAGE is envisaged as a central player in glial reactivity 2, 3, 4, our observations rule out a chief role for S100B/RAGE axis in sustaining striatal reactive gliosis. Our data further exclude a direct involvement of RAGE in DAergic axonopathy under the present experimental setting.
Previous works have demonstrated an early upregulation of striatal S100B contents returning to basal levels at 7 days, in an acute MPTP model 35, 37. These findings are aligned with normal S100B reported here 7 days postchronic MPTP model. However, initial increase in S100B reported by former authors suggests earlier changes in ligands/RAGE levels are likely to occur in striatum in the acute phase of MPTP toxic insult. Notably, a long‐lasting repression of S100B and RAGE gene expression after a transient increase was also reported in mesencephalon of MPTP‐treated mice 16. S100B and RAGE gene are both under complex transcriptional regulation as negative regulatory elements within promoters are responsible of suppressing their expression, maintaining low levels of protein 11, 38. Altogether, these findings imply the existence of active compensatory processes tightly regulating S100B/RAGE axis toward homeostasis within striatonigral injury.
RAGE upregulation in chronically injured tissues led to a large number of studies focusing on RAGE‐mediated neurotoxicity. For instance, a link between increased neuronal RAGE staining and neuronal death in preclinical and clinical HD settings is already established in striatum 36, 39. Nevertheless, RAGE's protective effects in homeostasis and repair after injury have been recently highlighted, substantiating the pleiotropism of this receptor 9, 40. In this chronic MPTP model, basal levels of S100B, endowed with neurotrophic activity 11, 41, and normal neuronal RAGE density are more likely to be engaged in striatal neuroprotective mechanisms rather than in toxic events. For example, S100B/RAGE axis was recently linked to dendritic rearborization in cortical neurons following an excitotoxic glutamatergic insult 42. Curiously, dendritic remodeling in striatal spiny projection neurons was correlated with mice locomotor spontaneous recovery 7 days postacute MPTP 29. Therefore, RAGE prevalent neuronal expression in striatum after MPTP exposure warrants future studies to explore whether S100B/RAGE axis is playing a role in presymptomatic compensatory responses in PD neuropathology.
Nevertheless, increased levels of RAGE and ligands reported in patients with PD 14, 15 necessarily imply failure of these overwhelmed repair‐driven pathways in the course of human PD neuropathology. The next imperative question is as follows: Which RAGE isoform is we talking about when addressing RAGE axis? In fact, RAGE signaling is dependent upon the interplay between the membrane‐bound isoform (mRAGE, which induces cellular signaling) and the soluble one (sRAGE, which is able to scavenge RAGE ligands thereby antagonizing mRAGE signaling) 5, 6, 7. In fact, RAGE ligands may either accumulate in PD due to upregulated biosynthesis or impaired clearance systems (e.g., sRAGE). Unfortunately, most of previous studies used antibodies, which are incapable of distinguishing both RAGE isoforms, thus neglecting this RAGE complexity.
We newly characterize the biology of mRAGE isoform and its astrocytic S100B ligand in striatum of experimental PD. Our findings rule out the involvement of S100B/mRAGE axis in striatal gliosis and DAergic axonopathy, early events of PD neuropathology. Instead, neurotrophic S100B/mRAGE protein levels observed herein warrant further exploration of its putative neuroprotective role in a presymptomatic compensatory PD stage, a fundamental period for successful implementation of therapeutic strategies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This research was supported by FCT, Portugal, Strategic Project PEst‐C/SAU/UI3282/2013, UID/NEU/04539/2013, COMPETE‐FEDER, and EXPL/DTP‐DES/0104/2013 under the frame of “Programa Operacional Temático Fatores de Competitividade (COMPETE/QREN)” and “Fundo Comunitário Europeu (FEDER).” SDV is a recipient of a PhD grant from Fundação para a Ciência e a Tecnologia (FCT, Portugal, SFRH/BD/78166/2011). The experiments comply with the current laws of Portugal.
References
- 1. Angelo MF, Aguirre A, Aviles Reyes RX, et al. The proinflammatory RAGE/NF‐kappaB pathway is involved in neuronal damage and reactive gliosis in a model of sleep apnea by intermittent hypoxia. PLoS ONE 2014;9:e107901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Origlia N, Bonadonna C, Rosellini A, et al. Microglial receptor for advanced glycation end product‐dependent signal pathway drives beta‐amyloid‐induced synaptic depression and long‐term depression impairment in entorhinal cortex. J Neurosci 2010;30:11414–11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Origlia N, Criscuolo C, Arancio O, Yan SS, Domenici L. RAGE inhibition in microglia prevents ischemia‐dependent synaptic dysfunction in an amyloid‐enriched environment. J Neurosci 2014;34:8749–8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Villarreal A, Seoane R, Gonzalez Torres A, et al. S100B protein activates a RAGE‐dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J Neurochem 2014;131:190–205. [DOI] [PubMed] [Google Scholar]
- 5. Kalea AZ, Schmidt AM, Hudson BI. Alternative splicing of RAGE: Roles in biology and disease. Front Biosci (Landmark Ed) 2011;16:2756–2770. [DOI] [PubMed] [Google Scholar]
- 6. Herold K, Moser B, Chen Y, et al. Receptor for advanced glycation end products (RAGE) in a dash to the rescue: Inflammatory signals gone awry in the primal response to stress. J Leukoc Biol 2007;82:204–212. [DOI] [PubMed] [Google Scholar]
- 7. Bierhaus A, Nawroth PP. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009;52:2251–2263. [DOI] [PubMed] [Google Scholar]
- 8. Alexiou P, Chatzopoulou M, Pegklidou K, Demopoulos VJ. RAGE: A multi‐ligand receptor unveiling novel insights in health and disease. Curr Med Chem 2010;17:2232–2252. [DOI] [PubMed] [Google Scholar]
- 9. Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 2013;1833:101–109. [DOI] [PubMed] [Google Scholar]
- 10. Kleindienst A, Hesse F, Bullock MR, Buchfelder M. The neurotrophic protein S100B: Value as a marker of brain damage and possible therapeutic implications. Prog Brain Res 2007;161:317–325. [DOI] [PubMed] [Google Scholar]
- 11. Donato R, Sorci G, Riuzzi F, et al. S100B's double life: Intracellular regulator and extracellular signal. Biochim Biophys Acta 2009;1793:1008–1022. [DOI] [PubMed] [Google Scholar]
- 12. Luo KR, Hong CJ, Liou YJ, Hou SJ, Huang YH, Tsai SJ. Differential regulation of neurotrophin S100B and BDNF in two rat models of depression. Prog Neuropsychopharmacol Biol Psychiatry 2010;34:1433–1439. [DOI] [PubMed] [Google Scholar]
- 13. Kato J, Svensson CI. Role of extracellular damage‐associated molecular pattern molecules (DAMPs) as mediators of persistent pain. Prog Mol Biol Transl Sci 2015;131:251–279. [DOI] [PubMed] [Google Scholar]
- 14. Sathe K, Maetzler W, Lang JD, et al. S100B is increased in Parkinson's disease and ablation protects against MPTP‐induced toxicity through the RAGE and TNF‐alpha pathway. Brain 2012;135(Pt 11):3336–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dalfo E, Portero‐Otin M, Ayala V, Martinez A, Pamplona R, Ferrer I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol 2005;64:816–830. [DOI] [PubMed] [Google Scholar]
- 16. Teismann P, Sathe K, Bierhaus A, et al. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiol Aging 2012;33:2478–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langston JW, Irwin I. MPTP: Current concepts and controversies. Clin Neuropharmacol 1986;9:485–507. [PubMed] [Google Scholar]
- 18. Przedborski S, Jackson‐Lewis V, Naini AB, et al. The parkinsonian toxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP): A technical review of its utility and safety. J Neurochem 2001;76:1265–1274. [DOI] [PubMed] [Google Scholar]
- 19. Luchtman DW, Shao D, Song C. Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson's disease. Physiol Behav 2009;98:130–138. [DOI] [PubMed] [Google Scholar]
- 20. Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y. A simple quantitative bradykinesia test in MPTP‐treated mice. Res Commun Chem Pathol Pharmacol 1985;50:435–441. [PubMed] [Google Scholar]
- 21. Monville C, Torres EM, Dunnett SB. Comparison of incremental and accelerating protocols of the rotarod test for the assessment of motor deficits in the 6‐OHDA model. J Neurosci Methods 2006;158:219–223. [DOI] [PubMed] [Google Scholar]
- 22. Pereira FC, Cunha‐Oliveira T, Viana SD, et al. Disruption of striatal glutamatergic/GABAergic homeostasis following acute methamphetamine in mice. Neurotoxicol Teratol 2012;34:522–529. [DOI] [PubMed] [Google Scholar]
- 23. Silva CD, Neves AF, Dias AI, et al. A single neurotoxic dose of methamphetamine induces a long‐lasting depressive‐like behaviour in mice. Neurotox Res 2014;25:295–304. [DOI] [PubMed] [Google Scholar]
- 24. Kohutnicka M, Lewandowska E, Kurkowska‐Jastrzebska I, Czlonkowski A, Czlonkowska A. Microglial and astrocytic involvement in a murine model of Parkinson's disease induced by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Immunopharmacology 1998;39:167–180. [DOI] [PubMed] [Google Scholar]
- 25. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pain S, Gochard A, Bodard S, Gulhan Z, Prunier‐Aesch C, Chalon S. Toxicity of MPTP on neurotransmission in three mouse models of Parkinson's disease. Exp Toxicol Pathol 2013;65:689–694. [DOI] [PubMed] [Google Scholar]
- 27. Kozina EA, Khakimova GR, Khaindrava VG, et al. Tyrosine hydroxylase expression and activity in nigrostriatal dopaminergic neurons of MPTP‐treated mice at the presymptomatic and symptomatic stages of parkinsonism. J Neurol Sci 2014;340:198–207. [DOI] [PubMed] [Google Scholar]
- 28. Ugrumov MV, Khaindrava VG, Kozina EA, et al. Modeling of presymptomatic and symptomatic stages of parkinsonism in mice. Neuroscience 2011;181:175–188. [DOI] [PubMed] [Google Scholar]
- 29. Kim W, Im MJ, Park CH, Lee CJ, Choi S, Yoon BJ. Remodeling of the dendritic structure of the striatal medium spiny neurons accompanies behavioral recovery in a mouse model of Parkinson's disease. Neurosci Lett 2013;557 (Pt B):95–100. [DOI] [PubMed] [Google Scholar]
- 30. Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: An update. J Parkinsons Dis 2011;1:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blesa J, Pifl C, Sanchez‐Gonzalez MA, et al. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: A PET, histological and biochemical study. Neurobiol Dis 2012;48:79–91. [DOI] [PubMed] [Google Scholar]
- 32. Nandhagopal R, Kuramoto L, Schulzer M, et al. Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson's disease. Brain 2011;134(Pt 11):3290–3298. [DOI] [PubMed] [Google Scholar]
- 33. Halliday GM, Stevens CH. Glia: Initiators and progressors of pathology in Parkinson's disease. Mov Disord 2011;26:6–17. [DOI] [PubMed] [Google Scholar]
- 34. O'Callaghan JP, Sriram K, Miller DB. Defining “neuroinflammation”. Ann N Y Acad Sci 2008;1139:318–330. [DOI] [PubMed] [Google Scholar]
- 35. Yasuda Y, Shimoda T, Uno K, et al. The effects of MPTP on the activation of microglia/astrocytes and cytokine/chemokine levels in different mice strains. J Neuroimmunol 2008;204:43–51. [DOI] [PubMed] [Google Scholar]
- 36. Anzilotti S, Giampa C, Laurenti D, et al. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington's disease. Brain Res Bull 2012;87:350–358. [DOI] [PubMed] [Google Scholar]
- 37. Muramatsu Y, Kurosaki R, Watanabe H, et al. Expression of S‐100 protein is related to neuronal damage in MPTP‐treated mice. Glia 2003;42:307–313. [DOI] [PubMed] [Google Scholar]
- 38. Riehl A, Nemeth J, Angel P, Hess J. The receptor RAGE: bridging inflammation and cancer. Cell Commun Signal 2009;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma L, Nicholson LF. Expression of the receptor for advanced glycation end products in Huntington's disease caudate nucleus. Brain Res 2004;1018:10–17. [DOI] [PubMed] [Google Scholar]
- 40. Saleh A, Smith DR, Tessler L, et al. Receptor for advanced glycation end‐products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp Neurol 2013;249:149–159. [DOI] [PubMed] [Google Scholar]
- 41. Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: An update. Biochim Biophys Acta 2009;1793:993–1007. [DOI] [PubMed] [Google Scholar]
- 42. Villarreal A, Aviles RR, Angelo MF, Reines AG, Ramos AJ. S100B alters neuronal survival and dendrite extension via RAGE‐mediated NF‐kappaB signaling. J Neurochem 2011;117:321–332. [DOI] [PubMed] [Google Scholar]