

Summary
Background
Parkinson disease (PD) is a neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra (SN) and diminished dopamine content in the striatum, which is at least partly associated with α‐synuclein protein overexpression in these neurons. Recent reports show that 7,8‐dihydroxyflavone (DHF), a TrkB agonist, has beneficial effects in animal model of PD. However, it is unclear whether the therapeutic effects of DHF are associated with the expression of α‐synuclein.
Aims
In this study, we investigated the protective effects of DHF on 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced deficit of motor functions, the loss of dopaminergic neurons and the expression of α‐synuclein as well as antioxidative activity in the C57BL/6 mice.
Results
Mice were treated with MPTP (30 mg/kg, i.p.) once a day for 5 days to induce dopaminergic neuron death in the SN. DHF (5 mg/kg, i.p.) was administrated once a day from the first day of MPTP injection until 9 days after the last injection of MPTP. Behavioral tests showed that DHF succeeded in ameliorating the impaired motor functions in the MPTP‐treated mice. The immunohistochemical assay showed that the amelioration of motor function was accompanied by a reduction in the loss of dopaminergic neurons in the SN and striatum. Western blot analyses showed that DHF prevented the inactivation of TrkB and suppressed α‐synuclein overexpression in the SN and striatum following MPTP treatment. Antioxidative activity detection revealed that DHF prevented MPTP‐induced reduction in glutathione and total superoxide dismutase activity in the SN and striatum.
Conclusion
Taken together, these results indicate that DHF treatment may suppress the accumulation of α‐synuclein and oxidative stress via activating TrkB and subsequently block the loss of dopaminergic neurons in the SN and striatum, thereby ameliorating MPTP‐induced motor deficits in the C57BL/6 mice.
Keywords: 7,8‐dihydroxyflavone; Antioxidative activity; Dopaminergic neuron; MPTP; Parkinson disease; α‐synuclein
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease worldwide affecting 1% at the age of 60 and 5% for people aged over 85 years 1, 2, 3. The pathological feature of PD is characterized by the loss of dopaminergic neurons in the substantia nigra (SN) and the loss of dopamine content in the striatum 1, 2, 3. Previous studies have proposed that the loss of dopaminergic neurons in PD is at least partly due to the overexpression of α‐synuclein in the cytoplasm of these neurons. Indeed, nigral α‐synuclein expression increases with age in humans and rhesus monkeys 4. Further genetic studies have shown that the overexpression of wild type or a mutated form of α‐synuclein increases copper‐induced dopaminergic cell death 5. On the contrary, downregulation of α‐synuclein expression can rescue dopaminergic cells from MPTP‐induced cell death both in vitro and in vivo 6, 7, 8, 9, 10. Alternatively, oxidative injury is also considered as a pivotal role in pathogenesis of PD 11, 12. Human postmortem 13, 14 and animal model 15 studies have suggested that oxidative damage occurred in the development of PD. These observations suggest that the inhibition of nigral α‐synuclein expression and oxidative stress may be a potential therapeutic for PD.
Brain‐derived neurotrophic factor (BDNF) and its receptor tropomyosin‐related kinase receptor type B (TrkB) are actively produced and trafficked in multiple regions in the brain, where they influence neuronal activity, survival, synaptic plasticity, and neurogenesis throughout life 16, 17. Given its neurotrophic actions on neuronal populations, BDNF is of particular therapeutic interest involved in neurodegenerative diseases including PD. Indeed, BDNF treatment prevents the loss of dopaminergic neurons induced by MPTP in the SN of rats 18. Furthermore, intrathecal infusion of BDNF exhibits beneficial anatomical and behavioral effects in MPTP‐induced parkinsonism in monkeys, by reducing nigral cell loss and enhancing striatal reinnervation 19. Unfortunately, the key challenge in the BDNF therapy is drug delivery to the central nervous system, as it is a moderately sized and charged protein, and only minimal amount of BDNF crosses the blood–brain barrier via peripheral administration 20.
Recently, 7,8‐dihydroxyflavone (DHF) has been identified as a small‐molecule compound that binds with high affinity and specificity to the TrkB receptor. As a flavone derivative, DHF has been shown to promote TrkB receptor dimerization and autophosphorylation and activate PI3K/Akt, MAPK, and Erk1/2 signaling cascades just as well as BDNF 21. A more recent study has shown intervention with DHF blocks further loss of dopaminergic terminals and ameliorates motor deficits in the progressive MPTP mouse model 22. As aforementioned, the loss of dopaminergic neurons in PD is partly due to the overexpression of α‐synuclein and oxidative stress in these neurons, and we therefore propose that DHF may suppress α‐synuclein and exert antioxidative activity, and subsequently reduce the loss of dopaminergic neurons, thereby rescuing motor deficits. In this study, we investigated this hypothesis using behavioral, immunohistochemical, and Western blot assessments in the MPTP‐induced mouse model of PD.
Materials and Methods
C57BL/6 mice (25–30 g) were obtained from Charles River (Beijing Office, China) and maintained at Children's Hospital of Chongqing Medical University Animal Care Centre. Animals were housed in plastic cages with free access to food and water and maintained in a temperature‐controlled room (21°C) with a 12‐h/12‐h light/dark cycle. All experiments and procedures were approved by Chongqing Medical University Animal Care and Use Committee, and every effort was made to minimize both the animal suffering and the number of animals used.
Drugs and Treatments
MPTP was purchased from Sigma‐Aldrich (St. Louis, MO, USA) and dissolved in sterile saline. DHF was purchased from Tokyo Chemical Industry (Tokyo, Japan) and dissolved in sterile saline containing 10% ethanol. Anti‐α‐synuclein and anti‐tyrosine hydroxylase (TH) antibodies were obtained from BD Transduction Laboratories. Anti‐TrkB antibody was obtained from Cell Signaling. Anti‐phospho‐TrkB (p‐TrkB) antibody was obtained from Santa Cruz. Anti‐β‐actin antibody was obtained from Abcam. Glutathione (GSH) and superoxide dismutase (SOD) test Kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Mice received intraperitoneal (i.p.) injection of 30 mg/kg parkinsonian toxin MPTP hydrochloride once a day for 5 days to induce dopaminergic neuron death in the substantia nigra, while the control mice received equal amount of saline injection. DHF (5 mg/kg, i.p.) was injected into the MPTP‐treated mice once a day from the first day of MPTP injection until 9 days after the last injection of MPTP (Figure 1).
Figure 1.
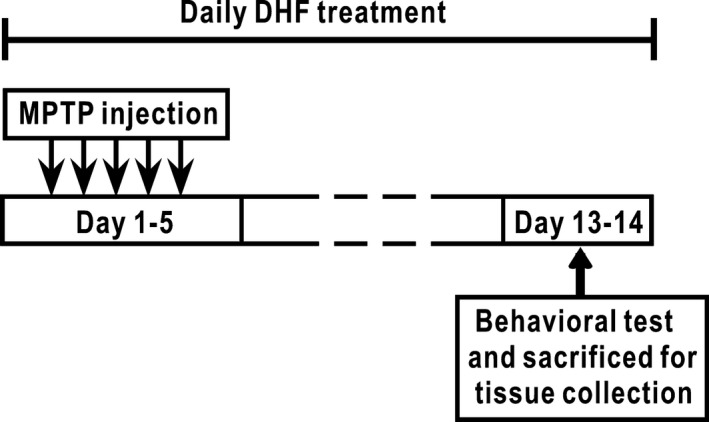
Experimental design. Mice received a chronic MPTP treatment (30 mg/kg per day, i.p.) or the same volume of sterile saline for 5 days, and DHF (5 mg/kg, i.p.) was administrated daily throughout the experiment. Seven days after the last injection of MPTP, behavioral tests were performed, and brain tissues were collected immediately after behavioral tests.
Rotarod Test
The rotarod test was performed as previously described, with modifications 23. In brief, 1 week after the last MPTP injection, mice received 4 rounds of training on the rotarod (Stoelting Co.). In the first two rounds of training, the rotarod was maintained at constant speed of 20 rpm for 3 min. In the second two rounds of training, the rotarod reversed rotation direction every 3 turns at the constant speed of 20 rpm for 3 min. Twenty‐four hours after the last round of training, mice received formal rotarod testing in which the rotarod reversed rotation direction every 3 turns at the constant speed of 20 rpm. Mice were tested 10 times at 20‐min intervals, and the time that they remained on the rotarod during each test was recorded. Maximum test time (cut‐off limit) was 300 s. The motor performance of the mouse was expressed as the latency to fall off the rotarod. The rotarod was cleaned with 70% ethanol and water between tests.
Pole Test
The pole consisted of a thin wooden cylinder (length: 50 cm, diameter: 1.5 cm) and a cross‐shaped wooden base placed in a clean cage. Rubber bands were wrapped around the cylinder at intervals of approximately 1.5 inches to increase traction. Mice were placed at the top of the pole facing downwards, and latency to descend the pole was measured. Trials were excluded if the mouse jumped or slid down the pole rather than climbed down. On the first day, each mouse was trained with two trials. On the second day, each mouse was given five trials before the rotarod test and the lowest latency to descend the pole was analyzed. The pole was cleaned with 70% ethanol and water between tests.
Wire Suspension Test
The wire (length: 80 cm, height: 25 cm) was fixed horizontally between two platforms. Each animal was hung with its paws on the middle of the wire, and the time needed to reach one platform was recorded. The maximal time allowed was set at 120 s. On the first day, each mouse was trained with two trials. On the second day, each mouse was given five trials before the rotarod test and the lowest latency to reach the platform was analyzed.
Western Immunoblotting
Brain tissues were lysed on ice in the lysis buffer, and then the solution was centrifuged at 18,000 × g for 10 min at 4°C. Supernatant was collected, and protein concentration was determined by BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amount of protein samples was mixed with 4 × sample buffer, boiled at 95°C for 5 min, and separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). Proteins were then transferred to Immobilon‐PTM polyvinylidene fluoride (PVDF) membranes (Bio‐Rad, Hercules, CA, USA). The membranes were blocked with 5% nonfat milk in Tris‐buffered saline containing 0.1% Tween‐20 (TBST) for 1 h at room temperature and then incubated overnight at 4°C with primary antibody. After washing 3 × 5 min in TBST, membranes were incubated with horseradish peroxidase‐conjugated secondary antibody for one hour at room temperature. After another three washes with TBST, protein was visualized in the Bio‐Rad Imager using ECL Western blotting substrate (Pierce). Immunoblotting with anti‐β‐actin was used to control equal loading and protein quality. The band intensity of each protein was quantified by the Bio‐Rad Quantity One software.
TH Staining and Quantification
Mice were deeply anesthetized with urethane (1.5 g/kg, i.p.) and then perfused with 0.9% saline and 4% paraformaldehyde. Brains were taken out and fixed in 4% paraformaldehyde for one more day before they were transferred to 30% sucrose/PBS solution for cryoprotection. After the brains had sunk to the bottom of sucrose solution, they were sectioned into 30‐μm slices using Leica cryostat. After blocking and permeabilization using PBS solution containing 1% BSA and 0.2% Triton X‐100 for 30 min at room temperature, the brain slices were incubated with anti‐tyrosine hydroxylase antibody (1:800 dilution) for 24 hours at 4 degree. Finally, the tyrosine hydroxylase‐positive neurons on the slices were stained and visualized under light microscope using the anti‐mouse Ig HRP detection kit according to the manufacturer's instruction.
The number of TH‐positive cells was quantified in brain sections with typical morphology of the SN, as described previously 24. Briefly, the number of TH‐positive neurons was counted manually at six‐section intervals throughout the entire extent of SN by bright‐field microscopy using ImageJ software. To quantify changes in the number of TH‐positive neurons in the SN, the number of TH‐positive neurons in control mice was normalized to 100%, and the number of TH‐positive neurons in other groups was expressed as a percentage of the control.
The optical density of the TH‐positive neuronal terminal staining in the mouse dorsolateral striatum was quantified using NIH ImageJ software. The optical density from the overlying corpus callosum was used as a background and subtracted from every measurement in the striatum 25. The optical density in the experimental group was normalized to the value from the control group.
Antioxidative Activity Analysis
Brain tissues of SN or striatum were homogenized with PBS or SOD detection buffer, followed by centrifugation at 4000 g for 10 min at 4°C. The supernatant was used to test glutathione (GSH) level and total superoxide dismutase (SOD) activity using dithio‐binitrobenzoic acid method and xanthine/xanthine oxidase method, respectively. Reaction product was determined by microplate reader (BioTek Instruments).
Statistical Analysis
All data were expressed as mean ± SEM. The differences of rotarod test among different groups of mice were analyzed by two‐way ANOVA, followed by Tukey's post hoc test. The data of all other experiments were analyzed by one‐way ANOVA, followed by Tukey's post hoc test. Significance level was set at P < 0.05.
Results
DHF Ameliorates MPTP‐induced Motor Deficits
It has been well documented that a core symptom of PD is motor abnormality 26, 27, and a recent study shows that the intervention with DHF ameliorates MPTP‐induced motor deficit in gait analysis 22. To further determine whether DHF can rescue MPTP‐induced motor deficits, we here introduced another 3 behavioral tasks: rotarod test, pole test and wire suspension test. In rotarod test, the mice treated with MPTP spent significantly less time on the rod during test compared with those treated with saline (Figure 2A), indicating an impairment of motor balance and coordination. Importantly, daily DHF treatment (5 mg/kg, i.p.) fully rescued the MPTP‐induced motor deficit, as reflected by a dramatic increase in the latency to fall off (saline: n = 16; DHF: n = 16; MPTP + saline: n = 16; MPTP + DHF: n = 18; Figure 2A). In pole test, the mice treated with MPTP spent much more time descending the pole compared with those treated with saline, whereas DHF treatment significantly shortened the descending time during test (saline: n = 16, 6.1 ± 0.5 s; DHF: n = 16, 5.8 ± 0.4 s, P > 0.05 vs. saline; MPTP + saline: n = 16, 7.6 ± 0.3 s, P < 0.05 vs. saline, P < 0.01 vs. DHF; MPTP + DHF: n = 18, 5.9 ± 0.2 s, P > 0.05 vs. saline, P < 0.01 vs. MPTP + saline; Figure 2B). Similarly, the mice treated with MPTP spent much more time reaching the platform during the wire suspension test, and DHF treatment ameliorated the motor capacity to control levels, as reflected by a similar time in reaching the platform (saline: n = 16, 10.5 ± 0.7 s; DHF: n = 16, 10.4 ± 0.9 s, P > 0.05 vs. saline; MPTP + saline: n = 16, 14.9 ± 1.0 s, P < 0.01 vs. saline, P < 0.01 vs. DHF; MPTP + DHF: n = 18, 11.3 ± 0.8 s, P > 0.05 vs. saline, P < 0.05 vs. MPTP + saline; Figure 2C).
Figure 2.
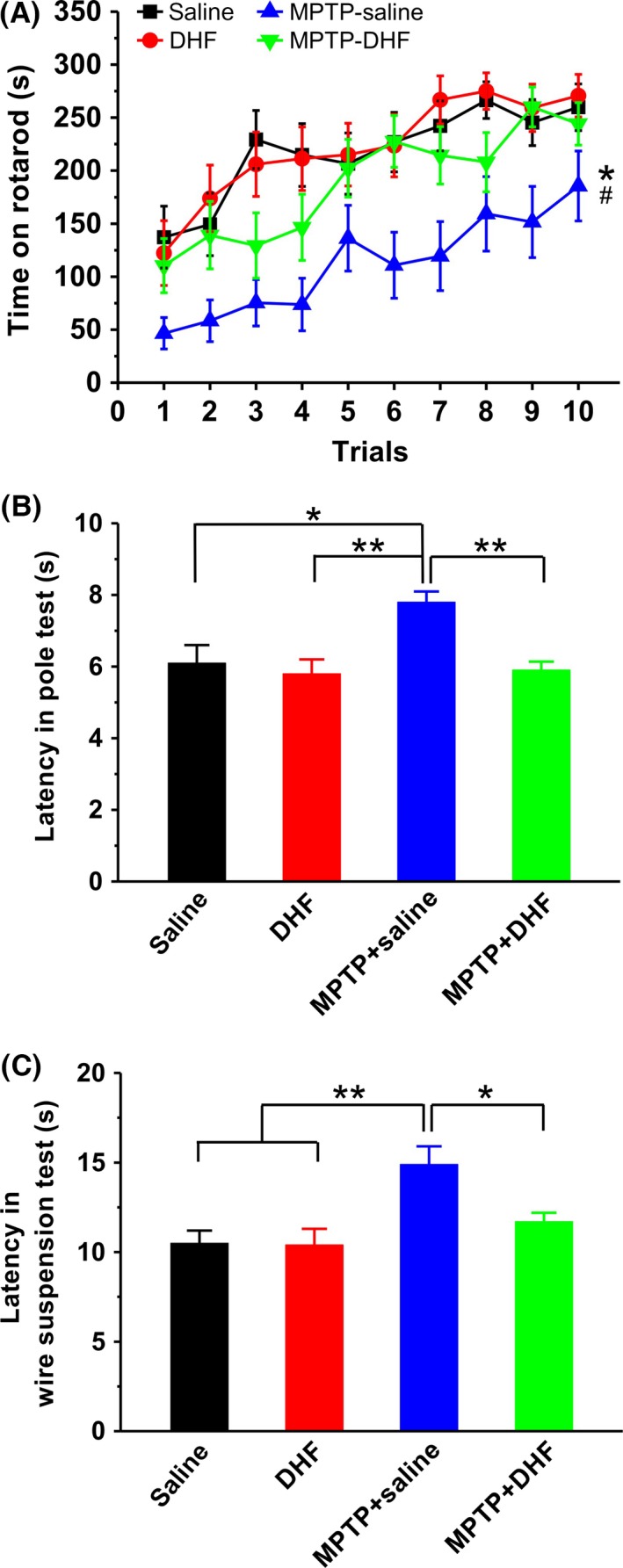
Behavioral analysis of motor functions. (A) Latency to fall off the rod in the rotarod test was significantly reduced by MPTP administration, and DHF treatment restored the latency to control level. Two‐way ANOVA was used in this experiment (group: F 3, 620 = 70.111, P < 0.001; trial: F 9, 620 = 22.352, P < 0.001; group × trial, F 27, 620 = 0.491, P = 0.987). (B) Latency to descend in the pole test was markedly increased by MPTP administration, and DHF treatment restored the latency to control level. One‐way ANOVA was used in this experiment (F 3, 62 = 6.366, P = 0.001). (C) Latency to reach the platform in the wire suspension test was significantly increased by MPTP administration, and DHF treatment restored the latency to control level. One‐way ANOVA was used in this experiment (F 3, 62 = 5.702, P = 0.002). Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01.
DHF Reduces MPTP‐induced Loss of Dopaminergic Neurons
To confirm the protective effects of DHF on dopaminergic neurons after MPTP insult, we examined the level of TH‐positive neurons in the SN and neuronal terminals in the striatum. The results showed that the MPTP administration markedly decreased the number of TH‐positive neurons in the SN compared with saline control, and DHF treatment significantly blocked the loss of TH‐positive neurons (saline: n = 7; DHF: n = 7, 104.7 ± 2.5% saline, P > 0.05 vs. saline; MPTP + saline: n = 7, 61.2 ± 2.8% saline, P < 0.01 vs. saline; MPTP + DHF: n = 7, 90.9 ± 4.0% saline, P < 0.05 vs. saline, P < 0.01 vs. MPTP + saline; Figure 3A). Similarly, MPTP administration significantly decreased the neuronal terminals in the striatum, as reflected by a dramatic decrease in the optical density of TH, whereas DHF treatment significantly ameliorated it although the optical density of TH is still lower than control level (saline: n = 7; DHF: n = 7, 106.4 ± 10.8% saline, P > 0.05 vs. saline; MPTP + saline: n = 7, 33.0 ± 6.2% saline, P < 0.01 vs. saline; MPTP + DHF: n = 7, 70.9 ± 7.9% saline, P < 0.05 vs. saline, P < 0.01 vs. MPTP + saline; Figure 3B).
Figure 3.
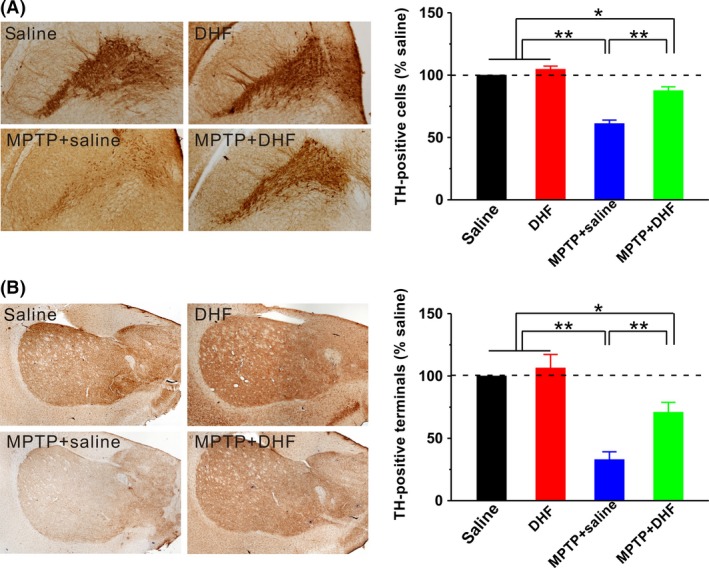
Immunohistochemical analysis of TH‐positive neurons. Representative photomicrographs on the left panels, along with bar graph summarizing group data on the right panel. The number of dopaminergic neurons in the SN (A) and neuronal terminals in the striatum (B) was significantly decreased by MPTP administration, and DHF treatment significantly restored it although the number is still less than control level. One‐way ANOVA was used in this experiment (SN: F 3, 24 = 87.347, P < 0.001; striatum: F 3, 24 = 20.614, P < 0.001). Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01.
DHF Rescues the Decrease in MPTP‐induced TH Expression
To further determine the protective effects of DHF on dopaminergic neurons, we next examine the TH expression in the SN and striatum using Western immunoblotting. The results showed that the MPTP administration significantly decreased TH expression in the SN (saline: n = 10; MPTP + saline: n = 10, 62.9 ± 7.3% saline, P < 0.01 vs. saline; Figure 4A) and striatum (saline: n = 10; MPTP + saline: n = 10, 56.8 ± 5.5% saline, P < 0.01 vs. saline; Figure 4B) compared with saline control. Consistent with recent reports 21, 22, DHF treatment fully rescued the decrease in TH expression in both the SN (DHF: n = 10, 114.6 ± 7.7% saline, P > 0.05 vs. saline; MPTP + DHF: n = 10, 91.0 ± 8.4% saline, P > 0.05 vs. saline, P < 0.05 vs. MPTP + saline; Figure 4A) and striatum (DHF: n = 10, 113.1 ± 14.4% saline, P > 0.05 vs. saline; MPTP + DHF: n = 10, 94.7 ± 17.5% saline, P > 0.05 vs. saline, P < 0.05 vs. MPTP + saline; Figure 4B).
Figure 4.
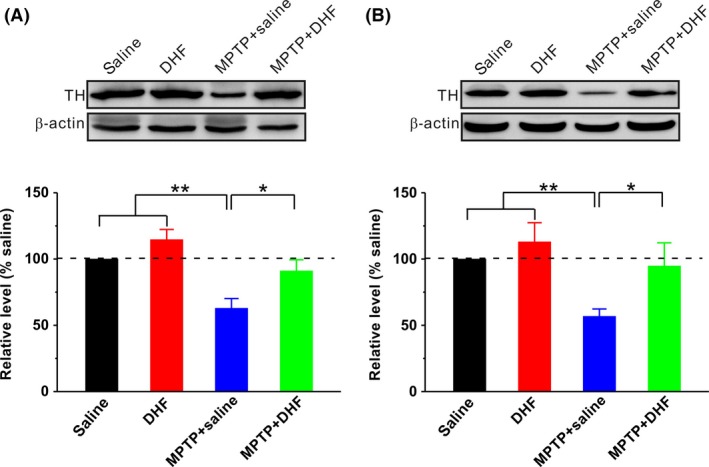
Western blot analysis of TH. Representative blots on the upper panels, along with bar graph summarizing group data on the bottom panel. MPTP administration significantly decreased TH expression in the SN (A) and striatum (B), and DHF treatment restored the expression of TH to control level. One‐way ANOVA was used in this experiment (SN: F 3, 36 = 10.347, P < 0.001; striatum: F 3, 36 = 4.294, P = 0.011). Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01.
DHF Inhibits the MPTP‐Induced α‐synuclein Overexpression
Previous studies have reported that the loss of dopaminergic neurons in PD is associated with the overexpression of α‐synuclein in the cytoplasm of these neurons 4, 5, 6, 7, 8, 9, 10. We next wanted to determine the effects of DHF treatment on the expression of α‐synuclein. As shown in Figure 5, MPTP administration significantly increased the expression of α‐synuclein in both the SN (saline: n = 6; MPTP + saline: n = 6, 149.2 ± 18.2% saline, P < 0.05 vs. saline; Figure 5A) and striatum (saline: n = 6; MPTP + saline: n = 6, 144.3 ± 16.9% saline, P < 0.05 vs. saline; Figure 5B) compared with saline control. More importantly, DHF treatment reduced the expression of α‐synuclein to control level in both the SN (DHF: n = 6, 85.5 ± 7.2% saline, P > 0.05 vs. saline; MPTP + DHF: n = 6, 112.5 ± 9.7% saline, P > 0.05 vs. saline, P < 0.05 vs. MPTP + saline; Figure 5A) and striatum (DHF: n = 6, 100.8 ± 2.5% saline, P > 0.05 vs. saline; MPTP + DHF: n = 6, 95.5 ± 7.7% saline, P > 0.05 vs. saline, P < 0.01 vs. MPTP + saline; Figure 5B).
Figure 5.
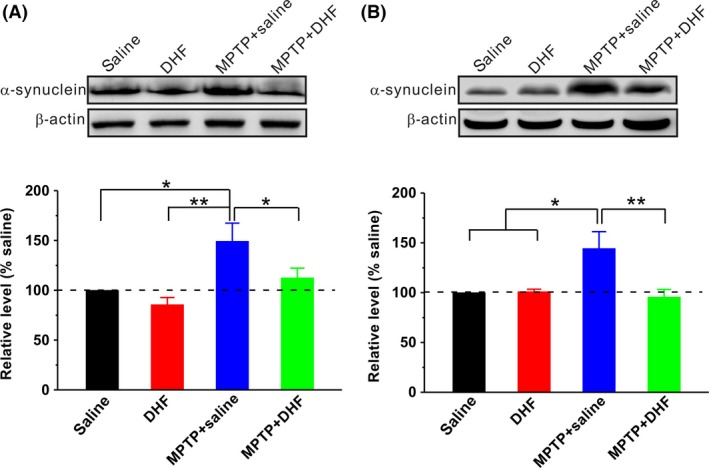
Western blot analysis of α‐synuclein. Representative blots on the upper panels, along with bar graph summarizing group data on the bottom panel. MPTP administration significantly increased α‐synuclein expression in the SN (A) and striatum (B), and DHF treatment restored the expression of α‐synuclein to control level. One‐way ANOVA was used in this experiment (SN: F 3, 20 = 6.258, P = 0.004; striatum: F 3, 20 = 5.944, P = 0.005). Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01.
DHF Reverses MPTP‐induced Decrease in p‐TrkB
Previous study has shown that DHF exerts neuroprotective effects by acting as a selective TrkB agonist 21. There are two forms of TrkB exist, full‐length isoform (TrkB‐FL) and a truncated protein lacking the tyrosine‐kinase domain, which is strikingly similar to the inactive TrkB‐T1 isoform (TrkB‐T1). Thus, we next examined the effects of DHF on all TrkB isoforms (panTrkB, including TrkB‐FL and TrkB‐T1) and p‐TrkB expression. The results showed that both MPTP and DHF had no effect on the expression of panTrkB in the SN (saline: n = 8; DHF: n = 8, 102.4 ± 9.5% saline, P > 0.05 vs. saline; MPTP + saline: n = 8, 99.7 ± 6.3% saline, P > 0.05 vs. saline; MPTP + DHF: n = 8, 104.0 ± 8.7% saline, P > 0.05 vs. saline, P > 0.05 vs. MPTP + saline; Figure 6A and C) and striatum (saline: n = 8; DHF: n = 8, 104.7 ± 7.7% saline, P > 0.05 vs. saline; MPTP + saline: n = 8, 95.6 ± 8.2% saline, P > 0.05 vs. saline; MPTP + DHF: n = 8, 96.2 ± 7.3% saline, P > 0.05 vs. saline, P > 0.05 vs. MPTP + saline; Figure 6B and D). However, the expression of p‐TrkB dramatically decreased after MPTP treatment in the SN (saline: n = 8; MPTP + saline: n = 8, 66.5 ± 3.2% saline, P < 0.05 vs. saline; Figure 6A and E) and striatum (saline: n = 8; MPTP + saline: n = 8, 68.9 ± 10.1% saline, P < 0.05 vs. saline; Figure 6B and F). As expected, DHF treatment reversed the expression of p‐TrkB in the SN (DHF: n = 8, 169.2 ± 16.1% saline, P < 0.05 vs. saline; MPTP + DHF: n = 8, 97.1 ± 5.7% saline, P > 0.05 vs. saline, P < 0.05 vs. MPTP + saline; Figure 6A and E) and striatum (DHF: n = 8, 131.92 ± 10.8% saline, P < 0.05 vs. saline; MPTP + DHF: n = 8, 107.2 ± 7.6% saline, P > 0.05 vs. saline, P < 0.01 vs. MPTP + saline; Figure 6B and F). These results suggest that DHF induces the TrkB activation in MPTP‐treated mice.
Figure 6.
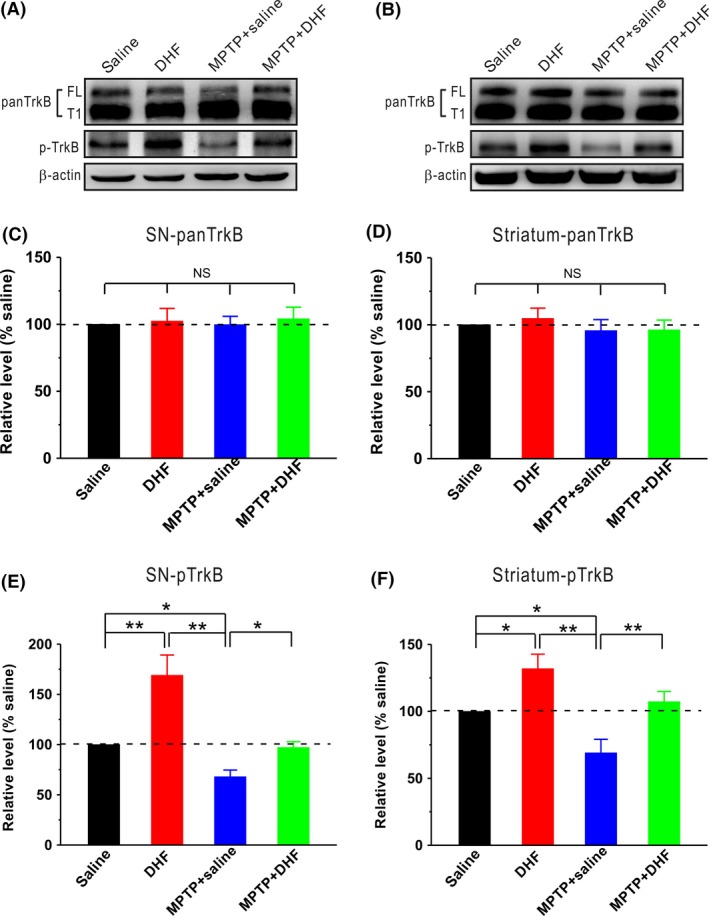
Western blot analysis of TrkB activation. (A–B) Representative blots showed the expressions of panTrkB and p‐TrkB in the SN (A) and striatum (B). (C–D) MPTP and DHF had no effect on panTrkB expression in the SN (C) and striatum (D) compared with control. One‐way ANOVA was used in this experiment (SN: F 3, 28 = 0.084, P = 0.968; striatum: F 3, 28 = 0.387, P = 0.763). (E–F) p‐TrkB expression was dramatically reduced in the SN (E) and striatum (F) following MPTP treatment, and DHF treatment restored the expression of p‐TrkB to control level (SN: F 3, 28 = 12.819, P < 0.001; striatum: F 3, 28 = 9.764, P < 0.001). Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01.
DHF Reverses MPTP‐induced Decrease in GSH and SOD
In addition, it has been well documented that MPTP destroys the defense of antioxidant enzymes in mice brain 28, whereas DHF has been shown to exert antioxidant action in in vitro model of PD 29. We therefore examined whether DHF can suppress MPTP‐induced oxidative stress in vivo. GSH was an important antioxidant, and SOD was a critical antioxidant enzyme in the body. As shown in Table 1, MPTP treatment significantly led to a decrease in GSH and SOD activity. Compared with saline treatment, DHF dramatically rescued the GSH and SOD activity following MPTP treatment in the SN and striatum (Table 1). These results indicated a powerful antioxidative capacity of DHF in MPTP‐treated mice.
Table 1.
Effect of DHF on GSH level (n = 8 in each group) and SOD activity (n = 6 in each group) in the SN and striatum
| Groups | GSH level (mg/g protein) | SOD activity (U/mg protein) | ||
|---|---|---|---|---|
| SN | striatum | SN | striatum | |
| Saline | 14.98 ± 1.09 | 19.73 ± 0.97 | 36.17 ± 1.84 | 42.27 ± 1.58 |
| DHF | 19.60 ± 1.77 | 22.31 ± 0.84 | 40.25 ± 1.23 | 43.42 ± 1.68 |
| MPTP + saline | 6.05 ± 0.91a, b, c | 15.02 ± 0.43a, b, c | 27.41 ± 1.82 a, b, c | 29.51 ± 1.61 a, b, c |
| MPTP + DHF | 18.33 ± 2.03 | 19.96 ± 1.08 | 35.12 ± 1.89 | 37.81 ± 1.21 |
Data are expressed as mean ± SEM, a P < 0.05 vs. saline, b P < 0.05 vs. DHF, c P < 0.05 vs. MPTP + DHF.
Discussion
In the present study, we confirm that DHF, a TrkB agonist, blocks the loss of dopaminergic neurons in the SN and neuronal terminals in the striatum and ameliorates motor deficits in different behavioral paradigms in the MPTP‐induced mouse model of PD. We further demonstrate that the beneficial effects of DHF on PD are associated with the inhibition of α‐synuclein expression and the activation of TrkB, as well as antioxidative activity in the SN and striatum. We have therefore provided evidence that DHF may suppress MPTP‐induced toxic effects of α‐synuclein overexpression and oxidative stress via activating TrkB and subsequently block the loss of dopaminergic neurons, thereby ameliorating behavioral decline.
The pathological hallmark of PD is a selective and progressive loss of dopaminergic neurons in the SN 1, 2, 3. It has been reported that BDNF promotes neuronal survival and differentiation and modulates synaptic plasticity by activating the TrkB receptor tyrosine kinase 30. In contrast, results obtained from genetic studies have shown that the deletion of either BDNF or TrkB in excitatory neurons causes dendritic degeneration and neuronal loss 31, 32. In addition, postmortem studies show that the level of BDNF is markedly reduced in the SN of PD patients 33, 34, 35, whereas the application of exogenous BDNF promotes the survival of dopaminergic neurons in PD animal models 19, 36. However, BDNF is difficult to use for PD treatment in clinic as mature BDNF does not cross the blood–brain barrier. Recent reports show that DHF as a bioactive high‐affinity TrkB agonist exerts neuroprotective effects in animal model of PD 21, 22. In full agreement with these findings, we here reported that systemic treatment with DHF dramatically increased the TrkB activation following MPTP treatment (Figure 6) and subsequently reduced MPTP‐induced loss of dopaminergic neurons in the SN and neuronal terminals in the striatum (Figures 3 and 4), and further behavioral experiments showed that DHF significantly ameliorated motor functions in different behavioral paradigms (Figure 2).
Besides the loss of dopaminergic neurons in the SN, another pathological change in PD is the overexpression of α‐synuclein, especially main pathologic form of phosphorylated and insoluble α‐synuclein 37, 38. We here reported that repeated MPTP application dramatically increased the expression of α‐synuclein in the SN and striatum (Figure 5), which may contribute to the deficits of motor function. Indeed, this hypothesis is supported by some previous reports. For example, chronic MPTP treatment can induce α‐synuclein oligomerization and such oligomers produce most pathological actions of α‐synuclein, including mediating neuronal death and motor dysfunction 39, 40. In addition, MPTP application markedly increases α‐synuclein expression in the animal model of PD 41, and the overexpression of α‐synuclein causes oxidative stress and mitochondrial structural abnormalities 42. Importantly, we here also found that chronic DHF treatment significantly inhibited MPTP‐induced α‐synuclein overexpression, which may reduce oxidative stress and mitochondrial dysfunction, thereby contributing to the amelioration of motor function. Indeed, we here found that DHF succeeded in reversing the decrease in the antioxidant GSH and antioxidant enzyme SOD in MPTP‐treated mice (Table 1). These results were in agreement with previous reports that oxidative stress‐mediated neuronal cell death may be associated with PD 43. Although a large number of reports have shown that MPTP/MPP+ administration causes cell death in vitro and in vivo 44, 45, 46, Aznavour and colleagues have recently reported that MPTP (5 mg/kg i.p. during 5 days) destroys TH protein rather than dopamine cells in a cat model of PD 47. These contradictory results may be caused in part by different species and/or different doses of MPTP, which were used in these studies. For example, previous study has shown that the percentage cell death at 60 mg/kg of total MPTP used was lower than at 80 mg/kg in mice 46. Although possible explanation for this controversy has been provided by previous research, further studies are required to determine the exact reason for the different results. Thus, future experiments examining mitochondrial function and neuronal cell death in MPTP‐treated animals with or without DHF treatment will help determine whether DHF's therapeutic effect in the MPTP‐induced mouse model of PD can be attributed to its amelioration of mitochondrial function and inhibition of cell death.
Conclusion
In summary, our study shows that DHF treatment suppresses MPTP‐induced α‐synuclein expression and oxidative stress, and then reduces the loss of dopaminergic neurons in the SN and striatum, thereby ameliorating motor deficits in a murine model of PD.
Conflict of Interest
The authors state no conflict of interest.
Acknowledgments
This work was supported by 973 Program of the Ministry of Science and Technology of the People's Republic of China (2014CB548100 to Z.D.), the National Natural Science Foundation of China (81271221 and 81571042 to Z.D., 81501143 to H.H.), and the Natural Science Foundation of Chongqing (cstc2015jcyjA00037 to H.H.).
References
- 1. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014;311:1670–1683. [DOI] [PubMed] [Google Scholar]
- 2. Buchman AS, Shulman JM, Nag S, et al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 2012;71:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non‐human primates. Nat Rev Neurosci 2011;12:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chu Y, Kordower JH. Age‐associated increases of alpha‐synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson's disease? Neurobiol Dis 2007;25:134–149. [DOI] [PubMed] [Google Scholar]
- 5. Anandhan A, Rodriguez‐Rocha H, Bohovych I, et al. Overexpression of alpha‐synuclein at non‐toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol Dis 2015;81:76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fountaine TM, Wade‐Martins R. RNA interference‐mediated knockdown of alpha‐synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J Neurosci Res 2007;85:351–363. [DOI] [PubMed] [Google Scholar]
- 7. Hayashita‐Kinoh H, Yamada M, Yokota T, Mizuno Y, Mochizuki H. Down‐regulation of alpha‐synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson's disease rat model. Biochem Biophys Res Commun 2006;341:1088–1095. [DOI] [PubMed] [Google Scholar]
- 8. Klivenyi P, Siwek D, Gardian G, et al. Mice lacking alpha‐synuclein are resistant to mitochondrial toxins. Neurobiol Dis 2006;21:541–548. [DOI] [PubMed] [Google Scholar]
- 9. McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA. Alpha‐synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE 2010;5:e12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thomas B, Mandir AS, West N, et al. Resistance to MPTP‐neurotoxicity in alpha‐synuclein knockout mice is complemented by human alpha‐synuclein and associated with increased beta‐synuclein and Akt activation. PLoS ONE 2011;6:e16706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol 2003;53(Suppl 3):S26–S36; discussion S‐8. [DOI] [PubMed] [Google Scholar]
- 12. Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 1996;47(6 Suppl 3):S161–S170. [DOI] [PubMed] [Google Scholar]
- 13. Alam ZI, Jenner A, Daniel SE, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8‐hydroxyguanine levels in substantia nigra. J Neurochem 1997;69:1196–1203. [DOI] [PubMed] [Google Scholar]
- 14. Zhang J, Perry G, Smith MA, et al. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 1999;154:1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feng G, Zhang Z, Bao Q, et al. Protective effect of chinonin in MPTP‐induced C57BL/6 mouse model of Parkinson's disease. Biol Pharm Bull 2014;37:1301–1307. [DOI] [PubMed] [Google Scholar]
- 16. Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 2011;10:209–219. [DOI] [PubMed] [Google Scholar]
- 17. Noble EE, Billington CJ, Kotz CM, Wang C. The lighter side of BDNF. Am J Physiol Regul Integr Comp Physiol 2011;300:R1053–R1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frim DM, Uhler TA, Galpern WR, Beal MF, Breakefield XO, Isacson O. Implanted fibroblasts genetically engineered to produce brain‐derived neurotrophic factor prevent 1‐methyl‐4‐phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc Natl Acad Sci USA 1994;91:5104–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsukahara T, Takeda M, Shimohama S, Ohara O, Hashimoto N. Effects of brain‐derived neurotrophic factor on 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced parkinsonism in monkeys. Neurosurgery 1995;37:733–739; discussion 9‐41. [DOI] [PubMed] [Google Scholar]
- 20. Ochs G, Penn RD, York M, et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:201–206. [DOI] [PubMed] [Google Scholar]
- 21. Jang SW, Liu X, Yepes M, et al. A selective TrkB agonist with potent neurotrophic activities by 7,8‐dihydroxyflavone. Proc Natl Acad Sci USA 2010;107:2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sconce MD, Churchill MJ, Moore C, Meshul CK. Intervention with 7,8‐dihydroxyflavone blocks further striatal terminal loss and restores motor deficits in a progressive mouse model of Parkinson's disease. Neuroscience 2015;290:454–471. [DOI] [PubMed] [Google Scholar]
- 23. Heldermon CD, Hennig AK, Ohlemiller KK, et al. Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS ONE 2007;2:e772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo L, Xiong H, Kim JI, et al. Dynamic rewiring of neural circuits in the motor cortex in mouse models of Parkinson's disease. Nat Neurosci 2015;18:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Antzoulatos E, Jakowec MW, Petzinger GM, Wood RI. MPTP neurotoxicity and testosterone induce dendritic remodeling of striatal medium spiny neurons in the C57Bl/6 mouse. Parkinsons Dis 2011;2011:138471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia Ruiz PJ, Catalan MJ, Fernandez Carril JM. Initial motor symptoms of Parkinson disease. Neurologist 2011;6(Suppl 1):S18–S20. [DOI] [PubMed] [Google Scholar]
- 27. Hallett M. Parkinson revisited: pathophysiology of motor signs. Adv Neurol 2003;91:19–28. [PubMed] [Google Scholar]
- 28. Sankar SR, Manivasagam T, Krishnamurti A, Ramanathan M. The neuroprotective effect of Withania somnifera root extract in MPTP‐intoxicated mice: an analysis of behavioral and biochemical variables. Cell Mol Biol Lett 2007;12:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han X, Zhu S, Wang B, et al. Antioxidant action of 7,8‐dihydroxyflavone protects PC12 cells against 6‐hydroxydopamine‐induced cytotoxicity. Neurochem Int 2014;64:18–23. [DOI] [PubMed] [Google Scholar]
- 30. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001;24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain‐derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci 2003;23:6856–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu B, Zang K, Ruff NL, et al. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 2000;26:233–245. [DOI] [PubMed] [Google Scholar]
- 33. Howells DW, Porritt MJ, Wong JY, et al. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol 2000;166:127–135. [DOI] [PubMed] [Google Scholar]
- 34. Mogi M, Togari A, Kondo T, et al. Brain‐derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci Lett 1999;270:45–48. [DOI] [PubMed] [Google Scholar]
- 35. Parain K, Murer MG, Yan Q, et al. Reduced expression of brain‐derived neurotrophic factor protein in Parkinson's disease substantia nigra. NeuroReport 1999;10:557–561. [DOI] [PubMed] [Google Scholar]
- 36. Levivier M, Przedborski S, Bencsics C, Kang UJ. Intrastriatal implantation of fibroblasts genetically engineered to produce brain‐derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson's disease. J Neurosci 1995;15:7810–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou J, Broe M, Huang Y, et al. Changes in the solubility and phosphorylation of alpha‐synuclein over the course of Parkinson's disease. Acta Neuropathol 2011;121:695–704. [DOI] [PubMed] [Google Scholar]
- 38. Braak H, Del Tredici K. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology 2008;70:1916–1925. [DOI] [PubMed] [Google Scholar]
- 39. Fornai F, Schluter OM, Lenzi P, et al. Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin‐proteasome system and alpha‐synuclein. Proc Natl Acad Sci USA 2005;102:3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shioda N, Yabuki Y, Kobayashi Y, Onozato M, Owada Y, Fukunaga K. FABP3 protein promotes alpha‐synuclein oligomerization associated with 1‐methyl‐1,2,3,6‐tetrahydropiridine‐induced neurotoxicity. J Biol Chem 2014;289:18957–18965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rekha KR, Selvakumar GP, Santha K, Inmozhi Sivakamasundari R. Geraniol attenuates alpha‐synuclein expression and neuromuscular impairment through increase dopamine content in MPTP intoxicated mice by dose dependent manner. Biochem Biophys Res Commun 2013;440:664–670. [DOI] [PubMed] [Google Scholar]
- 42. Hsu LJ, Sagara Y, Arroyo A, et al. Alpha‐synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 2000;157:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jenner P. Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord 1998;13(Suppl 1):24–34. [PubMed] [Google Scholar]
- 44. Nicotra A, Parvez S. Apoptotic molecules and MPTP‐induced cell death. Neurotoxicol Teratol 2002;24:599–605. [DOI] [PubMed] [Google Scholar]
- 45. Nicotra A, Parvez SH. Cell death induced by MPTP, a substrate for monoamine oxidase B. Toxicology 2000;153:157–166. [DOI] [PubMed] [Google Scholar]
- 46. Jackson‐Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Neurodegeneration 1995;4:257–269. [DOI] [PubMed] [Google Scholar]
- 47. Aznavour N, Cendres‐Bozzi C, Lemoine L, et al. MPTP animal model of Parkinsonism: dopamine cell death or only tyrosine hydroxylase impairment? A study using PET imaging, autoradiography, and immunohistochemistry in the cat. CNS Neurosci Ther 2012;18:934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]