

Summary
Aims
Here, we investigate the pharmacology of NS383, a novel small molecule inhibitor of acid‐sensing ion channels (ASICs).
Methods
ASIC inhibition by NS383 was characterized in patch‐clamp electrophysiological studies. Analgesic properties were evaluated in four rat behavioral models of pain.
Results
NS383 inhibited H+‐activated currents recorded from rat homomeric ASIC1a, ASIC3, and heteromeric ASIC1a+3 with IC 50 values ranging from 0.61 to 2.2 μM. However, NS383 was completely inactive at homomeric ASIC2a. Heteromeric receptors containing AISC2a, such as ASIC1a+2a and ASIC2a+3, were only partially inhibited, presumably as a result of stoichiometry‐dependent binding. NS383 (10–60 mg/kg, i.p.), amiloride (50–200 mg/kg, i.p.), acetaminophen (100–400 mg/kg, i.p.), and morphine (3–10 mg/kg, i.p.) all dose‐dependently attenuated nocifensive behaviors in the rat formalin test, reversed pathological inflammatory hyperalgesia in complete Freund's adjuvant‐inflamed rats, and reversed mechanical hypersensitivity in the chronic constriction injury model of neuropathic pain. However, in contrast to acetaminophen and morphine, motor function was unaffected by NS383 at doses at least 8‐fold greater than those that were effective in pain models, whilst analgesic doses of amiloride were deemed to be toxic.
Conclusions
NS383 is a potent and uniquely selective inhibitor of rat ASICs containing 1a and/or 3 subunits. It is well tolerated and capable of reversing pathological painlike behaviors, presumably via peripheral actions, but possibly also via actions within central pain circuits.
Keywords: Acid‐sensing ion channel, Acid‐sensing ion channel blockers, Electrophysiology, Hyperalgesia, Morphine
Introduction
Acid‐sensing ion channels (ASICs) are the principal molecular substrates responsible for transducing the excitatory actions of protons (H+) and belong to the amiloride‐sensitive epithelial Na+ channel‐degenerin superfamily of ion channels 1. Four rodent ASIC genes have been identified (ACCN1‐4), which encode six different subunits (ASIC1a/b, ASIC2a/b, ASIC3, ASIC4) 2. These can assemble into both homomeric and heteromeric channels consisting of three subunits surrounding a central pore permeable to Na+ ions, and in some instances Ca2+ 3, 4. Subunit composition also influences basic channel characteristics such as pH sensitivity, activation, and desensitization kinetics, with ASIC1 and ASIC 3 the most sensitive to H+ 5.
It can be readily appreciated that ASICs localized within cutaneous structures are well placed to serve a protective function by sensing changes in the external environment that could prove to be potentially damaging to tissue. However, the synaptic localization of ASICs within CNS limbic structures such as the amygdala combined with observations that ASIC1a knockout mice have impaired fear‐ and depression‐related behaviors suggests these channels might also contribute to psychiatric illnesses. Moreover, prolonged acidosis occurring as a consequence of disrupted synaptic transmission, inflammation, or ischemia can be neurotoxic linking ASICs in turn to neurological diseases such as epilepsy, multiple sclerosis, Parkinson's disease, and ischemic stroke 6, 7.
When damage to tissue does occur, tissue acidosis can also contribute to pain of diverse etiology with the local release of H+ and other modulators acting to sensitize the peripheral nerve endings of peripheral sensory neurons 8, 9, 10, 11, 12. These neurons variously express ASIC subunits 13, 14 indicating the likely existence of multiple heteromeric and homomeric combinations 15, 16, presumably enabling activity‐dependent responses activated by different sensory modalities to be engaged over a wide pH range in native tissues after injury. Accordingly, the firing rate of rat spinal dorsal horn neurons is increased by low pH solutions applied onto their peripheral receptive fields 17, consistent with human studies where intradermal acid injection increases pain scores 18, 19.
Although the availability of small molecule pharmacological tools that can modulate ASICs is somewhat limited, a number of toxins have contributed significantly to target validation efforts in pain 2. The sea anemone toxin APETx2 inhibits ASIC3 subunit‐containing channels 20, 21, while Psalmotoxin 1 (PcTx1) exerts its action by blocking homomeric ASIC1a 22. More recently, mambalgin toxins isolated from mamba venom have been shown to inhibit homomeric ASIC1a and heteromeric ASIC1a and ASIC1b containing channels 23. In keeping with the peripheral expression of ASIC3, attenuation of inflammatory and postoperative hypersensitivity in rodents is accomplished by local (e.g. intramuscular or intraplantar) injection of APETx2, but not PcTx1 24, 25. Yet, when administered intrathecally, PcTx1 and mambalgin‐1 markedly attenuate pathological pain hypersensitivity 26, 27, in alignment with ASIC1a subunit expression within the spinal dorsal horn 27, 28. However, the recent demonstration that APETx2 also inhibits TTX‐resistant Nav1.8 channels 29 reinforces the need for novel tools with which to interrogate ASIC involvement in pain pathophysiology.
Here, we characterize the novel small molecule NS383 (Figure 1). It possesses submicromolar potency for cloned rat ASIC1a and ASIC3, with a putative mechanism and profile of action distinct from other known ASIC inhibitors such as amiloride. After systemic administration, NS383 markedly attenuated nociceptive behaviors in rat models of inflammatory and neuropathic pain with a benign tolerability profile. Given that NS383 distributes poorly across the blood–brain barrier, we believe these actions were primarily mediated via peripheral ASICs containing 1a and/or 3 subunits, albeit we cannot exclude that actions within central pain circuits are contributing.
Figure 1.
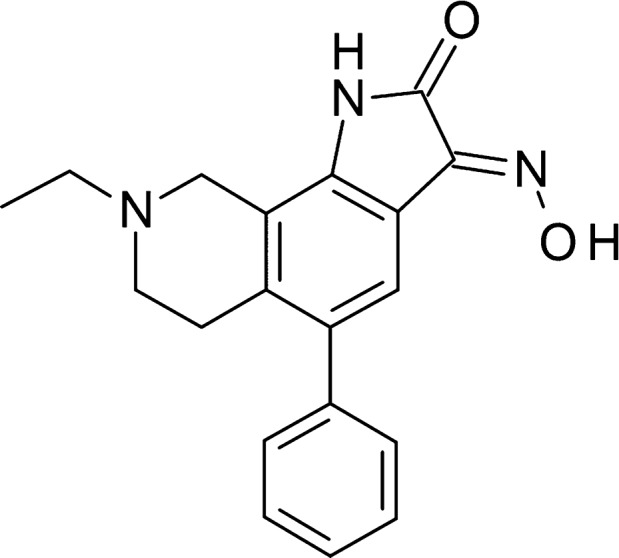
Chemical structure of NS383.
Materials and Methods
Drugs
NS383, (3E)‐8‐ethyl‐3‐(hydroxyimino)‐5‐phenyl‐1H,2H,3H,6H,7H,8H,9H‐pyrrolo[3,2‐h]isoquinolin‐2‐one‐3‐hydroxy‐4‐mycinide (Figure 1), was synthesized at NeuroSearch A/S, Ballerup, Denmark (WO2007/059608), and at Syngene, Bengaluru, India. Amiloride hydrochloride and acetaminophen were purchased from Sigma‐Aldrich, Brøndby, Denmark. Morphine hydrochloride was obtained from Nomeco A/S, Copenhagen, Denmark.
ASIC Expression in CHO Cells
Cloning and expression of rat ASIC1a, ASIC2a, and ASIC3 was described in detail previously 5. For heterologous expression, ASIC constructs were transiently transfected into CHO‐K1 cells (ATCC no. CCL61) using the lipofectamine PLUS transfection kit (Life Technologies) according to manufacturer's protocol. With each transfection, an amount of cDNA that would yield whole‐cell currents within a 0.5–10 nA range was used, to avoid saturation of the patch‐clamp amplifier (approximately 100 ng cDNA). Electrophysiological measurements were performed 16‐ to 48‐h posttransfection. On the measurement day, cells were detached from the culture flask using trypsin and seeded on 3.5‐mm glass coverslips, precoated with poly‐D‐lysine.
Electrophysiology
All experiments were performed under voltage clamp using conventional whole‐cell patch‐clamp methods as described previously 5. In brief, Pulse software was used to control an EPC‐9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) and data were sampled at 2 kHz and low‐pass filtered at 667 Hz. Recording pipettes were pulled from borosilicate glass (Vitrex Medical, Herlev, Denmark) using a horizontal puller (Zeitz‐Instrumente, Augsburg, Germany). Pipettes were backfilled with an intracellular solution containing 120 mM KCl, 31 mM KOH, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES at pH 7.2. For experiments requiring symmetrical sodium concentrations, the intracellular solution contained 10 mM KCl, 110 mM NaOH, 30 mM NaOH, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES at pH 7.2. For recordings, a coverslip with transfected cells was placed in the recording chamber (RC‐25F; Warner Instruments, Hamden, CT, USA), which was continuously perfused at a rate of ~2.5 mL/min with an extracellular solution (ECS) containing 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 5 mM MES at pH 7.4. When immerged in the bath solution, open‐pipette resistances were in the range of 1.5–3.0 MΩ and amplifier series‐resistance compensation was set at 80%. After gigaseal formation, whole‐cell configuration was attained by suction.
Acid‐sensing ion channel currents were induced by rapid exchange of the solution surrounding the patched cell from neutral (pH 7.4) to acidic (pH 4.0–6.8) ECS. This was achieved using a piezo‐controlled double‐barreled application pipette (θ‐tube) with a continuous flow of neutral ECS from the one barrel and acidic ECS from the other barrel. Application pipettes were fabricated from theta glass tubes (1.5 mm outer diameter, World Precision Instruments, Sarasota, FL, USA), which were pulled in the electrode puller and broken to obtain a tip diameter of ~70 μM. An application pipette was mounted in a piezoelectric device (PZS‐100HS; Burleigh Instruments, Thorlabs, Newton, NJ, USA) connected to a piezo‐driver (PZ‐150M, Burleigh Instruments, Thorlabs, Newton, NJ, USA), which was again controlled by TTL pulses from the EPC‐9 amplifier. When performing experiments, the tip of the application pipette was positioned close to the patched cell in such a way that the cell was subjected only to neutral solution. Upon a TTL pulse, the application pipette was rapidly translocated thereby subjecting the cell to only acidic solution. With this system, complete solution exchange could be achieved in <1 ms, as measured by open‐pipette liquid junction potential shift.
Compound stock solutions, with a concentration of 30 mM, were prepared in 100% DMSO and used for preparing final test solutions. For each compound concentration tested, a neutral as well as an acidic ECS was prepared, that is one for each barrel of the application pipette. Each experiment was initiated with a set of control ASIC currents, which were induced by switching from control neutral to control acidic solution every 30 s until responses of repeatable amplitudes were obtained. Upon change to test solutions, the patched cell was perfused (preincubated) with compound for >1 min at neutral pH before evaluating compound effect by switching to the acidic test solution. The concentration of DMSO did not exceed 0.33% in the final test solutions, and applications with 0.33% DMSO were identical to those of control applications at all receptors tested.
Analysis of Electrophysiology Data
Background subtracted peak current amplitudes were measured from recorded current traces, and test compound effects were calculated as the current amplitude in the presence of compound divided by the current amplitude of a prior control response. Compounds were tested over a wide range of concentrations, and IC50 values and Hill coefficients were determined based on the equation Y = Bottom + (Top – Bottom)/(1 + (X/IC50)n, where Y is the fraction of the control response; Bottom is the maximal fitted inhibition level (in most cases a value close to 0); Top is the no inhibition level (value of 1); X is the concentration of the test compound; and n is the Hill coefficient. These nonlinear regression calculations were performed using GraphPad Prism version 6 (GraphPad Software Inc., San Diego, CA, USA). All results are presented as means ± SE.
Animals
Adult male Sprague‐Dawley rats (Harlan Scandinavia, Allerod, Denmark) were used unless stated otherwise. They were housed in Makrolon III cages (20 × 14 × 18 cm or 20 × 40 × 18 cm; in groups of 3–5 per cage according to weight) containing wood‐chip bedding material (3 × 1 × 4 mm). The environment was temperature (20 ± 2°C) and humidity (55 ± 15%) controlled and consisted of a light–dark cycle of 13:11 h (lights on at 06.00 h and off at 19.00 h). Food (Altromin®; Brogaarden, Lynge, Denmark) and water were available ad libitum. The rats were allowed to habituate to the housing facilities for at least 1 week prior to being assigned to behavioral experiments. At the end of each experiment, rats were immediately euthanized by cervical displacement. All experiments were performed according to the Ethical Guidelines of the International Association for the Study of Pain 30 and the Danish Committee for Experiments on Animals.
Measurement of NS383 Levels in Plasma and Brain Tissue
Whole blood and brain samples were collected at 0.5, 1, 1.5, 2, and 4 h after administration of NS383 hydrochloride (30 mg/kg, i.p.). Plasma was prepared by centrifugation of whole blood in EDTA‐K+ tubes at 1000 g for 20 min. Plasma and brains were stored at –18°C prior to analysis. Plasma (25 μL) was precipitated with acetonitrile (75 μL) containing 100 ng/mL internal standard. Samples were then centrifuged at 16,000 g for 25 min at 5°C and the supernatant transferred and diluted 1:1 with water. Brain tissue (0.1 g) was homogenized with 1‐mm zirconia beads in acetonitrile:water (80:20; 1 mL), containing 100 ng/mL internal standard, using a bead beater (Biospec Products, Inc., Minibeadbeater, 96+). The tissue homogenates were centrifuged at 16,000 g for 25 min at 5°C and the supernatant transferred and diluted 1:1 with water. All samples were subsequently analyzed by liquid chromatography UPLC (Acquity system; Waters) in combination with a triple quadrupole mass spectrometer (Quattro Ultima Platinum; Micromass). Detection of NS383 and the internal standard was performed by multiple reaction monitoring (MRM) in electrospray positive ion mode, fragmenting protonated parent ion to a specific prominent product ion. Quantification of NS383 was performed using linear regression (weighted 1/x). The calibration range was 10–5000 ng/mL in plasma and 30–20,000 ng/g in brain tissue.
Tail Flick Test
Individual rats (body weight 130 g) were tested as described previously 31, 32. A radiant heat source (Ugo Basile, Comerio, Italy) was focused on the underside of the tail 3 cm from its distal end. Two measurements separated by 5 min were made and an average obtained. Animals were then administered drug or vehicle and posttreatment latency responses were determined enabling the change in latency response (s) to be calculated. Raw data were expressed as a % of the maximal possible effect (%MPE) based on the assay cutoff point of 15 s.
Formalin Test
Assessment of formalin‐induced flinching behavior in naive rats (body weight 150–180 g) was made with the use of an Automated Nociception Analyzer (University of California, San Diego, USA; 33) as described previously 32. Each rat (four rats were included in each testing session) was administered drug or vehicle according to the experimental paradigm being followed. Next, it was gently restrained to allow formalin (5% in saline, 50 μL, s.c.) to be injected into the dorsal surface of the hindpaw using a 27G needle. Rats were then placed in separate observation chambers and recording of nocifensive flinching behaviors initiated. Three phases of nociceptive behavior were identified and scored; first phase (0–5 min), interphase (6–15 min), and second phase (16–40 min). Raw data were summed to obtain the total number of flinches occurring during that phase and expressed as a % of the vehicle response.
Complete Freunds Adjuvant‐Induced Inflammatory Pain
Rats (body weight 260–300 g) were given an injection of complete Freund's adjuvant (CFA, Sigma; 50% in saline, 100 μL; s.c.) into the plantar surface of the hindpaw under brief isoflurane anesthesia. Nociceptive behaviors were routinely assessed prior to and 24 h following CFA injection. Firstly, differences in hindpaw weight bearing were assessed using an Incapacitance tester (Linton Instruments, Diss, Norfolk, UK) as an index of ongoing or spontaneous painlike behavior. Three readings were obtained and averaged for each hindpaw and the weight‐bearing difference (g) calculated. Immediately thereafter tail flick latency measurements were measured as described above. Twenty‐four h after CFA injection, baseline weight bearing and tail flick responses were measured and then rats administered drug or vehicle. For weight‐bearing differences, raw data were subsequently expressed as a % of the corresponding post‐CFA baseline response.
Chronic Constriction Injury
A chronic constriction injury (CCI) was performed in rats (body weight 180–220 g at the time of surgery) as described previously 34. Anesthesia was induced and maintained by 2% isoflurane (Baxter A/S, Allerod, Denmark), combined with oxygen (30%) and nitrous oxide (68%), and all rats placed on heat pads during the operation. The skin was shaved at the mid‐thigh level and sterilized with a 0.4% iodine solution (Novartis Healthcare A/S, Copenhagen, Denmark). The sciatic nerve was exposed proximal to the sciatic trifurcation and four chromic gut ligatures (4/0; Ethicon, New Brunswick, NJ, USA) tied around the nerve, 1 mm apart. Following hemostasis, the overlying muscle was closed in layers with 4/0 synthetic absorbable surgical suture. The skin was closed and sutured with 4/0 silk thread. Rats recovered under infrared lightning and were carefully monitored until fully awake.
Chronic constriction injury rats were routinely tested for the presence of hindpaw mechanical hypersensitivity as described previously 32. Individual rats were briefly habituated in an openly ventilated Plexiglas testing chamber, placed upon an elevated metal grid allowing access to the plantar surface of the injured hindpaw. A series of calibrated von Frey filaments (0.06–22.9 g; Stoelting Co, Wood Dale, IL, USA) were then applied to the hindpaw with increasing force until the filament used just started to bend. Each filament was applied for a period of 1–2 s and was repeated five times at 1–2 s intervals. The filament that induced a reflex paw withdrawal in 3 of 5 applications was considered to represent the threshold level for a positive response to occur. Only CCI rats showing distinct neuropathic behaviors from 12 to 30 days postsurgery were included in drug testing experiments (approximately 80% in total).
Rotarod Test
Compound effects on activity‐induced motor function were evaluated in naïve rats (body weight 250–350 g) using an accelerating rotarod (Ugo Basile). The rotarod speed was increased from 3 to 30 rpm over a 180 s period, with the minimum and maximum time possible to spend on the rod designated as 0 s and 180 s, respectively. Rats received two training trials (separated by 3–4 h) 1–2 days prior to drug testing. Thereafter, a baseline response was obtained, with rats subsequently administered drug or vehicle and effects on motor performance evaluated. Raw data were subsequently expressed as a % of the corresponding baseline response.
Drug Formulation and Protocols for In Vivo Administration
For in vivo administration, NS383, amiloride, morphine, and acetaminophen were dissolved in 30% hydroxypropyl‐β‐cyclodextrin (Sigma), 10% Tween 80, 5% cremophor and saline, respectively. NS383, amiloride, and acetaminophen were administered i.p. in a dosing volume of 2 mL/kg. Morphine was administered s.c. in a dosing volume of 1 mL/kg. NS383, amiloride, and morphine doses are expressed as mg weight salt per kg body weight, with acetaminophen doses expressed as mg weight free base per kg body weight. Unless stated otherwise, drugs were preadministered using times expected to coincide with maximal effects on behavior within a given assay (60–120 min for NS383, 60–120 min for amiloride, 30–60 min for morphine, and 60–120 min for acetaminophen). Prior to experiments, the rats were randomly distributed into treatment groups. The investigator was blinded to drug treatment for evaluation of effects on behavior, except for formalin‐induced nocifensive behaviors which were performed unblinded.
Data Analysis and Statistical Comparisons on Behavioral Data
All data are presented as mean ± standard error (SE). Statistical analysis was performed with SigmaPlot 11.2.0.5 (Systat Software Inc., Chicago, IL, USA) and GraphPad Prism 4 for Windows (GraphPad Software Inc.). Unless stated otherwise either parametric or nonparametric analysis of variance (ANOVA) was used to analyze the overall effects of treatments depending upon distribution of the data around the mean. For normally distributed data when the F value was significant, this was followed by Bonferroni's t‐test, and for abnormally distributed data when the H value was significant, this was followed by Dunn's test. P < 0.05 was considered to be statistically significant.
Results
NS383 (Figure 1) was identified as a potent inhibitor of ASIC1a in HEK293 cells stably overexpressing human ASIC1a using real‐time Ca2+‐imaging via a fluorescent imaging plate reader (FLIPR). To further characterize NS383, the compound was tested along with the archetypical ASIC inhibitor amiloride in patch‐clamp electrophysiology and in vivo behavioral studies.
Effect of Amiloride and NS383 at Homomeric Rat ASICs
As expected, amiloride concentration‐dependently inhibited H+‐evoked ASIC1a, ASIC2a, and ASIC3 currents obtained by lowering pH from 7.4 to 6.5 (ASIC1a and ASIC3) or 4.5 (ASIC2a). When plotting average inhibition values as a function of compound concentrations, resulting curves were well approximated by the Hill equation (Figure 2A, Table 1). IC50 values were found to be in the range of 12–35 μM, which correlates well with previous reports 35, 36. NS383 likewise concentration‐dependently inhibited ASIC1a and ASIC3 currents with IC50 values of 0.44 and 2.1 μM, respectively. However, no robust inhibition was observed at homomeric ASIC2a (Figure 2B, Table 1). Compared with amiloride, NS383 is thus 10‐ to 30‐fold more potent and displays selectivity between ASIC subtypes. Interestingly, the inhibition by NS383 at ASIC1a and ASIC3 was affected by the stimulatory H+ concentration (Figure 2D). In a pH range from 6.8 to 5, the efficacy of a fixed submaximal concentration of NS383 (1 μM) decreased with lower stimulatory pH values (Figure 2E,F). As a result of this, the potency of NS383 varies with the pH value used for activation with the highest potencies observed at pH values close to neutral. In contrast, a fixed concentration of amiloride (30 μM) showed similar inhibitory efficacy in a pH range from 6.5 to 5 (I30μM_Amiloride/Icontrol for ASIC1a: 0.35 ± 0.04, n = 3 and for ASIC3: 0.55 ± 0.04, n = 3).
Figure 2.
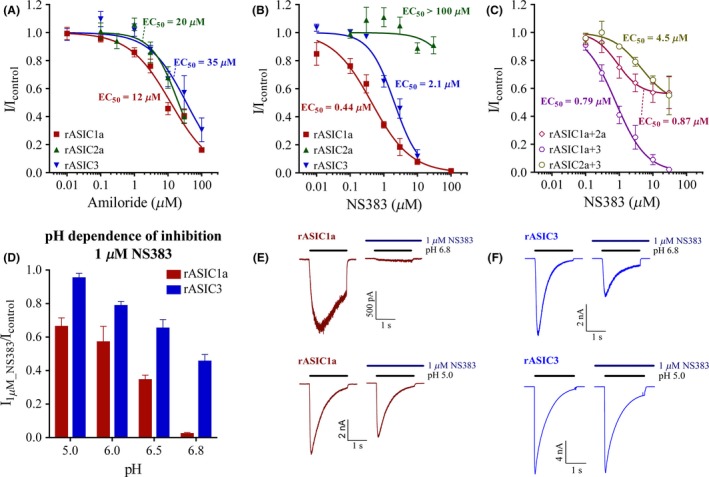
Effects of amiloride and NS383 on homomeric and heteromeric rat acid‐sensing ion channels (ASICs). CHO cells were transiently transfected with cDNA coding for the indicated ASICs, and experiments were performed using conventional whole‐cell patch‐clamp studies with the membrane potential clamped at −60 mV. Rapid (ms scale) pH changes were obtained using a piezo‐controlled θ‐tube as described in Materials. Inhibitory effects of (A) amiloride and (B, C) NS383 at H+‐induced currents from homomeric rat ASIC1a, ASIC2a or ASIC3, or heteromeric rat ASIC1a+2, ASIC1a+3, or ASIC2a+3 plotted as a function of compound concentration. Peak current amplitudes were expressed as fractions of the control peak current amplitude induced by a lowering pH from 7.4 to 6.5 (ASIC1a, ASIC3, ASIC1a+3), or to 5.5 (ASIC1a+2a, ASIC2a+3), or to 4.5 (ASIC2a). Chosen stimulatory pH values approximately represent half‐maximal activation (pH50) at each channel 5. Data points are plotted as mean ± SE, and fitted to the Hill equation using nonlinear regression. Curves represent the fitting, and derived values are seen in Table 1. (D) The inhibitory efficacy of 1 μM NS383 at ASIC1a and ASIC3 showed pH dependency. Data are plotted as mean ± SE from n = 3–22 individual cells. (E, F) Representative traces of ASIC1a and ASIC3 in the presence or absence of NS383. Bars above each trace indicate the pH stimulations or presence of NS383 as indicated.
Table 1.
Fitting values of amiloride and NS383 at rat and human acid‐sensing ion channels (ASICs). Amiloride and NS383 inhibition concentration response relationships were obtained as described in the Figure 2 legend. Data points were fitted to the Hill equation using nonlinear regression where the top value (representing infinite compound dilution) was set to 1 and the bottom value to 0 for ASIC1a, ASIC2a, ASIC3, and rASIC1a+3.Variable bottom was used for NS383 at ASIC1a+2a, ASIC2a+3, and hASIC1a+3 as saturating compound concentrations clearly did not cause complete inhibition. Data are presented as EC50 in μM ± SE, inhibition level in % (where bottom = 0 reflect 100% inhibition) and Hill slopes ± SE
| Amiloride (rat receptors) | NS383 (rat receptors) | NS383 (human receptors) | ||||
|---|---|---|---|---|---|---|
| EC50 (inhibition) | Hill slope | EC50 (inhibition) | Hill slope | EC50 (inhibition) | Hill slope | |
| μM (%) | μM (%) | μM (%) | ||||
| ASIC1a | 12 ± 2 (100) | −0.71 ± 0.07 | 0.44 ± 0.06 (100) | −0.75 ± 0.09 | 0.12 ± 0.02 (100) | −1.0 ± 0.1 |
| ASIC2a | 20 ± 3 (100) | −1.1 ± 0.2 | no effect | no effect | ||
| ASIC3 | 35 ± 8 (100) | −0.79 ± 0.15 | 2.1 ± 0.2 (100) | −1.2 ± 0.1 | no effect | |
| ASIC1a+2a | not determined | 0.87 ± 0.44 (44) | −1.3 ± 0.6 | 0.33 ± 0.06 (59) | −1.3 ± 0.3 | |
| ASIC1a+3 | not determined | 0.79 ± 0.11 (100) | −0.96 ± 0.12 | 0.69 ± 0.24 (81) | −1.2 ± 0.4 | |
| ASIC2a+3 | not determined | 4.5 ± 4.3 (51) | −1.0 ± 0.5 | no effect | ||
Effect of NS383 at Heteromeric Rat ASICs
NS383 concentration‐dependently inhibited H+‐evoked (pH 6.5) ASIC1a+3 currents (Figure 2C, Table 1). The fitted IC50 value was found to be 0.79 μM which is in between the IC50 values observed at homomeric ASIC1a and ASIC3. NS383 also displayed concentration‐dependent inhibition at H+‐evoked (pH 5.5) ASIC1a+2a and ASIC2a+3 (Figure 2C, Table 1), albeit surprisingly the presence of saturating concentrations of NS383 only caused partial inhibitions of 44–51%. Fitted IC50 values were found to be 0.87 and 4.5 μM, respectively. Hence, the lack of NS383 activity at ASIC2a homomeric receptors results in a complex inhibition pattern in heteromeric subunit combinations including ASIC2a.
Further Profiling of NS383
To determine whether inhibitory characteristics of amiloride and NS383 show membrane polarity dependence, inhibition was compared with the membrane potential clamped at −60 mV and +60 mV. ASIC3 was selected for this experiment as it generally has less run‐down associated issues. Amiloride (30 μM) inhibited ASIC3 current responses by 54 ± 2% at −60 mV, a finding that is significantly different (P < 0.005) from the 11 ± 2% inhibition seen at +60 mV (Figure 3A,B). In sharp contrast, NS383 (10 μM) caused 95 ± 2% inhibition at −60 mV, which was not significantly different from the 96 ± 1% inhibition seen at +60 mV. Hence, the inhibiting mechanism of NS383 differentiates from the channel blocker amiloride.
Figure 3.
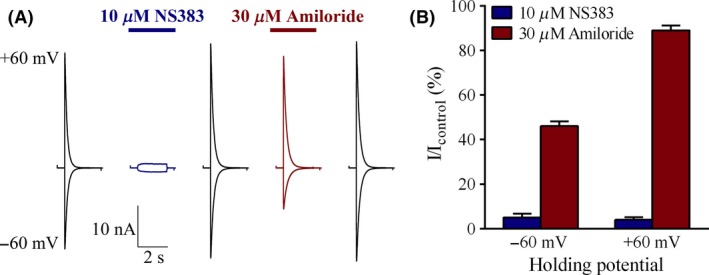
Mechanism of NS383 inhibitory action. A set of intra‐ and extracellular solutions, symmetrical for the primary conduction ion, sodium, was used to obtain a reversal potential of approximately 0 mV. (A) Representative traces of H+‐evoked currents (pH 6.5) in an ASIC3 expressing cell at −60 mV and at +60 mV, respectively. After attainment of responses of a repeatable amplitude (only one response shown), 10 μM NS383 or 30 μM amiloride was included in the solutions as indicated by horizontal bars. The interval between stimulations was 30 s. (B) Calculated percent remaining ASIC3 current in the presence of 10 μM NS383 and 30 μM amiloride at −60 mV and +60 mV, respectively. Each bar represents the mean ± SE.
Finally, the selectivity profile of NS383 was determined at human receptors (Table 1). NS383 is highly potent at hASIC1a, hASIC1a+2a, and hASIC1a+3 but has no inhibitory efficacy at the remaining receptors tested (hASIC2a, hASIC3, and hASIC2a+3). As with rat 2a containing heteromeric ASICs, a substantial residual current was observed for hASIC1a+2a. The selectivity profile of NS383 thus displays species variance, and the lack of activity at hASIC3 underscores the significant difference between this subunit and rat ASIC3 at the protein level.
NS383 Receptor Profiling Screen, Plasma, and Brain Tissue Levels
NS383 (1 μM) was tested using MDS Pharma's Lead Profiling Screen. In binding assays against a panel of 63 G‐protein‐coupled receptors, ion channels, and transporters, only binding to the 5‐Hydroxytryptamine 2A receptor surpassed the 50% effect threshold level, which is the criteria for significance. Subsequently, when NS383 (30 μM) was tested in functional studies against three related receptor subtypes (adrenergic α 1D, adrenergic α 1A, adrenergic α 2A) from rat aorta or vas deferens, no significant activity (≥50%) was reported.
To obtain a correlation between levels of NS383 in the plasma and central nervous system with in vivo behavioral observations, pharmacokinetic and tissue organ analysis studies were performed. NS383 (30 mg/kg) reached a maximum concentration (Cmax) in plasma of 7.0 μM within 1 h after administration. The Cmax achieved in brain tissue was 0.53 μM occurring within 1.5 h after administration. NS383 had a considerably longer half‐life (first‐order kinetics) in brain tissue compared with plasma, 6.8 h versus 0.9 h, and a brain/plasma ratio of 0.1 (based on area under the curve).
Effect of NS383 on Acute Nocifensive Behaviors in Naïve Rats
To test for possible effects upon acute nociceptive processing NS383, amiloride, and morphine were administered to naive rats in the tail flick test (data not shown). One‐way ANOVA showed no effect of NS383 (10–60 mg/kg) treatment on the latency to respond to noxious thermal stimulation of the tail, F(3, 31) = 1.043, P = 0.389. In contrast, both amiloride (50–200 mg/kg) and morphine (6–24 mg/kg) significantly reversed the tail flick response, F(3, 31) = 6.665, P = 0.002 and F(3, 31) = 60.253, P < 0.001 respectively. However, when expressed as a %MPE, the relative efficacy achieved for the highest dose of amiloride (17.6%, P < 0.001 versus vehicle) was considerably less than that obtained even with the lowest dose of morphine (53.2%, P < 0.001 versus vehicle).
Effect of NS383 on Persistent Nocifensive Behaviors Induced by Formalin
The formalin test although limited in aspects of face validity still encompasses multiple sequelae of injury‐induced hyperexcitability that contribute to clinical signs and symptoms of chronic pain. Moreover, painlike behaviors induced by capsaicin or formalin in rats can be augmented under acidic conditions 37. While administration of NS383 (10–60 mg/kg) 30 min prior to formalin injection had no effect on first‐phase flinching behavior (Figure 4A), nocifensive behaviors were attenuated during both interphase, F(3, 31) = 3.636, P < 0.05, and the second phase of the test, F(3, 31) = 21.159, P < 0.001. Similar results were obtained with amiloride (50–00 mg/kg) during these latter periods, F(3, 31) = 9.447, P < 0.05, and, F(3, 31) = 8.301, P < 0.001 (Figure 4B). As expected, morphine (3–10 mg/kg) dose‐dependently attenuated nocifensive behaviors throughout all 3 phases, F(3, 34) = 10.425, P < 0.001; F(3, 34) = 2.984, P < 0.05; F(3, 34) = 20.274, P < 0.001 (Figure 4C). Finally, acetaminophen (100–400 mg/kg) attenuated flinching during both the first, F(3, 31) = 2.962, P < 0.05, and the second phase of the test, F(3, 31) = 12.414, P < 0.001 (Figure 4D).
Figure 4.
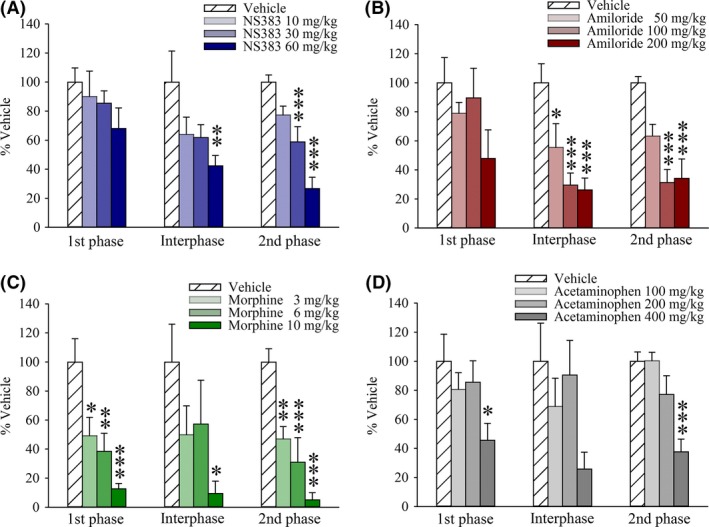
NS383 reduces persistent nocifensive behaviors in the formalin test. Rats were administered either drug or vehicle at the time (t) indicated prior to intraplantar injection of formalin (5% in saline, 50 μL, s.c.) into the hindpaw. % Vehicle values are shown for the first phase (0–5 min), interphase (6–15 min) or second phase (16–40 min) of the test. Effects of (A) NS383 (10–60 mg/kg, i.p., t = −30 min), (B) amiloride (50–200 mg/kg, i.p., t = −60 min), (C) morphine (3–10 mg/kg, s.c., t = −30 min), (D) acetaminophen (100–400 mg/kg i.p., t = −30 min) or vehicle. All groups n = 8 rats except for morphine 6 mg/kg n = 9 rats, morphine 10 mg/kg n = 9 rats. Data are presented as mean ± SE *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding vehicle group (one‐way ANOVA followed by Bonferroni's test).
NS383 Attenuates Pathophysiological Nociceptive Behaviors in CFA‐Inflamed and CCI Rats
Injection of CFA in the rat hindpaw produces a persistent inflammatory hyperalgesia that is associated with increased ASIC subunit expression within peripheral and central pain circuits and that can be inhibited by blockers such as amiloride and A‐317567 16, 27. Twenty‐four hours after hindpaw CFA injection, a marked alteration in hindpaw weight bearing indicative of spontaneous nonevoked pain was observed (44.4 ± 2.3 g vs. 5.2 ± 2.2 g prior to injection, n = 128, P < 0.001, Student's t‐test). This deficit was completely reversed by NS383 (10–60 mg/kg; F(3, 30) = 9.677, P < 0.001, amiloride (50–200 mg/kg; F(3, 31) = 4.958, P < 0.01), and acetaminophen (100–400 mg/kg; F(3, 31) = 4.195, P < 0.05) (Figure 5A–C). However, whereas the tail flick response in the same rats was unaffected by NS383, small but significant analgesic effects were associated with both amiloride and acetaminophen treatment in CFA rats, F(3, 31) = 4.031, P < 0.05 and F(3, 31) = 6.554, P < 0.01, which essentially mirror the data obtained in naïve rats. Hence, this suggests that NS383 selectively reverses pathophysiological pain after injury rather than possessing intrinsic analgesic actions per se.
Figure 5.
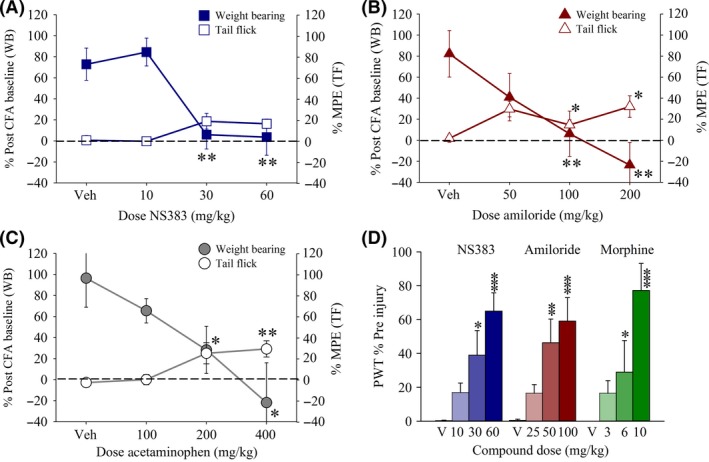
NS383 attenuates inflammatory hyperalgesia and neuropathic hypersensitivity in injured rats. (A–C) Prior to the establishment of hindpaw inflammation, the weight‐bearing difference (WB) and tail flick (TF) latency response was measured. Rats were then given a s.c. injection of complete Freund's adjuvant (CFA) into the hindpaw, and 24 h later post‐CFA, baseline responses were obtained. Thereafter, rats were administered either NS383 (10–60 mg/kg, i.p.), amiloride (50–200 mg/kg, i.p.), acetaminophen (100–400 mg/kg, i.p.) or vehicle and effects on weight bearing and tail flick responses expressed as a % post‐CFA baseline (WB) and %MPE (TF), respectively. The dashed line indicates the normalized WB response prior to CFA injection. All groups n = 7–8 rats. (D) Immediately after a second baseline response had been obtained, chronic constriction injury rats were administered either NS383 (10–60 mg/kg, i.p.; all groups n = 6 rats), amiloride (25–100 mg/kg, i.p.; all groups n = 8 rats), morphine (3–10 mg/kg, s.c.; all groups n = 6 rats), or vehicle (V; n = 6, 8, 6 rats for NS383, amiloride and morphine, respectively) and effects on threshold to mechanical stimulation of the injured hindpaw with von Frey filaments measured and expressed as % Pre‐injury. All data are presented as mean ± SE. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding vehicle group (one‐way ANOVA followed by Bonferonni's test).
Next, we proceeded to evaluate effects upon reflex nociceptive responses of the injured hindpaw in the CCI model of peripheral nerve injury. Following surgery, CCI rats developed behavioral signs of mechanical allodynia, observed as a decrease in the paw withdrawal threshold to 1.1 ± 0.1 g in response to von Frey hair stimulation compared to presurgery levels that typically ranged from 8.4 to 19.7 g. NS383 dose‐dependently attenuated mechanical allodynia, F(3, 23) = 10.512, P < 0.001 (Figure 5D). Similarly, both amiloride (25–100 mg/kg) and morphine (3–10 mg/kg) reversed mechanical responses in CCI rats, F(3, 31) = 7.607, P < 0.001 and, F(3, 23) = 8.040, P < 0.01.
Effect of NS383 on Motor Function and Coordination
To be able to confidently attribute the antihyperalgesic effects of the compounds tested here in inflamed and neuropathic rats as being mediated via direct actions on nociceptive circuits, rather than via indirect effects on motor circuits, all compounds were tested for possible impairment of motor function and coordination in the rotarod test (Figure 6). As initial experiments with NS383 (10–60 mg/kg) revealed no effects, much higher doses were tested. Motor function was mildly but significantly, H(3) = 9.209, P = 0.027, impaired by NS383 (120, 240 and 480 mg/kg). However, ataxia was only observed for the highest dose of NS383 (480 mg/kg) tested (P < 0.05 vs. vehicle). Administration of amiloride (50, 100 and 200 mg/kg) had no effect on motor function as compared with corresponding baseline values. As expected, morphine (6, 12 and 24 mg/kg) produced a marked dose‐dependent impairment of motor performance, H(3) = 24.060, P < 0.001. Motor function was also significantly impaired, H(3) = 8.619, P = 0.035, by administration of acetaminophen (200, 400, and 800 mg/kg).
Figure 6.
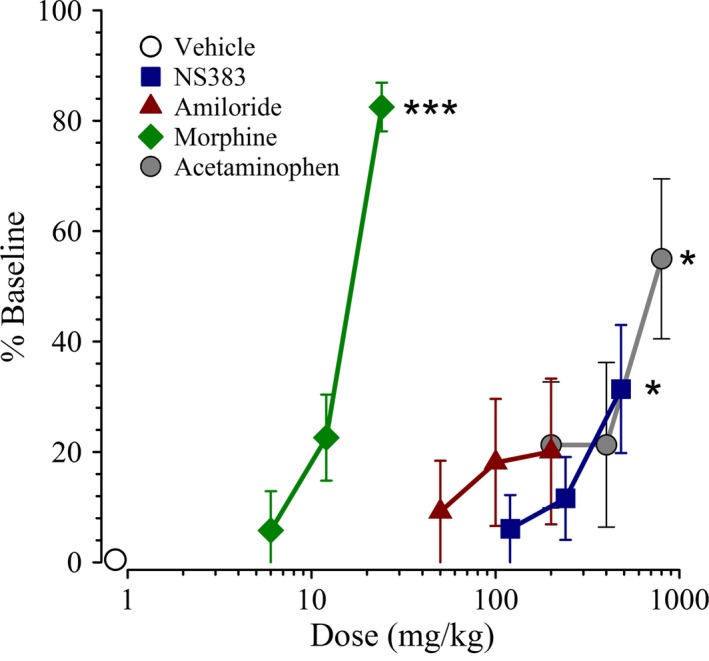
The effects of NS383 on motor function and coordination in naive rats. Normal, uninjured rats were administered either NS383 (120–480 mg/kg, i.p.), amiloride (50–200 mg/kg, i.p.), morphine (6–24 mg/kg, s.c.), acetaminophen (200–800 mg/kg, i.p.), or vehicle immediately after a baseline response had been obtained and the effects on motor performance (represented as % Baseline) determined. Data are presented as mean ± SE. All groups n = 8 rats except for NS383 120 mg/kg n = 7 rats, NS383 240 mg/kg n = 7 rats. Note that the Vehicle group indicated is comprised of rats from each individual experiment and is shown for reference purposes only. *P < 0.05, ***P < 0.001 versus corresponding vehicle (one‐way ANOVA followed by Dunn's test).
Discussion
Our electrophysiology data clearly show that both NS383 and amiloride inhibit recombinantly expressed homomeric ASIC1a and ASIC3 activated by lowering the pH. Notably, NS383 inhibition displayed half‐maximal inhibition (IC50) values that were of approximately 10‐fold higher potencies than those obtained with amiloride 35, 36. Although amiloride also inhibited H+‐evoked ASIC2a currents, NS383 had no robust inhibitory effects at concentrations up to 30 μM. To date, a limited number of other small molecule inhibitors of ASICs such as A‐317567 and antiprotozoal diarylamidines have been reported in the literature 16, 38, 39, albeit these compounds like amiloride are nonselective. Moreover, a series of amiloride derivatives, together with an indole amidine class and nonamidine class of compounds, have been described by Merck as potent blockers of ASIC3 (IC50 <1 μM). However, no mention of selectivity to other ASICs was given 40, 41. NS383 thus possesses a unique selectivity profile at homomeric ASICs, and this was confirmed by screening against a diverse panel of G‐protein‐coupled receptors, ion channels and transporters.
The selectivity profile of NS383 needs to be juxtaposed against native channels that also exist in various heteromeric combinations within the peripheral and central nervous systems 15. To this end, we have shown elsewhere that heteromeric receptor complexes can be distinguished from homomeric counterparts with respect to parameters such as pH sensitivity and desensitization kinetics 5. Here, we observed that H+‐evoked currents recorded from ASIC1a+3 complexes were fully inhibited by NS383 with a potency similar to that obtained for ASIC1a or ASIC3 homomers. However, the presence of ASIC2a with either ASIC1a or ASIC3 subunits markedly affected the ability of saturating concentrations of NS383 to inhibit H+‐evoked currents through these channels. This partial inhibition upon transfection of 2a containing cDNA mixtures into CHO cells could be speculated to arise from a mixed pool of receptors with variant subunit involvement, for example ASIC1a+2a mixed with homomeric ASIC2a. However, homomeric ASIC2a are only marginally activated at pH 5.5 5 making it unlikely that these are responsible for the substantial (~50%) remaining current. Another intriguing, and more likely, possibility is a pool of receptors with variant stoichiometries. Such has been observed for several classes of receptors, for example α4β2 nicotinic receptors, and in fact also for ASIC1a+2a in Xenopus laevis oocytes 42, 43. In this case, NS383 could potentially only inhibit one stoichiometry (e.g. (ASIC1a)2(ASIC2a)1) and not the other (e.g. (ASIC1a)1(ASIC2a)2) leaving approximately 50% residual current. The significance of the partial inhibition is difficult to pinpoint from a therapeutic perspective given that intrathecal administration of mambalgin toxins has recently been shown to mediate a robust analgesia in mice by targeting spinal ASIC1a+ASIC2a 23.
Amiloride is a weak base at neutral pH and has been shown to be a potent pore blocker of epithelial sodium channels (ENaCs) 44. Although NS383 is not particularly basic (calculated pKa 7.9), a possible site of interaction with ASICs could still be within the channel pore. To investigate this, we compared the inhibitory actions of NS383 with amiloride at negative versus positive membrane potentials, to determine whether pore block is involved; whereas 30 μM amiloride was capable of inhibiting more than half of the H+‐evoked currents at −60 mV, a five‐fold lower inhibition was observed at +60 mV. In contrast, 10 μM NS383 almost completely inhibited H+‐evoked currents at both membrane potentials. Of itself, this indicates that NS383 is not a pore blocker and in combination with its selectivity profile, suggests that NS383 binds to a region of ASIC protein subunits distinct from amiloride.
Small molecule blockers of ASICs such as amiloride and A‐317567 attenuate hyperalgesia in rodent pain models of diverse etiology, a number of which are associated with reduced pH at the site of injury 4, 16, 45. In our experiments, a lack of effect of NS383 and amiloride against formalin‐induced flinching during the first phase indicates that ASICs targeted by these compounds do not to contribute to afferent firing after direct stimulation of nociceptors. This would appear to rule out an effect of either compound being mediated via 1b‐containing ASICs 23, albeit this specific aspect of NS383 pharmacology has not been tested herein. In contrast, NS383 and amiloride clearly reduced second‐phase flinching behavior indicating that ASICs targeted by these compounds are preferentially involved in facilitated states of nociceptive processing. In direct comparison with morphine, the analgesic efficacy levels observed with NS383 and amiloride were lower; however, morphine also substantially inhibited first‐phase flinching. Furthermore, both NS383 and amiloride fully reversed hindpaw weight‐bearing deficits in the CFA‐injected rats. This is consistent with reports that hindpaw CFA increases the expression of ASIC1a and ASIC3 subunits within rat sensory neurons and ASIC1a within spinal pain circuits 13, 46. Accordingly, local administration of APETx2 into the hindpaw or spinal infusion with PcTx1 or antisense oligonucleotides diminishes CFA‐induced hypersensitivity in rats 27, 47. Notably, of the tested compounds, only NS383 attenuated inflammatory pain at doses that had no effect on the tail flick response in the same animals. This separation aligns with the requirement to enable preservation of normal nociceptive processing in accordance with its biological protective function.
After peripheral nerve injury, painlike behaviors are accompanied by an increased sensitivity to acetic acid injection, with a corresponding increase in the number of ASIC3 expressing cells within the ipsilateral L4 DRG 48. Although these observations indicate that peripheral ASICs contribute to neuropathic hypersensitivity, the ability of intrathecal PcTx1 to reverse hypersensitivity in neuropathic rats clearly suggests that central 1a‐containing ASICs also contribute 26. Our data show that NS383 and amiloride have pronounced effects on mechanical allodynia in CCI rats, comparing favorably with morphine. However, whereas morphine markedly impaired motor function at a dose just 4‐fold greater than the antiallodynic minimal effective dose in neuropathic rats 49, the therapeutic index obtained for NS383 was much greater.
Amiloride appeared to be well tolerated in all behavioral experiments at the time of testing. However, we observed in preliminary neuropathic experiments that CCI rats administered 200 mg/kg amiloride died 24–48 h later, likely as a result of excessive diuresis (hyperkalemia). Such adverse events were never observed with NS383, which overall appeared well tolerated. PK analysis revealed that NS383 can enter the brain albeit poorly. This is consistent with compromised motor function in the rotarod test at very high doses (480 mg/kg). Although the calculated brain to plasma ratio was only 0.1, the half‐life of NS383 in brain was 7‐fold greater than in plasma. Moreover, the Cmax in brain tissue (0.53 μM) following administration of the lowest efficacious dose (30 mg/kg) could indicate that it lies close to the IC50 required to inhibit homomeric ASIC1a and heteromeric ASIC1a+3. A caveat with such an inference is that a Cmax value does not indicate the free concentration of a compound within brain. Free concentrations can be orders of magnitude lower than a Cmax value depending on the compound characteristics. On balance, we think that NS383‐mediated analgesia probably occurs peripherally via channels containing ASIC1a and ASIC3 subunits. A future comparison of antinociceptive efficacy conferred by intrathecally and systemically administered NS383, and indeed other selective ASIC blockers would help clarify this important issue.
We are only aware of one ASIC blocker currently in clinical development for alleviating pain in humans. PPC‐5650 is described as a selective and potent peripherally acting ASIC1a antagonist, with minimal oral bioavailability, and to exert a significant effect on heat and mechanical pain sensitivity induced by UV burn. Aros Pharma have conducted two Phase 1 trials in volunteers with experimental esophageal pain and in patients with irritable bowel syndrome with mixed outcomes to investigate the efficacy and safety of this compound after local administration 50, 51. We cannot comment on the selectivity profile of this compound at ASICs compared with that shown here for NS383, making it difficult to conclude whether NS383‐like compounds might be more effective in alleviating clinical pain. Moreover, as discussed above, with its limited CNS penetration we cannot easily judge whether such compounds might have utility in other neurological or psychiatric illnesses.
In summary, we show that NS383 is a relatively potent blocker of ASIC currents, and in contrast to other known small molecule inhibitors of ASICs, exhibits intriguing selectivity for channels containing ASIC1a and ASIC3 subunits versus those containing ASIC2a. Importantly, this translated into efficacious actions after systemic administration in rat pain models coupled with a benign tolerability profile. Together, our data indicate that NS383‐like molecules will be useful tools for understanding ASIC function in preclinical and possibly even clinical pain models.
Disclosure
The authors were all paid employees at NeuroSearch A/S when this work was performed.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We would like to thank Nete Ibsen and Helene Dyhr for expert technical assistance. This work was supported by IMK Almene Fond, Denmark.
References
- 1. Waldmann R, Lazdunski M. H(+)‐gated cation channels: Neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol 1998;8:418–424. [DOI] [PubMed] [Google Scholar]
- 2. Deval E, Gasull X, Noël J, et al. Acid‐sensing ion channels (ASICs): Pharmacology and implication in pain. Pharmacol Ther 2010;128:549–558. [DOI] [PubMed] [Google Scholar]
- 3. Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid‐sensing ion channel 1 at 1.9 A resolution and low pH. Nature 2007;449:316–323. [DOI] [PubMed] [Google Scholar]
- 4. Baron A, Lingueglia E. Pharmacology of acid‐sensing ion channels ‐Physiological and therapeutical perspectives. Neuropharmacology 2015;94:19–35. [DOI] [PubMed] [Google Scholar]
- 5. Hesselager M, Timmermann DB, Ahring PK. pH Dependency and desensitization kinetics of heterologously expressed combinations of acid‐sensing ion channel subunits. J Biol Chem 2004;279:11006–11015. [DOI] [PubMed] [Google Scholar]
- 6. Liu S, Cheng XY, Wang F, Liu CF. Acid‐sensing ion channels: Potential therapeutic targets for neurologic diseases. Transl Neurodegener 2015;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wemmie JA, Taugher RJ, Kreple CJ. Acid‐sensing ion channels in pain and disease. Nat Rev Neurosci 2013;14:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Immke DC, McCleskey EW. Lactate enhances the acid‐sensing Na+ channel on ischemia‐sensing neurons. Nat Neurosci 2001;4:869–870. [DOI] [PubMed] [Google Scholar]
- 9. Berdiev BK, Xia J, McLean LA, et al. Acid‐sensing ion channels in malignant gliomas. J Biol Chem 2003;278:15023–15034. [DOI] [PubMed] [Google Scholar]
- 10. Sugiura T, Dang K, Lamb K, Bielefeldt K, Gebhart GF. Acid‐sensing properties in rat gastric sensory neurons from normal and ulcerated stomach. J Neurosci 2005;25:2617–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan J, Edelmayer RM, Wei X, De Felice M, Porreca F, Dussor G. Dural afferents express acid‐sensing ion channels: A role for decreased meningeal pH in migraine headache. Pain 2011;152:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deval E, Lingueglia E. Acid‐Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology 2015;94:49–57. [DOI] [PubMed] [Google Scholar]
- 13. Voilley N, de Weille J, Mamet J, Lazdunski M. Nonsteroid anti‐inflammatory drugs inhibit both the activity and the inflammation‐induced expression of acid‐sensing ion channels in nociceptors. J Neurosci 2001;21:8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM. Functional implications of the localization and activity of acid‐sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci USA 2002;99:2326–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benson CJ, Xie J, Wemmie JA, et al. Heteromultimers of DEG/ENaC subunits form H+‐gated channels in mouse sensory neurons. Proc Natl Acad Sci USA 2002;99:2338–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dubè GR, Lehto SG, Breese NM, et al. Electrophysiological and in vivo characterization of A‐317567, a novel blocker of acid sensing ion channels. Pain 2005;117:88–96. [DOI] [PubMed] [Google Scholar]
- 17. Carpenter KJ, Nandi M, Dickenson AH. Peripheral administration of low pH solutions causes activation and sensitisation of convergent dorsal horn neurones in the anaesthetised rat. Neurosci Lett 2001;298:179–182. [DOI] [PubMed] [Google Scholar]
- 18. Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S. Amiloride‐blockable acid‐sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest 2002;110:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jones NG, Slater R, Cadiou H, McNaughton P, McMahon SB. Acid‐induced pain and its modulation in humans. J Neurosci 2004;24:10974–10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diochot S, Baron A, Rash LD, et al. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid‐sensitive channel in sensory neurons. EMBO J 2004;23:1516–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jensen JE, Durek T, Alewood PF, Adams DJ, King GF, Rash LD. Chemical synthesis and folding of APETx2, a potent and selective inhibitor of acid sensing ion channel 3. Toxicon 2009;54:56–61. [DOI] [PubMed] [Google Scholar]
- 22. Escoubas P, De W Jr, Lecoq A, et al. Isolation of a tarantula toxin specific for a class of proton‐gated Na+ channels. J Biol Chem 2000;275:25116–25121. [DOI] [PubMed] [Google Scholar]
- 23. Diochot S, Baron A, Salinas M, et al. Black mamba venom peptides target acid‐sensing ion channels to abolish pain. Nature 2012;490:552–555. [DOI] [PubMed] [Google Scholar]
- 24. Karczewski J, Spencer RH, Garsky VM, et al. Reversal of acid‐induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br J Pharmacol 2010;161:950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deval E, Noël J, Gasull X, et al. Acid‐sensing ion channels in postoperative pain. J Neurosci 2011;31:6059–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mazzuca M, Heurteaux C, Alloui A, et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat Neurosci 2007;10:943–945. [DOI] [PubMed] [Google Scholar]
- 27. Duan B, Wu LJ, Yu YQ, et al. Upregulation of acid‐sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. J Neurosci 2007;27:11139–11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alvarez de la Rosa D, Krueger SR, Kolar A, Shao D, Fitzsimonds RM, Canessa CM. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol 2003;546:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blanchard MG, Rash LD, Kellenberger S. Inhibition of voltage‐gated Na(+)currents in sensory neurones by the sea anemone toxin APETx2. Br J Pharmacol 2012;165:2167–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983;16:109–110. [DOI] [PubMed] [Google Scholar]
- 31. Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev 2001;53:597–652. [PubMed] [Google Scholar]
- 32. Munro G, Lopez‐Garcia JA, Rivera‐Arconada I, et al. Comparison of the novel subtype‐selective GABAA receptor‐positive allosteric modulator NS11394 [3′‐[5‐(1‐hydroxy‐1‐methyl‐ethyl)‐benzoimidazol‐1‐yl]‐biphenyl‐2‐carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther 2008;327:969–981. [DOI] [PubMed] [Google Scholar]
- 33. Yaksh TL, Ozaki G, McCumber D, et al. An automated flinch detecting system for use in the formalin nociceptive bioassay. J Appl Physiol 2001;90:2386–2402. [DOI] [PubMed] [Google Scholar]
- 34. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988;33:87–107. [DOI] [PubMed] [Google Scholar]
- 35. Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non‐inactivating proton‐gated Na+ channel specific for sensory neurons. J Biol Chem 1997;272:20975–20978. [DOI] [PubMed] [Google Scholar]
- 36. Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton‐gated cation channel involved in acid‐sensing. Nature 1997;386:173–177. [DOI] [PubMed] [Google Scholar]
- 37. Rocha‐González HI, Herrejon‐Abreu EB, López‐Santillán FJ, García‐López BE, Murbartián J, Granados‐Soto V. Acid increases inflammatory pain in rats: Effect of local peripheral ASICs inhibitors. Eur J Pharmacol 2009;603:56–61. [DOI] [PubMed] [Google Scholar]
- 38. Chen X, Qiu L, Li M, et al. Diarylamidines: High potency inhibitors of acid‐sensing ion channels. Neuropharmacology 2010;58:1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen X, Orser BA, MacDonald JF. Design and screening of ASIC inhibitors based on aromatic diamidines for combating neurological disorders. Eur J Pharmacol 2010;648:15–23. [DOI] [PubMed] [Google Scholar]
- 40. Kuduk SD, Chang RK, Di Marco CN, et al. Identification of non‐amidine inhibitors of acid‐sensing ion channel‐3 (ASIC3). Bioorg Med Chem Lett 2011;21:4255–4258. [DOI] [PubMed] [Google Scholar]
- 41. Kuduk SD, Di Marco CN, Chang RK, et al. Amiloride derived inhibitors of acid‐sensing ion channel‐3 (ASIC3). Bioorg Med Chem Lett 2009;19:2514–2518. [DOI] [PubMed] [Google Scholar]
- 42. Harpsøe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T. Unraveling the high‐ and low‐sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci 2011;31:10759–10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bartoi T, Augustinowski K, Polleichtner G, Gründer S, Ulbrich MH. Acid‐sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci USA 2014;111:8281–8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol Rev 2002;82:735–767. [DOI] [PubMed] [Google Scholar]
- 45. Woo YC, Park SS, Subieta AR, Brennan TJ. Changes in tissue pH and temperature after incision indicate acidosis may contribute to postoperative pain. Anesthesiology 2004;101:468–475. [DOI] [PubMed] [Google Scholar]
- 46. Wu LJ, Duan B, Mei YD, et al. Characterization of acid‐sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem 2004;279:43716–43724. [DOI] [PubMed] [Google Scholar]
- 47. Deval E, Noël J, Lay N, et al. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J 2008;27:3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Omori M, Yokoyama M, Matsuoka Y, et al. Effects of selective spinal nerve ligation on acetic acid‐induced nociceptive responses and ASIC3 immunoreactivity in the rat dorsal root ganglion. Brain Res 2008;1219:26–31. [DOI] [PubMed] [Google Scholar]
- 49. Erichsen HK, Hao JX, Xu XJ, Blackburn‐Munro G. Comparative actions of the opioid analgesics morphine, methadone and codeine in rat models of peripheral and central neuropathic pain. Pain 2005;116:347–358. [DOI] [PubMed] [Google Scholar]
- 50. Nielsen LM, Olesen AE, Andresen T, Simrén M, Törnblom H, Drewes AM. Efficacy and safety of PPC‐5650 on experimental rectal pain in patients with irritable bowel syndrome. Basic Clin Pharmacol Toxicol 2015;16:140–145. [DOI] [PubMed] [Google Scholar]
- 51. Olesen AE, Nielsen LM, Larsen IM, Drewes AM. Randomized clinical trial: Efficacy and safety of PPC‐5650 on experimental esophageal pain and hyperalgesia in healthy volunteers. Scand J Gastroenterol 2015;50:138–144. [DOI] [PubMed] [Google Scholar]