

Summary
Aims
Sigma‐1 receptors are involved in the pathophysiological process of several neuropsychiatric diseases such as epilepsy, depression. Allosteric modulation represents an important mechanism for receptor functional regulation. In this study, we examined antidepressant activity of the latest identified novel and selective allosteric modulator of sigma‐1 receptor 3‐methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668).
Methods and Results
A single administration of SOMCL‐668 decreased the immobility time in the forced swimming test (FST) and tailing suspended test in mice, which were abolished by pretreatment of sigma‐1 receptor antagonist BD1047. In the chronic unpredicted mild stress (CUMS) model, chronic application of SOMCL‐668 rapidly ameliorated anhedonia‐like behavior (within a week), accompanying with the enhanced expression of brain‐derived neurotrophic factor (BDNF) and phosphorylation of glycogen synthase kinase 3β (GSK3β) (Ser‐9) in the hippocampus. SOMCL‐668 also rapidly promoted the phosphorylation of GSK3β (Ser‐9) in an allosteric manner in vitro. In the cultured primary neurons, SOMCL‐668 enhanced the sigma‐1 receptor agonist‐induced neurite outgrowth and the secretion of BDNF.
Conclusion
SOMCL‐668, a novel allosteric modulator of sigma‐1 receptors, elicits a potent and rapid acting antidepressant effect. The present data provide the first evidence that allosteric modulation of sigma‐1 receptors may represent a new approach for antidepressant drug discovery.
Keywords: Allosteric modulation, Antidepressant, Brain‐derived neurotrophic factor, Glycogen synthase kinase 3β, Sigma‐1 receptors
Introduction
Major depressive disorder (MDD) affects large population worldwide 1. Serotonin selective reuptake inhibitors (SSRIs) and dual serotonin and norepinephrine reuptake inhibitors (SNRIs), such as fluoxetine and venlafaxine, are the most highly prescribed medications in drug therapy for depression. However, there are significant limitations such as the delayed therapeutic response and low response rates. Thus, there is clearly an urgent need for the development of new targets and faster‐acting antidepressants.
As a kind of chaperone proteins comprising 223 amino acids, sigma‐1 receptors bind with immunoglobulin‐binding protein (Bip) (GRP78, the marker protein of the endoplasmic reticulum) and reside more specifically in the endoplasmic reticulum–mitochondrion interface 2. When the receptor is activated, sigma‐1 receptors dissociate from Bip and translocate to the plasma membrane, where sigma‐1 receptors interact with other proteins to elicit various biological responses 3.
Recently, growing evidence has indicated that the sigma‐1 receptor may serve as a novel target for antidepressant development. It has been reported that sigma‐1 receptor knockout mice exhibit gender‐related anxiety and depressive‐like behaviors 4. Moreover, many of the marketed antidepressants, including fluoxetine, fluvoxamine, sertraline, imipramine, and amitriptyline, possess a high‐to‐moderate affinity for the sigma‐1 receptor 5. Additionally, the acute antidepressant effect of fluvoxamine can be blocked by N'‐[2‐(3,4‐dichlorophenyl)ethyl]‐N,N,N'‐trimethylethane‐1,2‐diamine (BD1047), an antagonist of sigma‐1 receptors 6. Sigma‐1 receptor agonists, 1‐(2‐(3, 4‐dimethoxyphenyl)ethyl)‐4‐(3‐phenylpropyl)piperazine (SA4503) and PRE‐084 (2‐morpholin‐4‐ylethyl 1‐phenylcyclohexane‐1‐carboxylate), exhibit antidepressant effects 7, 8, which may associate with sigma‐1 receptor‐ regulated production of brain‐derived neurotrophic factor (BDNF) 9, 10. It appears that modulation of sigma‐1 receptors may provide a potential novel approach for MDD treatment.
In addition to direct agonists, the sigma‐1 receptor is subject to allosteric modulation 11. As an alternative approach in regulating receptor function, allosteric modulators depend on the types of agonists 12. Allosteric modulation of receptors represents an important mechanism for the functional regulation of receptors. Unlike receptor agonists, allosteric modulation requires the presence of endogenous or exogenous agonists to take effect, the allosteric modulator is known to enhance the selectivity of agonistic action 13. Although the antidepressant effects of sigma‐1 receptor agonists have been reported, the potential unwanted effects for the treatment of depression remain a challenge. Considering the wide distribution and complex biological functions of sigma‐1receptors, allosteric modulation may be particularly important for receptors such as sigma‐1 that are widely distributed. Recently, two novel sigma‐1 receptor allosteric modulators, SKF83959 and ER1, have been identified in our laboratory 14 and by another group 15. Although SKF83959 is a very potent sigma‐1 receptor allosteric modulator, the selectivity is relatively weak because SKF83959 has D1 dopamine receptor affinity and other activities 16, 17, 18, 19, 20. To eliminate the potential effects of SKF83959 on the D1 and serotonin receptors, we conducted a series of structural modifications and developed the new compound 3‐methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668), referred to as SOMCL‐668 (Figure S1). The compound exhibits potent sigma‐1 receptor allosteric modulation but exhibits no significant affinity for the D1, D2, D3, 5‐HT1A, and 5‐HT2A receptors 21, 22. In this study, we took advantage of the new selective sigma‐1 receptor allosteric modulator and investigated the potential effects of the allosteric modulator of the sigma‐1 receptor in antidepressant therapy.
Materials and Methods
Materials
SOMCL‐668 was synthesized in the Shanghai Institute of Materia Medic at the Chinese Academy of Sciences with a purity higher than 98%. BD1047 and venlafaxine hydrochloride were obtained from Sigma‐Aldrich, Co (St. Louis, MO, USA). Recombined BDNF was purchased from Cell Signaling Technology, Inc (Danvers, MA, USA). SOMCL‐668, BD1047 and [2S‐(2α,6α,11R*]‐1,2,3,4,5,6‐hexahydro‐6,11‐dimethyl‐3‐(2‐propenyl)‐2,6‐methano‐3‐benzazocin‐8‐ol hydrochloride (N‐allylnormetazocine) hydrochloride (SKF)] ((+)SKF‐10047) were initially dissolved in dimethyl sulfoxide (DMSO) for stock solutions and diluted with PBS or saline (DMSO ≤0.2%). PRE‐084 and venlafaxine hydrochloride were directly dissolved in PBS or saline.
Animals
Male C57BL/6 mice 6–8 weeks old were maintained at 20–22°C with free access to water and food under a 12:12 h light/dark cycle (with lights on at 8:00 a.m.). The animals were used according to the NIH Guide for the Care and Use of Laboratory Animals, and the experiments were performed following approval of the protocol by the Ethics Committee of the Institution of Soochow University. All efforts were made to minimize animal suffering and to reduce the number of animals used in the experiments.
Behavioral Tests
The forced swimming test (FST), tail suspension test (TST) and open field test was performed as previously described 23, 24, 25. In FST and TST, the mice were randomly divided into six groups: control (saline, n = 10), SOMCL‐668 treatments (5, 10, 20 mg/kg, respectively. n = 10 mice in each group), venlafaxine 10 mg/kg (n = 10 mice), PRE‐084 10 mg/kg (n = 10 mice). The dose of venlafaxine and PRE‐084 was adopted from previous reports 8, 26. The unpredictable chronic mild stress (CUMS) protocol was based on previously published literature 27. In this study, the mice were randomly divided into five groups each with 10 animals: control; CUMS treatment; CUMS + SOMCL‐668 (5, 10 mg/kg); and CUMS + venlafaxine (10 mg/kg). The drug (SOMCL‐668, venlafaxine) was applied once animals developed depression‐like behavioral with 8 weeks of CUMS treatment. All the saline or drugs were administrated by i.p injection. The sucrose preference tests were performed every week during CUMS. The details for the behavior test are given in Supporting Information.
Cell Culture
HT22 murine hippocampal neuronal cells were maintained in Dulbecco's minimal essential medium (DMEM) supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin (100 units and 100 μg/mL). Primary cortical/hippocampal neurons were prepared from C57BL/6 mice at embryonic day 17 or 18 and cultured as described previously 28. Details about primary neuron culture are given in Supporting Information.
Immunostaining
The cells were fixed in ice‐cold 4% paraformaldehyde in 0.01 M phosphate‐buffer for 1 h prior to a wash in 0.1% Triton X‐100 PBS (PBST). Followed by blocking the cells with PBST containing 0.3% Triton X‐100 and 3% BSA for 1 h, the slides were incubated overnight (4°C) with primary antibodies against sigma‐1 receptors (1:300; Santa Cruz, Santa Cruz, CA, USA) and Bip (1:300; Beyotime, Nantong, China). After three washes with PBST, Alexa Fluor 488 (green)‐conjugated anti‐goat IgG or Alexa Fluor 594 (red)‐conjugated anti‐rabbit IgG antibodies (1:1000; Cell Signaling) were added to the slices. The samples were incubated at room temperature for another 1 h, the slices were then rinsed with PBST and incubated with Hoechst (1:10,000; Invitrogen, Carlsbad, CA, USA). Immunostaining images were captured with a confocal microscope (LSM 700; Zeiss, Oberkochen, Germany).
Neurite Outgrowth
Primary neurons were seeded at 5 × 104 cells/well in 6‐well plates. After treatment with (+)SKF‐10047 (1 μM), SOMCL‐668 (10 μM) or a combination of both drugs for 48 h, the cells were fixed and immunostained with a MAP‐2 antibody (1:200; Sigma‐Aldrich) as described in the Immunocytochemistry section. Image J software was used to detect the lengths of the longest cell neurites (at least longer than the diameter of its soma). At least 20 cells were measured in each experiment, and each experiment was replicated three times.
Enzyme‐Linked Immunosorbent Assay
Primary neurons were seeded at 1 × 106 cells/well in 6‐well plates. The cells received the same treatment for 48 h as the treatment described in the neurite outgrowth section. Following the treatment, the conditioned media was collected and centrifuged at 1300 g for 10 min at 4°C. The BDNF content of the conditioned media was determined using and Enzyme‐linked immunosorbent assay (ELISA) kit (Boster Bio, Wuhan, China). Each experiment was replicated three times.
Knockdown of the Sigma‐1 Receptor
The sigma‐1 receptor siRNA (sc‐42251) and control siRNA (sc‐37007) were purchased from Santa Cruz Biotechnology. The siRNA was transfected into HT22 cells using Lipofectamine RNAiMAX (Invitrogen). After 48 h, the transfected HT22 cells were collected.
Western Blotting
The hippocampus tissue or cells samples were mechanically homogenized in lysis buffer (Tris 50 mM pH 7.0, EDTA 1 mM, NaF 100 mM, PMSF 0.1 mM, Na3VO4 2 mM, 1% Triton X‐100, 10% glycerol and Amresco Protease Inhibitor Cocktail) for 30 min on ice. Western blotting was performed by standard protocols using anti‐BDNF (1:200; Santa Cruz), anti‐sigma‐1 receptor (1:200; Santa Cruz), the anti‐pGSK3β (1:1000; Cell Signaling Technology), glycogen synthase kinase 3β (GSK3β) (1:1000; Cell Signaling Technology) antibodies, the anti‐Bip (1:1000; Beyotime) and anti‐α‐tubulin antibody (1:10,000, Sigma‐Aldrich). After washing, the membrane was incubated for 1 h with secondary antibodies conjugated with horseradish peroxidase (Sigma‐Aldrich). The membrane was developed with ECL plus solution (ECL kit; Millipore, Bedford, MA, USA), exposed with the ChemiScope Mini system (Clinx Science, Shanghai, China) and analyzed quantitatively through densitometry with Image J software. Each experiment was replicated at least three times.
Statistics
Data were presented as mean ± SEM. For comparisons of two groups, the two‐tailed Student's t‐test was used. The differences between multiple groups were tested with a one‐way ANOVA followed by Tukey's multiple comparison test. In some cases, the two‐way ANOVA was used and followed by a Bonferroni post hoc test. For data with weight gain in the CUMS test, ANOVA for repeated measurements was employed. The significance level was set at P < 0.05.
Results
SOMCL‐668 Reduces the Immobility Time in the FST and the TST
The antidepressant effects of SOMCL‐668 were examined in the FST and TST, which are highly reliable behavioral assays for detecting antidepressant potential 29. As shown in the Figure 1A,B, the treatment with SOMCL‐668 (i.p., 1 h) significantly reduced the immobility time in the FST [F(5, 54) = 11.60, P < 0.01; one‐way ANOVA, n = 10 in each group] and TST [F(5, 54) = 6.235, P < 0.01; one‐way ANOVA, n = 10 in each group]. For example, in post hoc analysis in the TST, a single injection of SOMCL‐668 at the 10 or 20 mg/kg significantly reduced the immobility time by 25.9% and 31.4% (P < 0.01 vs. control, respectively). A similar effect was observed in the groups receiving the PRE‐084, a sigma‐1 receptor agonist (P < 0.01 vs. control respectively) or conventional antidepressant drug venlafaxine treatment (P < 0.01 vs. control respectively). The antidepressant effects of SOMCL‐668 in both the FST and TST were blocked by pretreatment of the sigma‐1 receptor antagonist BD1047 (2 mg/kg, i.p., 15 min before SOMCL‐668 administration, P < 0.01 respectively) (Figure 1C–D). The two‐way ANOVA revealed a significant effect of SOMCL‐668 × BD1047 treatment interaction on immobility time in the FST [F(1, 36) = 9.401, P < 0.01] and TST [F(1, 36) = 13.37, P < 0.01]. Furthermore, SOMCL‐668 alone did not elicit any significant locomotor activity [Figure S1B, F(6, 63) = 0.6652, P > 0.05]. The results revealed that SOMCL‐668, as a sigma‐1 receptor allosteric modulator, possessed a potent the antidepressant‐like effects of SOMCL‐668 in both the FST and TST, and that activation of sigma‐1 receptor contributed to the effects.
Figure 1.
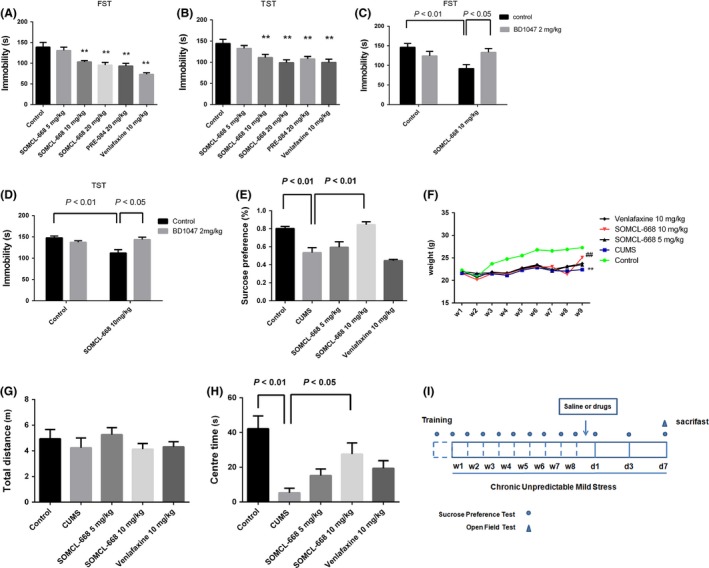
Effects of 3‐methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668) on depressive behavioral action in the forced swimming test (FST), tail suspension test (TST) and the chronic unpredicted mild stress (CUMS) model. (A, B) One hour before FST and TST, male C57BL/6J mice were injected i.p with saline (n = 10), 5 mg/kg SOMCL‐668 (n = 10), 10 mg/kg SOMCL‐668 (n = 10), 20 mg/kg SOMCL‐668 (n = 10), 10 mg/kg PRE‐084 (n = 10), and 10 mg/kg venlafaxine (n = 10), respectively. (C, D) Effects of BD1047 on the antidepressant activity of SOMCL‐668 in FST and TST, n = 10 in each group. BD 1047 (2 mg/kg) was administered i.p. 15 min prior to the injection of SOMCL‐668. (E–H) Effect of SOMCL‐668 on the depressive‐like behaviors in CUMS‐treated mice. Mice were subjected to CUMS as described in methods. Animals were then randomly divided into five groups, each with 10 mice: control; CUMS; SOMCL‐668 5 mg/kg; SOMCL‐668 10 mg/kg; and venlafaxine 10 mg/kg. (E) the sucrose consumption, (F) the weight gain, (G) total distance in the locomotion test and (H) the time that mice stayed in the central field. (I) Schematic diagram indicates the timeline for drug treatment, CUMS exposure, and behavioral testing. The saline or drugs were administered i.p. once daily when the animals developed steady depressive‐like behaviors (8th week), and the tests were detected at 7th day after treatments. The data were expressed as mean ± SEM. (A, B, E, G, H) were analyzed with one‐way ANOVA analysis, (C, D) were analyzed with two‐way ANOVA analysis and (F) was analyzed with one‐way repetitive ANOVA. **P < 0.01 versus control, ## P < 0.01 versus CUMS.
SOMCL‐668 Blocks the Decrease in Sucrose Preference and Ameliorates Anxiety‐Like Behavior Caused by CUMS
The chronic stress‐ induced depression model is a reliable model for revealing the pathological mechanisms underlying depression and screening antidepressant drugs. In the present study, we examined the effects of SOMCL‐668 on sucrose preference in the CUMS model. As shown in Figure 1E, CUMS induced a strong decrease in the sucrose preference compared with control [F(4, 45) = 17.56, P < 0.01; control: 81.7 ± 2.1%, CUMS: 53.5 ± 5.5%, P < 0.01; one‐way ANOVA], and subchronic treatment with SOMCL‐668 (once daily for 1 week) at 10 mg/kg significantly reversed the decrease in the sucrose preference compared with CUMS (84.6 ± 3.2%, P < 0.01). Our results suggest that SOMCL‐668 may increase hedonic states in depressive mice. Depression is often accompanied by weight loss and anxiety. In this study, as shown in Figure 1G,F,H, CUMS caused decreases in weight gain [time × CUMS interaction: F(32, 288) = 12.59, P < 0.01; repeated measures ANOVA] and center time in the open field [F(4, 45) = 6.976, P < 0.01; CUMS vs. control, P < 0.01; one‐way ANOVA], and 1 week subchronic treatment with SOMCL‐668 at 10 mg/kg significantly reversed detrimental effects of CUMS (P < 0.01 vs. CUMS respectively). Indeed, in the naïve mice, administration of SOMCL‐668 at 10 mg/kg/day for 7 days also produced significant anxiolytic effects in the NSFT (t = 3.015, df = 18, P < 0.01 vs. control, Student's t‐test, Figure S1C).
It is interesting to note that the onset time at which SOMCL‐668 displayed the antidepressant effect was within 7 days of drug treatment compared with that of venlafaxine treatment.
SOMCL‐668 Inhibits GSK3β via the Sigma‐1 Receptor in Mice
The inactivation of GSK3β is thought to be a common mechanism that underlies many antidepressant drugs 30. We therefore investigated the effect of SOMCL‐668 on the activation of GSK3β. As shown in Figure 2A, a single injection of SOMCL‐668 or PRE‐084 (i.p., 1 h) increased the phosphorylation of GSK3β (ser‐9) in the hippocampus of naïve mice [F(3, 8) = 8.865, P < 0.01; P < 0.01 vs. control respectively; one‐way ANOVA, n = 4 in each group]. Two‐way ANOVA analysis revealed significant interaction effects for SOMCL‐668 × BD1047 on the phosphorylation of GSK3β (ser‐9) [F(1, 8) = 22.08, P < 0.01]. Stimulation of pGSK3β at ser‐9 by SOMCL‐668 was abolished by pretreatment with a sigma‐1 receptor antagonist BD1047 (i.p., 15 min before the administration of SOMCL‐668, P < 0.01, n = 4 in each group). A similar effect on GSK3β was also observed in HT22 cells. As shown in Figure 2B–D, a selective sigma‐1 receptor agonist (+)SKF‐10047 dose and time dependently increased the phosphorylation of GSK3β (ser‐9) [F(5, 12) = 30.51, P < 0.01; F(3, 8) = 52.00, P < 0.01; one‐way ANOVA], which was potentiated by SOMCL‐668 [the interaction effects for SOMCL‐668 × 0.1 μM (+)SKF‐10047: F(1, 8) = 17.33, P < 0.01; for SOMCL‐668 × 0.3 μM (+)SKF‐10047, F(1, 8) = 20.67, P < 0.01; two‐way ANOVA]. However, this effect of SOMCL‐668 disappeared while the sigma‐1 receptor was knockdown by the siRNA (Figure 2E; the interaction effects for SOMCL‐668 × 0.1 μM (+)SKF‐10047: F(1, 8) = 0.1905, P > 0.05; two‐way ANOVA). In addition, application of another sigma‐1 receptor agonist PRE‐084 or the putative endogenous sigma‐1 receptor ligand dehydroepiandrosterone (DHEA) also inhibited the phosphorylation of GSK3β (ser‐9) [Figure 2F, F(3, 8) = 26.55, P < 0.01; one‐way ANOVA]. As BDNF‐Trk B receptor‐AKT plays an important role in GSK3β regulation 31. Sigma‐1 receptor agonist SA4503 was showed to stimulate the expression of BDNF 9.We wonder if the observed effect of sigma‐1 receptor‐ regulated GSK3β activity is dependent on BDNF‐Trk B receptor‐AKT. As shown in Figure 3A, 50 ng/mL BDNF treatment elicited rapidly phosphorylation of GSK3β at ser‐9 in the HT22 cells [F(2, 6) = 14.19, P < 0.01; one‐way ANOVA]. (+)SKF‐10047‐ induced phosphorylation of GSK3β (ser‐9) was blocked by either k252a (a Trk B receptor inhibitor, P < 0.01) or LY294002 (a PI3K/AKT pathway inhibitor, P < 0.01). Two‐way ANOVA analysis revealed significant interaction effects for (+)SKF‐10047 × k252a [F(1, 8) = 15.78, P < 0.01] or (+)SKF‐10047 × LY294002 [F(1, 8) = 8.258, P < 0.05] on the pGSK3β (ser‐9) (Figure 3B, C). Taken together, our data demonstrated that the activation of sigma‐1 receptors elicited the phosphorylation of GSK3β (ser‐9), which appears to depend on Trk B receptor‐AKT signal‐pathway.
Figure 2.
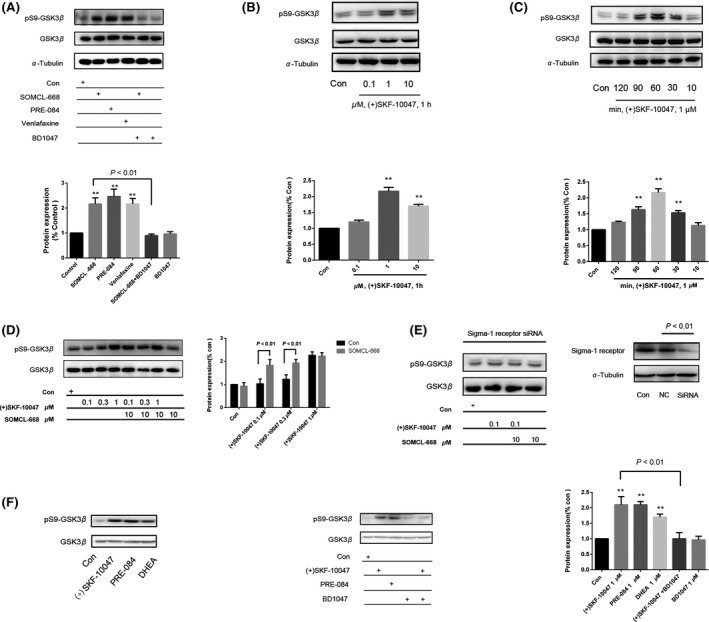
Effect of 3‐methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668) on the phosphorylation of GSK3β. (A) Male C57BL/6J mice (each group with four mice) were injected i.p with saline, 10 mg/kg SOMCL‐668, 10 mg/kg PRE‐084, or 10 mg/kg venlafaxine, respectively for 1 h before sacrificed. Brain tissues were rapidly dissected and hippocampus were collected and kept for Western blotting assays as described in methods. Data were expressed as mean ± SEM and analyzed with one‐way ANOVA analysis. **P < 0.01, compared with the control group. BD1047 2 mg/kg was administered i.p. 15 min before the injection of SOMCL‐668. Data were expressed as mean ± SEM and analyzed with two‐way ANOVA analysis. (B–E) Effect of (+)SKF‐10047 and SOMCL‐668 on the in the cultured HT22 cells. (B) the dose–response of (+)SKF10047 (0.1–10 μM), (C) the time ‐response of (+)SKF‐10047 1 μM, (D) effect of SOMCL‐668 (10 μM, 30 min pretreatment) on (+)SKF‐10047‐induced phosphorylation of GSK3β and (E) the sigma‐1 receptor knockdown cells by the siRNA. (F) The HT22 cells were treated with respective agonists of sigma‐1 receptors 1 μM (+)SKF10047, 1 μM PRE‐084, or 1 μM dehydroepiandrosterone (DHEA) for 1 h. Cells were collected for western blot assays using anti‐GSK3β and anti‐phosphorylated GSK3β antibodies as described in methods. The levels of phosphorylation of GSK3β (Ser‐9) normalized by total GSK3β. Data were expressed as mean ± SEM and analyzed with one‐way ANOVA (B, C, F) or two‐way ANOVA analysis (D, E), **P < 0.01 vs. control. Each experiment was replicated three times.
Figure 3.
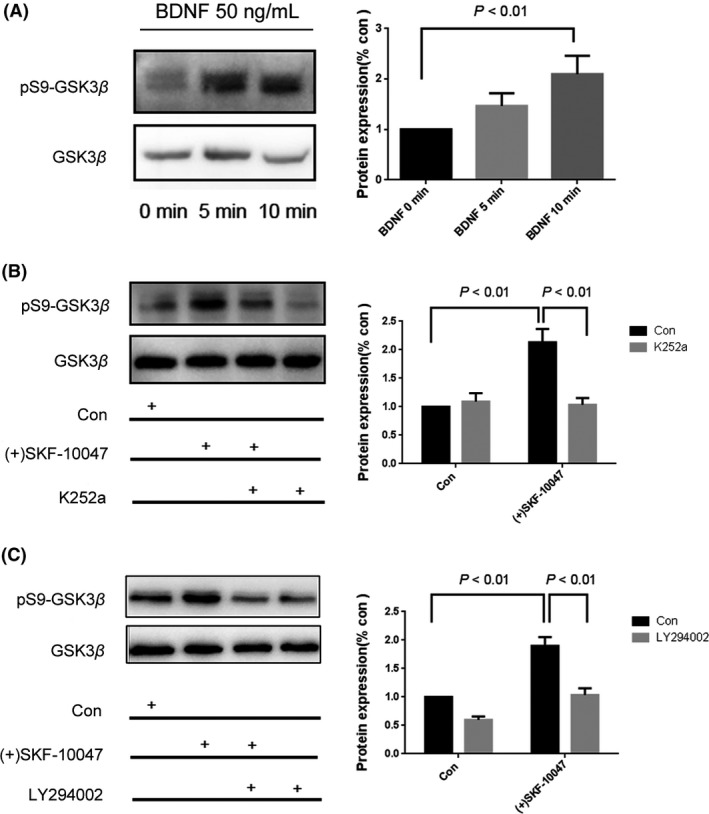
The sigma‐1 receptor increases phosphorylation of GSK3β (ser‐9) through the Trk B receptor‐AKT pathway. (A) HT22 cells were treated with 50 ng/mL brain‐derived neurotrophic factor (BDNF) for 5–10 min. Cells were then collected for western blot assays using anti‐GSK3β and anti‐phosphorylated GSK3β as described in methods. (B, C) The effects of k252a (100 nM, 30 min) or LY294002 (50 μM, 30 min) pretreatment on the (+)SKF‐10047 (1 μM, 1 h)‐ altered phosphorylation of GSK3β (ser‐9). The levels of phosphorylation of GSK3β (Ser‐9) normalized by total GSK3β. Data were expressed as mean ± SEM and analyzed with one‐way for (A) or two‐way ANOVA for (B, C). **P < 0.01 vs. the control. Each experiment was replicated three times.
SOMCL‐668 Rescues the Deficit of BDNF‐GSK3β Pathway in the Hippocampus in the CUMS Models
The elevated BDNF expression serves as a therapeutic biomarker for antidepressant efficacy, whereas the BDNF‐GSK3β pathway is known to be involved in the pathophysiological process and therapeutic response of depression. We further investigated the effect of SOMCL‐668 on the BDNF‐GSK3β pathway in the CUMS model. As expected, CUMS decreased the BDNF expression in the hippocampus [F(4, 10) = 20.92, P < 0.01; CUMS vs. control, P < 0.05; one‐way ANOVA, n = 4 in each group]. SOMCL‐668 (10 mg/kg, once daily for 1 week) restored CUMS‐suppressed BDNF expression in the hippocampus (Figure 4A, P < 0.01 vs. CUMS, n = 4). Moreover, CUMS also decreased the phosphorylation of GSK3β at ser‐9 in the hippocampus [F(4, 10) = 20.18, P < 0.01; CUMS vs. control, P < 0.01; one‐way ANOVA, n = 4 in each group]. Likewise, chronic SOMCL‐668 treatment robustly ameliorated the effect of CUMS on the phosphorylation of GSK3β (ser‐9) (P < 0.01 vs. CUMS, n = 4 in each group) (Figure 4B). In the contrast, similar treatment with venlafaxine (10 mg/kg, once daily for 1 week) failed to recover the expression of BDNF and phosphorylation of GSK3β (ser‐9) in the CUMS‐ induced depression‐like mice (Figure 4A, P > 0.05, n = 4 in each group). The effects of SOMCL‐668 on BDNF expression (t = 5.033, df = 4, P < 0.01) and the phosphorylation of GSK3β (ser‐9) (t = 5.029, df = 4, P < 0.01) were also observed in the naïve mice that received the drug daily for 1 week (Figure 4C).
Figure 4.
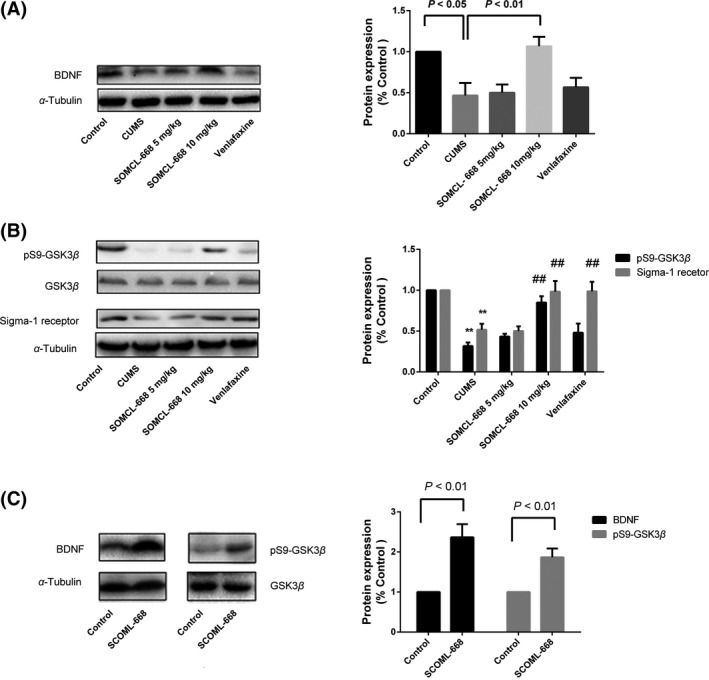
3‐Methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668) rescues impairment of brain‐derived neurotrophic factor (BDNF)‐GSK3β pathway induced by chronic unpredicted mild stress (CUMS). Mice were subjected to CUMS as described in methods. The animals developed steady depression‐like behaviors with 8 weeks CUMS. Animals were then randomly divided into five groups (each with four mice) and treated once daily with vehicle, SOMCL‐668 5 mg/kg, SOMCL‐668 10 mg/kg, and venlafaxine 10 mg/kg respectively, for 1 week before sacrifice. (A) The expression of BDNF in hippocampus in the mice. (B) The expression of phosphorylated GSK3β (ser‐9) and sigma‐1 receptor in hippocampus. Data were expressed as mean ± SEM and analyzed with one‐way ANOVA. **P < 0.01 vs. the control, ## P < 0.01 vs. the CUMS. (C) Effect of SOMCL‐668 treatment (10 mg/kg, daily for 1 week) on BDNF expression and phosphorylation of GSK3β (Ser‐9) in the hippocampus in the naïve mice. The levels of phosphorylation of GSK3β (Ser‐9) normalized by total GSK3β. Data were expressed as mean ± SEM and analyzed with Student's t‐test.
In the heart failure‐ induced depressant model, the expression of sigma‐1 receptors in the brain was found to be decreased 32. We also examined the expression of sigma‐1 receptors in the CUMS‐induced depressant animals. A significantly decrease in the expression of sigma‐1 receptor was evidenced in the CUMS‐treated mice [Figure 4B, F(4, 10) = 8.917, P < 0.01; CUMS vs. control, P < 0.01; n = 4 in each group]. Subchronic treatment with SOMCL‐668 (10 mg/kg, once daily for 1 week) restored the expression of sigma‐1 receptors in the hippocampus in CUMS (Figure 4B, P < 0.01, n = 4 in each group).
SOMCL‐668 Promotes the Release of BDNF and Neurite Outgrowth in an Allosteric Manner
Our previous work has confirmed that SOMCL‐668 can promote the binding of 3H‐(+)‐pentazocine with the sigma‐1 receptor in an allosteric manner 22. We further examined the functional allosteric effect of SOMCL‐668 on sigma‐1 receptors. The activated sigma‐1 receptor separated from Bip and translocated from the ER to the plasma membrane 2. As shown in Figure 5, although SOMCL‐668 treatment alone did not significantly alter the association between the sigma‐1 receptor and Bip at all tested doses (1, 10, 100 μM), the compound, however, significantly synergized the effect of (+)SKF‐10047 on sigma‐1 receptor disassociation from Bip. This observation was further confirmed by immunoprecipitation and the plasma membrane translocation assay (Figure S2).
Figure 5.
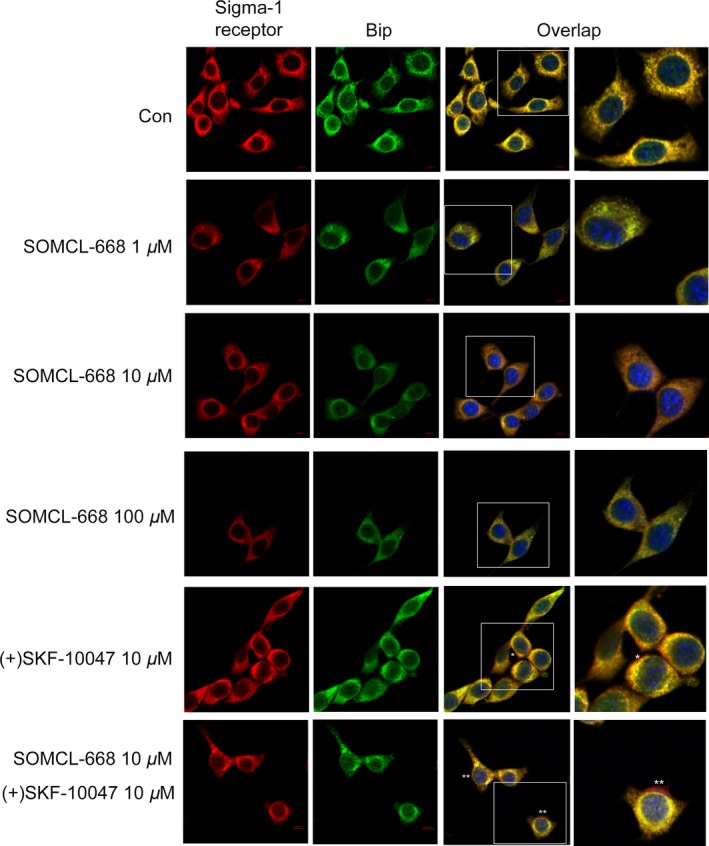
3‐Methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668) and (+)SKF‐10047 regulate the translocation of sigma‐1 receptors. The cultured HT22 cells were stimulated for 1 h by SOMCL‐668 (1, 10, 100 μM), (+)SKF‐10047 (10 μM) or the combination (30 min pretreatment for SOMCL‐668). The cells were then subjected to double immunostaining with anti‐sigma‐1 receptor and anti‐immunoglobulin‐binding protein (Bip) antibodies. Sigma‐1 receptor was marked in red, Bip, the marker of endoplasmic reticulum, in green. Nuclei were marked in blue from a Hoechst staining.
We also investigated the effects of SOMCL‐668 on neurite outgrowth and the secretion of BDNF in primary neurons. As shown in Figure 6, 10 μM SOMCL‐668 alone did not significantly alter neurite outgrowth or the secretion of BDNF. However, SOMCL‐668 significantly potentiated (+)SKF‐10047‐ stimulated neurite growth [the interaction effects for SOMCL‐668 × (+)SKF‐10047: F(1, 236) = 40.03, P < 0.01; (+)SKF‐10047 vs. SOMCL‐668+ (+)SKF‐10047, P < 0.01, two‐way ANOVA] and the production of BDNF [the interaction effects for SOMCL‐668 × (+)SKF‐10047: F(1, 16) = 5.004, P < 0.05; (+)SKF‐10047 vs. SOMCL‐668+ (+)SKF‐10047, P < 0.05, two‐way ANOVA]. Taken together, these data demonstrated that SOMCL‐668 functionally modulates sigma‐1 receptors in an allosteric manner.
Figure 6.
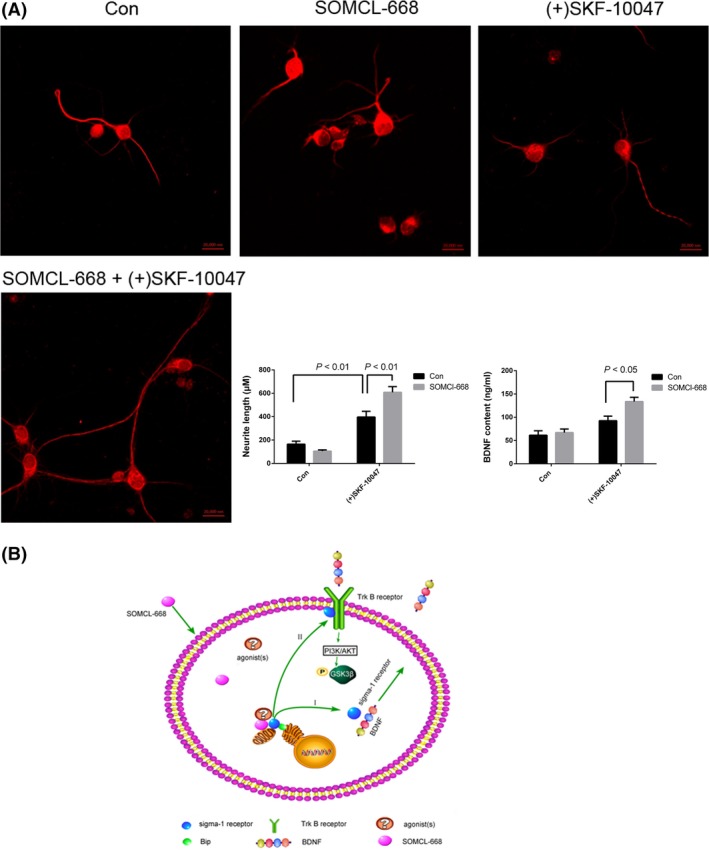
Effects of 3‐methyl‐phenyl‐2, 3, 4, 5‐tetrahydro‐1H‐benzo[d]azepin‐7‐ol (SOMCL‐668) and (+)SKF‐10047 on neurite outgrowth and secretion of brain‐derived neurotrophic factor (BDNF) in primary cultured neurons. (A) The cultured hippocampus neurons from mice were stimulated by 10 μM SOMCL‐668, 1 μM (+)SKF‐10047 or SOMCL‐668+ (+)SKF‐10047 for 48 h. Neurite length was defined as the length of the longest cell neurites (at least longer than the diameter of its soma). At least 20 cells were included in each measurement. The concentration of BDNF in the culture supernatant was detected by ELISA. Data were expressed as mean ± SEM and analyzed with two‐way ANOVA analysis. Each experiment was replicated three times. (B) The schematic diagram about possible mechanisms of SOMCL‐668 with potent antidepressant activity. Sigma‐1 receptors can be activated by SOMCL‐668 in an allosteric manner in the neuron. I. Activated sigma‐1 receptors to promote BDNF secretion, and then increase the phosphorylation of GSK3β at ser‐9. II. Additionally, activated sigma‐1 receptors acutely increase the phosphorylation of GSK3β at ser‐9 via cross talk with Trk B receptors.
Discussion
In this study, we described the antidepressant activity of SOMCL‐668, a novel allosteric modulator of the sigma‐1 receptor. SOMCL‐668 decreased the immobility time of mice in the FST and TST, which was blocked by the sigma‐1 receptor antagonist BD1047. SOMCL‐668 also attenuated the anhedonia‐like behavior in the CUMS models in a fast‐acting manner. The present data also suggested that allosteric modulating sigma‐1 receptors‐ mediated the BDNF‐GSK3β pathway, may associate with the antidepressant effect of SOMCL‐668.
One of interesting findings in our study was that SOMCL‐668 treatment efficiently improved anhedonia in a fast‐acting manner. The effectiveness was observed within a week of SOMCL‐668 treatment, which is faster than that of the serotonin transporter (SERT) and the norepinephrine transporter (NET) inhibitor venlafaxine. The exact mechanism for allosteric modulation of sigma‐1 receptors ‐mediated fast‐acting antidepressant action is currently not very clear. However, we detected a significant increase in BDNF expression in the depressive or naïve mice within 7 days of treatment with 10 mg/kg of SOMCL‐668 (Figure 4A,C). In agreement with previous study, we also demonstrated that inhibition of Trk B activation abolished sigma‐1 receptor‐ induced inhibition of the phosphorylation of GSK3β at ser‐9 (Figure 3B). Interestingly, chronic inhibition of GSK3β has been shown to increase the secretion of BDNF 33, 34.
Inactivation of GSK3β by phosphorylating ser‐9 is considered to be a critical signaling event associated with antidepressant effects of fluoxetine and ketamine 35, 36. We found that SOMCL‐668 alone promoted the phosphorylation of GSK3β (ser‐9) in vivo (Figure 2A) and enhanced the effect of sigma‐1 receptor agonist (+)SKF‐10047 in vitro (Figure 2D). To the best of our knowledge, this is the first report demonstrating that sigma‐1 receptors modulate GSK3β activity. Considering that GSK3β is a converging pathway for many antidepressants and antipsychotic drugs 37, 38, the current finding may reveal a novel mechanism for the antidepressant effect for sigma‐1 receptor allosteric modulators. Although a detailed mechanism for SOMCL‐668 to promote the phosphorylation of GSK3β at ser‐9 is not clear, we observed the elevated translocation of sigma‐1 receptors to the plasma membrane in response to sigma‐1 receptor allosteric stimulation (Figure S2). It has been reported that plasma membrane sigma‐1 receptors can directly interact with the Trk B receptor 39, we confirmed that the Trk B receptor‐AKT pathway is involved in the regulation of GSK3β activity (Figures 3 and 6B).
Alterations in sigma‐1 receptor expression in depression have not been thoroughly investigated. The present data demonstrated that there is a decreased sigma‐1 receptor expression in the hippocampus in the CUMS depression‐like mice. There was a report showing that decreased sigma‐1 receptor expression in experimental myocardial infarction that may associate with heart failure‐ induced depression 40, 41. Interestingly, a recent study showed that the plasma sigma‐1 receptor concentration is increased in response to antidepressant treatment in a small group of late‐life MDD patients 42, 43. Here we demonstrated that chronic treatment with either SOMCL‐668 or venlafaxine restored the CUMS‐suppressed sigma‐1 receptor expression. Whether decreased brain sigma‐1 receptor expression or the recovery of the sigma‐1 receptor can serve as a biomarker for MDD or be a measurement for antidepressant therapeutic effectiveness deserve further study.
Allosteric modulation requires the presence of a receptor agonist which is of ligand selection. There are a few putative endogenous molecules such as progesterone, DHEA and endogenous trace amines, that have been suggested as sigma‐1 receptor agonists 44, 45. It has been reported that DHEA and other neurosteroids may be potential antidepressants 46. Importantly, DHEA has been shown to promote BDNF production, inhibition of GSK3β activity and neurogenesis 47, 48, 49. As we previously has reported that allosteric sigma‐1 receptor modulator SKF83959 inhibited microglia‐ mediated inflammation via DHEA (Wu et al., 2015), in the present study, we failed to find that SOMCL‐668 promotes the phosphorylation of GSK3β at ser‐9 via DHEA (Figure S1D). Suggesting that the effects of SOMCL‐668 on the regulation of GSK3β activity is unlikely mediated by allosteric regulation on endogenous DHEA.
Conclusion
In conclusion, our study demonstrated that SOMCL‐668 is a rapid onset antidepressant compound that functions as a novel sigma‐1 receptor ligand with putative allosteric property. The antidepressant property of SOMCL‐668 may be associated with the sigma‐1 receptor‐regulated BDNF‐GSK3β pathway and upregulation of the sigma‐1 receptor. Due to the advantages of allosteric modulation, these findings may indicate that the development of an allosteric modulator for sigma‐1 receptors may provide a novel strategy for anti‐MDD drug discovery.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1 (A) The chemical structural of SOMCL‐668. (B) The total distance and average speed of SOMCL‐668, PRE‐084, venlafaxine and BD1047 in the open field test. All the drugs in this study did not alter the locomotor activity of mice (P > 0.05 respectively, one‐way ANOVA analysis, n = 10 in each group). (C) The novelty suppressed feeding test in the naïve mice. Chronic SOMCL‐668 (10 mg/kg, daily for 1 week) significantly decreased the latency to feed (P < 0.05, Student's t‐test, n = 10) (D) SOMCL‐668 did not promote DHEA‐induced increased phosphorylation of GSK3β (ser‐9) [the interaction effects for SOMCL‐668 × DHEA 0.1 μM: F(1, 8) = 0.6305, P = 0.4501; for SOMCL‐668 ×DHEA 1 μM: F(1, 8) = 0.129, P = 0.7287; for SOMCL‐668 × DHEA 10 μM: F(1, 8) = 0.01754, P = 0.8979; two‐way ANOVA].
Figure S2 SOMCL‐668 promotes sigma‐1 receptor agonist‐induced disassociation from Bip.
Appendix S1 Methods.
Acknowledgment
This work was supported by funds from the National Science Foundation of China (81402905, 81130023, 81373382, 81372688) and the National Basic Research Plan (973) of the Ministry of Science and Technology of China (2011CB504403). Support provided from the Priority Academic Program Development of Jiangsu Higher Education Institutes (PAPD) and the Jiangsu Key Laboratory Grant (BM2013003) is also appreciated.
Yun Wang and Lin Guo contributed equally to this work.
References
- 1. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS‐R). JAMA 2003;289:3095–3105. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi T, Su TP. Sigma‐1 receptor chaperones at the ER‐mitochondrion interface regulate Ca(2 + ) signaling and cell survival. Cell 2007;131:596–610. [DOI] [PubMed] [Google Scholar]
- 3. Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma‐1 receptor: Roles in neuronal plasticity and disease. Trends Neurosci 2012;35:762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabino V, Cottone P, Parylak SL, Steardo L, Zorrilla EP. Sigma‐1 receptor knockout mice display a depressive‐like phenotype. Behav Brain Res 2009;198:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fishback JA, Robson MJ, Xu YT, Matsumoto RR. Sigma receptors: Potential targets for a new class of antidepressant drug. Pharmacol Ther 2010;127:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Villard V, Meunier J, Chevallier N, Maurice T. Pharmacological interaction with the sigma1 (σ1)‐receptor in the acute behavioral effects of antidepressants. J Pharmacol Sci 2011;115:279–292. [DOI] [PubMed] [Google Scholar]
- 7. Wang D, Noda Y, Tsunekawa H, et al. Role of N‐methyl‐d‐aspartate receptors in antidepressant‐like effects of sigma 1 receptor agonist 1‐(3,4‐dimethoxyphenethyl)‐4‐(3‐phenylpropyl)piperazine dihydrochloride (SA‐4503) in olfactory bulbectomized rats. J Pharmacol Exp Ther 2007;322:1305–1314. [DOI] [PubMed] [Google Scholar]
- 8. Skuza G, Rogoz Z. Antidepressant‐like effect of PRE‐084, a selective sigma1 receptor agonist, in Albino Swiss and C57BL/6J mice. Pharmacol Rep 2009;61:1179–1183. [DOI] [PubMed] [Google Scholar]
- 9. Kikuchi‐Utsumi K, Nakaki T. Chronic treatment with a selective ligand for the sigma‐1 receptor chaperone, SA4503, up‐regulates BDNF protein levels in the rat hippocampus. Neurosci Lett 2008;440:19–22. [DOI] [PubMed] [Google Scholar]
- 10. Fujimoto M, Hayashi T, Urfer R, Mita S, Su TP. Sigma‐1 receptor chaperones regulate the secretion of brain‐derived neurotrophic factor. Synapse 2012;66:630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cobos EJ, Lucena G, Baeyens JM, Del Pozo E. Differences in the allosteric modulation by phenytoin of the binding properties of the sigma1 ligands [3H](+)‐pentazocine and [3H]NE‐100. Synapse 2006;59:152–161. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz TW, Holst B. Allosteric enhancers, allosteric agonists and ago‐allosteric modulators: Where do they bind and how do they act? Trends Pharmacol Sci 2007;28:366–373. [DOI] [PubMed] [Google Scholar]
- 13. Christopoulos A, Kenakin T. G protein‐coupled receptor allosterism and complexing. Pharmacol Rev 2002;54:323–374. [DOI] [PubMed] [Google Scholar]
- 14. Guo L, Zhao J, Jin G, et al. SKF83959 is a potent allosteric modulator of sigma‐1 receptor. Mol Pharmacol 2013;83:577–586. [DOI] [PubMed] [Google Scholar]
- 15. Zvejniece L, Vavers E, Svalbe B, et al. The cognition‐enhancing activity of E1R, a novel positive allosteric modulator of sigma‐1 receptors. Br J Pharmacol 2014;171:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou SL, Chu HY, Jin GZ, Cui JM, Zhen XC. Effects of SKF83959 on the excitability of hippocampal CA1 pyramidal neurons: A modeling study. Acta Pharmacol Sin 2014;35:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fang X, Guo L, Jia J, et al. SKF83959 is a novel triple reuptake inhibitor that elicits anti‐depressant activity. Acta Pharmacol Sin 2013;34:1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chu HY, Wu Q, Zhou S, et al. SKF83959 suppresses excitatory synaptic transmission in rat hippocampus via a dopamine receptor‐independent mechanism. J Neurosci Res 2011;89:1259–1266. [DOI] [PubMed] [Google Scholar]
- 19. Chu HY, Gu Q, Jin GZ, Hu GY, Zhen X. Electrophysiological effects of SKF83959 on hippocampal CA1 pyramidal neurons: Potential mechanisms for the drug's neuroprotective effects. PLoS One 2010;5:e13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen XQ, Zhang J, Neumeyer JL, et al. Arylbenzazepines are potent modulators for the delayed rectifier K+ channel: A potential mechanism for their neuroprotective effects. PLoS One 2009;4:e5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J, Huang J, Song Z, et al. Structural manipulation on the catecholic fragment of dopamine D(1) receptor agonist 1‐phenyl‐N‐methyl‐benzazepines. Eur J Med Chem 2014;85:16–26. [DOI] [PubMed] [Google Scholar]
- 22. Guo L, Chen Y, Zhao R, et al. Allosteric modulation of sigma‐1 receptors elicits anti‐seizure activities. Br J Pharmacol 2015;172:4052–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: A primary screening test for antidepressants. Arch Int Pharmacodyn Ther 1977;229:327–336. [PubMed] [Google Scholar]
- 24. Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology 1985;85:367–370. [DOI] [PubMed] [Google Scholar]
- 25. Kong H, Zeng XN, Fan Y, et al. Aquaporin‐4 knockout exacerbates corticosterone‐induced depression by inhibiting astrocyte function and hippocampal neurogenesis. CNS Neurosci Ther 2014;20:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ide S, Fujiwara S, Fujiwara M, et al. Antidepressant‐like effect of venlafaxine is abolished in mu‐opioid receptor‐knockout mice. J Pharmacol Sci 2010;114:107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elsayed M, Banasr M, Duric V, Fournier NM, Licznerski P, Duman RS. Antidepressant effects of fibroblast growth factor‐2 in behavioral and cellular models of depression. Biol Psychiatry 2012;72:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsai SY, Hayashi T, Harvey BK, et al. Sigma‐1 receptors regulate hippocampal dendritic spine formation via a free radical‐sensitive mechanism involving Rac1xGTP pathway. Proc Natl Acad Sci USA 2009;106:22468–22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: Recent developments and future needs. Trends Pharmacol Sci 2002;23:238–245. [DOI] [PubMed] [Google Scholar]
- 30. Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: Novel mechanisms for rapid‐acting agents. Trends Neurosci 2012;35:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mai L, Jope RS, Li X. BDNF‐mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem 2002;82:75–83. [DOI] [PubMed] [Google Scholar]
- 32. Ito K, Hirooka Y, Matsukawa R, Nakano M, Sunagawa K. Decreased brain sigma‐1 receptor contributes to the relationship between heart failure and depression. Cardiovasc Res 2012;93:33–40. [DOI] [PubMed] [Google Scholar]
- 33. Gimenez‐Cassina A, Lim F, Diaz‐Nido J. Chronic inhibition of glycogen synthase kinase‐3 protects against rotenone‐induced cell death in human neuron‐like cells by increasing BDNF secretion. Neurosci Lett 2012;531:182–187. [DOI] [PubMed] [Google Scholar]
- 34. Long ZM, Zhao L, Jiang R, et al. Valproic acid modifies synaptic structure and accelerates neurite outgrowth via the glycogen synthase kinase‐3beta signaling pathway in an Alzheimer's disease model. CNS Neurosci Ther 2015;21:887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polter AM, Yang S, Jope RS, Li X. Functional significance of glycogen synthase kinase‐3 regulation by serotonin. Cell Signal 2012;24:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase‐3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry 2011;16:1068–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Omata N, Chiu CT, Moya PR, et al. Lentivirally mediated GSK‐3beta silencing in the hippocampal dentate gyrus induces antidepressant‐like effects in stressed mice. Int J Neuropsychopharmacol 2011;14:711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsai SJ, Liou YJ, Hong CJ, Yu YW, Chen TJ. Glycogen synthase kinase‐3beta gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J 2008;8:384–390. [DOI] [PubMed] [Google Scholar]
- 39. Kimura Y, Fujita Y, Shibata K, Mori M, Yamashita T. Sigma‐1 receptor enhances neurite elongation of cerebellar granule neurons via TrkB signaling. PLoS One 2013;8:e75760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hashimoto K. Sigma‐1 receptor chaperone and brain‐derived neurotrophic factor: Emerging links between cardiovascular disease and depression. Prog Neurobiol 2013;100:15–29. [DOI] [PubMed] [Google Scholar]
- 41. Ito K, Hirooka Y, Sunagawa K. Brain sigma‐1 receptor stimulation improves mental disorder and cardiac function in mice with myocardial infarction. J Cardiovasc Pharmacol 2013;62:222–228. [DOI] [PubMed] [Google Scholar]
- 42. Shimizu H, Takebayashi M, Tani M, et al. Sigma‐1 receptor concentration in plasma of patients with late‐life depression: A preliminary study. Neuropsychiatr Dis Treat 2013;8:1867–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kishi T, Yoshimura R, Okochi T, et al. Association analysis of SIGMAR1 with major depressive disorder and SSRI response. Neuropharmacology 2010;58:1168–1173. [DOI] [PubMed] [Google Scholar]
- 44. Ramachandran S, Chu UB, Mavlyutov TA, Pal A, Pyne S, Ruoho AE. The sigma1 receptor interacts with N‐alkyl amines and endogenous sphingolipids. Eur J Pharmacol 2009;609:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Targeting ligand‐operated chaperone sigma‐1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets 2011;15:557–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peixoto C, Devicari Cheda JN, Nardi AE, Veras AB, Cardoso A. The effects of dehydroepiandrosterone (DHEA) in the treatment of depression and depressive symptoms in other psychiatric and medical illnesses: A systematic review. Curr Drug Targets 2014;15:901–914. [DOI] [PubMed] [Google Scholar]
- 47. Moriguchi S, Shinoda Y, Yamamoto Y, et al. Stimulation of the sigma‐1 receptor by DHEA enhances synaptic efficacy and neurogenesis in the hippocampal dentate gyrus of olfactory bulbectomized mice. PLoS One 2013;8:e60863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rahmani A, Shoae‐Hassani A, Keyhanvar P, Kheradmand D, Darbandi‐Azar A. Dehydroepiandrosterone stimulates nerve growth factor and brain derived neurotrophic factor in cortical neurons. Adv Pharmacol Sci 2013;2013:506191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pluchino N, Russo M, Santoro AN, Litta P, Cela V, Genazzani AR. Steroid hormones and BDNF. Neuroscience 2013;239:271–279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) The chemical structural of SOMCL‐668. (B) The total distance and average speed of SOMCL‐668, PRE‐084, venlafaxine and BD1047 in the open field test. All the drugs in this study did not alter the locomotor activity of mice (P > 0.05 respectively, one‐way ANOVA analysis, n = 10 in each group). (C) The novelty suppressed feeding test in the naïve mice. Chronic SOMCL‐668 (10 mg/kg, daily for 1 week) significantly decreased the latency to feed (P < 0.05, Student's t‐test, n = 10) (D) SOMCL‐668 did not promote DHEA‐induced increased phosphorylation of GSK3β (ser‐9) [the interaction effects for SOMCL‐668 × DHEA 0.1 μM: F(1, 8) = 0.6305, P = 0.4501; for SOMCL‐668 ×DHEA 1 μM: F(1, 8) = 0.129, P = 0.7287; for SOMCL‐668 × DHEA 10 μM: F(1, 8) = 0.01754, P = 0.8979; two‐way ANOVA].
Figure S2 SOMCL‐668 promotes sigma‐1 receptor agonist‐induced disassociation from Bip.
Appendix S1 Methods.