

Summary
Aims
Acute cerebral ischemia may lead to ischemic stroke, which is a major cause of death and disability worldwide. Hydrogen sulfide (H2S) functions importantly in mammalian systems. The present work was designed to study the effect of sodium sulfide, a donor of H2S, on acute cerebral ischemia.
Methods
Acute cerebral focal ischemia was produced by middle cerebral artery occlusion (MCAO) in Sprague‐Dawley (SD) rats. Bilateral vertebral arteries and common carotid arteries were blocked to establish cerebral global ischemia in SD rats. Acute cerebral anoxia was produced by hypobaric anoxia in C57BL/6 mice and hypoxic anoxia in SD rats. Nimodipine and aspirin were set as positive control separately.
Results
Infarct size after MCAO was decreased by sodium sulfide. Sodium sulfide improved cerebral energy metabolism after cerebral global ischemia and prolonged survival time of animals with acute cerebral anoxia. In addition, increased cerebral blood flow and decreased cerebrovascular resistance, blood viscosity, and thrombogenesis were observed in animals treated with sodium sulfide. In cultured neurons, sodium sulfide increased cell viability and decreased cell apoptosis induced by oxygen‐glucose deprivation.
Conclusion
Sodium sulfide, a H2S donor, presents protective effect on acute cerebral ischemia, and might be a promising therapeutic drug.
Keywords: Acute cerebral ischemia, Hydrogen sulfide, Sodium sulfide, Stroke
Introduction
Stroke has become the second most common cause of death worldwide and a major cause of adult chronic disability 1, 2. Acute cerebral ischemia is a condition in which there is insufficient blood flow to the brain to meet metabolic demand. This includes focal ischemia and global ischemia and leads to cerebral hypoxia and thus to the death of brain tissue or ischemic stroke 3, 4, 5, which is the largest subtype of stroke.
Sodium sulfide is a colorless water‐soluble salt with the formula Na2S, which gives strongly alkaline solution. When exposed to moist air, sodium sulfide emits hydrogen sulfide (H2S), which smells like rotten eggs. Increasing evidences have shown that H2S plays an important role in neurological, cardiovascular, and gastrointestinal disorders, particularly cerebral ischemia 6, 7, 8, 9, 10, 11. However, controversial results about the role of H2S in cerebral ischemia have been reported recently. Low dose of H2S protects cortical neurons against brain ischemia 12 as well as oxidative stress 13 in experimental models. In contrast, H2S showed a detrimental effect in cerebral ischemia in both patients and rat stroke model 14, 15.
In the current study, effect of sodium sulfide, a donor of H2S, on acute cerebral ischemia was investigated and compared with nimodipine, a well‐known drug for cerebral vasospasm and resultant ischemia 16, 17. Such effect was tested in animal models of middle cerebral artery occlusion (MCAO), cerebral global ischemia, and acute cerebral anoxia, as well as in cultured neurons under oxygen‐glucose deprivation (OGD), and further confirmed in examinations of cerebral energy metabolism and inflammation, cerebrovascular resistance (CVR) and permeability (CVP), and blood viscosity.
Materials and Methods
Animals and Reagents
Sprague‐Dawley (SD) rats (230–270 g) and C57BL/6 mice (18–20 g) were provided by Sino‐British SIPPR/BK Laboratory Animals (Shanghai, China). Beagle dogs (8–10 kg) and New Zealand white rabbits (1.8–2.2 kg) were provided by Shanghai Jiagan Biotechnology (Shanghai, China). Animals were housed at 22°C under a 12/12 light schedule (on: 08:00), with free access to tap water and standard chow. All experimental procedures were in accordance with institutional animal care guidelines and approved by the local ethics committee. Sodium sulfide was purchased from Sigma‐Aldrich (St. Louis, MO, USA) and dissolved in 0.3% citric acid solution (vehicle control) with PH <8.7. Nimodipine injection was purchased from Bayer HealthCare Pharmaceuticals (Berlin, Germany). Lactic acid (Lac) assay kit and adenosine triphosphate (ATP) assay kit were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Aspirin was purchased from Zhengzhou Chenxu Chemicals (Zhengzhou, China). Creatine phosphate (CP), tumor necrosis factor (TNF) α, and interleukin (IL) six ELISA kits were purchased from Shanghai Bangyi Biotechnology (Shanghai, China).
MCAO, Neurological Deficit Scoring, and 2, 3, 5‐triphenyltetrazolium Chloride (TTC) Staining
The rats were anesthetized with 15% chloral hydrate (300 mg/kg, i.p.). MCAO surgery was performed as described 18, 19, 20. The core temperature was maintained at 36.5–37.5°C by use of a temperature controller pad (Nanjing Xin Xiao Yuan Biotech, Nanjing, China) throughout the surgery. Left external jugular vein was isolated and catheterized for drug administration. Cerebral focal ischemia was produced by intraluminal occlusion of the left middle cerebral artery using a silicone rubber‐coated nylon monofilament. Drugs were given right after MCAO in rats within 15 min. Cessation of cerebral blood flow (CBF) to the area supplied by the left MCA was verified by using a laser Doppler computerized main unit (ADInstruments, Bella Vista, NSW, Australia). CBF must be reduced by at least 70% for inclusion in further experiments. Two hours after MCAO, the occluding filament was withdrawn to allow reperfusion. Twenty‐four hours after MCAO, rats were sacrificed for TTC staining after neurological deficit scoring as described 18, 19, 20.
The coronal slices of brains were prepared with brain‐cutting matrix (ASI Instruments, Warren, MI, USA) and incubated in 0.5% TTC solution (Sinopharm Chemical Reagent, Shanghai, China) at 37°C for 5 min. Then, the slices were photographed with a digital camera. The infarct size was traced and quantified with ImageJ software (National Institutes of Health, Bethesda, MD, USA) and expressed as a percentage of the contralateral hemisphere. The possible interference of a brain edema in assessing the infarct size was corrected with a standard method of subtracting the volume of the nonischemic ipsilateral hemisphere from that of the contralateral hemisphere.
Cerebral Global Ischemia and Electroencephalogram (EEG)
Rats were anesthetized with ketamine (50 mg/kg, i.p.) and diazepam (5 mg/kg, i.p.). Four‐vessel occlusions were performed as described 21. Briefly, skin incision was made above the spinous processes of the first cervical vertebral column to expose the vertebral arteries at the alar foramina of the atlas. The vertebral arteries were electrocoagulated before incision suturing. Animals were kept in rearing cages for 24 h with free access to water and food, and then anesthetized with 15% chloral hydrate (300 mg/kg, i.p.). Left femoral vein was isolated and catheterized for drug administration. Drugs were given 10 min before cerebral global ischemia surgery within 15 min. Needle electrodes were fixed on the back and both sides of the skull subcutaneously for EEG with PowerLab data acquisition system (ADInstruments). After a midline neck incision, bilateral common carotid arteries were isolated and simultaneously blocked using arteriole clamps for 30 second with suppressed waves of EEG, and then the occluders were removed to restore the blood flow. EEG was recorded until waves became normal. Rats were then sacrificed, and immunohistochemistry was performed with one hemisphere. The other hemisphere was homogenized and dried separately for measurement of Lac, ATP, and CP and brain water content detection, calculated as (wet brain weight – dry brain weight) / wet brain weight × 100%.
Measurement of CBF and CVR
Dogs were anesthetized with 3% pentobarbital sodium (30 mg/kg, i.v.). Left femoral artery and vein were isolated and catheterized for blood pressure recording with PowerLab data acquisition system (ADInstruments) and intravenous administration, respectively. Left external carotid artery and arteriolae around were ligated. And then skin incision was made above episternum to expose the left vertebral artery at the lower lateral sutura of the sixth cervical vertebra. Flow probes were attached to common carotid artery and vertebral artery with couplant, and blood flow was detected by a Laser Doppler blood flow meter (Transonic, Ithaca, NY, USA). After stabilization for 15 min, mean blood pressure (MBP), heart rate (HR), and blood flow were recorded at 0, 15, 30, 60, 90, and 120 min after drug administration. Animals were sacrificed, and brains were weighed. CBF (mL/min 100 g brain tissue) = [blood flow of common carotid artery (mL/min) + blood flow of vertebral artery (mL/min)] × 2 × 100 / weight of brain tissue (g). CVR (mmHg min 100 g brain tissue/mL) = MBP (mmHg) / CBF (mL/min 100 g brain tissue).
Measurement of Cerebrovascular Contraction (CVC)
Dogs were anesthetized with 3% pentobarbital sodium (30 mg/kg, i.v.), then killed by air injection through the femoral vein. After craniotomy, the brain was rapidly removed. Basilar arteries were dissected, immersed in cold oxygenated Krebs‐Henseleit solution (KHS) [(mol/L): NaCl, 123; KCl, 6.0; CaCl2, 2.5; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 20; and glucose, 11], and cut into ring segments 3 mm in length. Arterial rings were mounted vertically between two stainless steel hooks in KHS at 37°C aerated with 95% O2 and 5% CO2. One of the hooks was anchored and the other was connected to a force transducer of a biological signal analytical system (Shanghai Alcott Biotech, Shanghai, China). Changes in isometric force were amplified and displayed on a chart recorder of the same analytical system. The resting tension was adjusted to 0.5 g, and the arterial rings were allowed to equilibrate for 60 min, during which time the bathing solution was replaced every 20 min. After induction of contraction with 10−6 mol/L phenylephrine hydrochloride (Shanghaii Harvest Pharmaceutical, Shanghai, China), the arterial rings were rinsed twice and allowed to equilibrate for another 60 min with bathing solution replaced every 20 min. Drugs were added to bathing solution after tension stabilization from the minimum concentration.
Measurement of Ventricular Papillary Muscle Contraction (VPMC)
Dogs were anesthetized with 3% pentobarbital sodium (30 mg/kg, i.v.), then killed by air injection through the femoral vein. The heart was rapidly removed. The right ventricle was opened and anterior papillary muscle was dissected out, and immersed in KHS at 37°C aerated with 95% O2 and 5% CO2. The muscle preparation was vertically placed between parallel platinum plate electrodes and electrical field stimulation was carried out at the rate of 0.5 Hz, using square‐wave pulses lasting 3 ms. The electrical current intensity was set 20% greater than the minimum necessary to produce mechanical response. The metallic clip on the upper end of the muscle was attached to a force transducer of a biological signal analytical system (Shanghai Alcott Biotech) and the lower clip was firmly fixed to the bottom of the bath. Changes in isometric force were amplified and displayed on a chart recorder of the same analytical system. The resting tension was adjusted to 1.0 g, and the muscle preparation was allowed to equilibrate for 60 min, during which time the bathing solution was replaced every 20 min. Drugs were added to bathing solution after tension stabilization from the minimum concentration.
Measurement of Platelet Aggregation (PAG) and Blood Viscosity
Drugs were given i.v. in rabbits within 15 min for 7 days. Whole blood was collected through cardiac puncture. In vitro PAG was assessed by light transmittance aggregometry at 37°C with an Optical Lumi‐Aggregometer (Beijing Succeeder Technology, Beijing, China). Platelet rich plasma (PRP) was obtained from citrate blood after centrifugation at 800 r.p.m. for 10 min. After recovering the PRP, the blood samples were subjected to further centrifugation at 3000 r.p.m. for 10 min to recover platelet poor plasma (PPP). PAG in PRP was induced with Adenosine diphosphate Na2 (Amresco, Solon, OH, USA) at 5 μmol/L. PAG was expressed as the maximal percentage change in light transmittance from baseline, using PPP as a reference. Blood viscosity under shear rates of 10 and 200/second at 37°C was tested with a blood viscometer (Beijing Succeeder Technology).
Acute Cerebral Anoxia
For hypobaric anoxia, drugs were given through the caudal vein of mice within 15 min, and mice were put into a sealed jar with soda lime in it to absorb water and CO210 min later. Survival time of mice in the sealed jar was monitored. For hypoxic anoxia, drugs were given through the caudal vein of rats within 15 min, and rats were put into a relatively sealed organic glass cage with one inlet and one outlet to be continuously ventilated with a mixture of 97% N2 and 3% O2 10 min later. Survival time of rats in the cage was monitored.
Measurement of Thrombus Weight and CVP
For measurement of thrombus weight, rats were anesthetized with 15% chloral hydrate (300 mg/kg, i.p.), and left femoral vein was isolated and catheterized for drug administration. Drugs were given within 15 min. Thrombus formation was determined by measuring the change in wet weight of a silk thread placed in a shunt between right common carotid artery and left external jugular vein during a period of 15 min.
For measurement of CVP, rats were anesthetized with 15% chloral hydrate (300 mg/kg, i.p.), and left femoral vein was isolated and catheterized for drug administration. Drugs were given within 15 min, and bilateral common carotid arteries were isolated 1 h later. Evans blue (EB, 50 mg/kg, Sinopharm Chemical Reagent) was injected through sublingual vein, and bilateral common carotid arteries were ligated 5 min later. Animals were sacrificed 3 h later, and brains were weighed, immersed in 10 mL formamide (Sinopharm Chemical Reagent), and then incubated at 45°C for 72 h for extravasation of EB. EB concentration in formamide was quantitatively determined by measuring the absorbance at 570 nm with a spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). EB content was quantified as micrograms of EB per gram of wet brain, using a standard curve.
Neuron Culture and OGD Model
Primary neuronal cells were obtained from the cerebral cortex of neonatal SD rats within 6 h after birth, as described previously 22, 23. One day after isolation, the cultures were replenished with Neurobasal medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B27 (Invitrogen). Glial growth was suppressed by addition of 10 μ m uridine. The cultured cells were proved to contain > 90% neurons by staining for NeuN (neuron marker) with anti‐NeuN antibody (Millipore, Billerica, CA, USA). After 7‐day culture in vitro, the neurons were treated with drugs, 24 h prior to OGD. To establish OGD condition, the cultured neurons were washed three times, cultured in DMEM with no glucose and incubated for 12 h in a hypoxic chamber (Thermo Fisher Scientific) that was continuously flushed with 94% N2 and 5% CO2 at 37°C to obtain 1% O2. After 7‐day culture in vitro, cultured cells were exposed to OGD for 12 h prior to assay of cell viability and apoptosis and ELISA of TNF‐α and IL‐6 in conditioned medium.
Cell Viability and Apoptosis Assay
For cell viability assay, cells were treated with 5 mg/mL thiazolyl blue tetrazolium bromide (MTT) solution (Sigma‐Aldrich), and incubated at 37°C for 4 h. Formazan crystal was dissolved in 100 μL DMSO, and MTT reduction was measured at 570 nm using a spectrophotometer (Thermo Fisher Scientific) as cell viability 24, 25, 26. For cell apoptosis assay, cell survival and death were examined by manually counting the cells double‐stained with 1 μg/mL Hoechst (Beyotime Institute of Biotechnology, Haimen, China) and by TUNEL staining kit (Roche, Mannheim, Germany), respectively. The cell nuclei were counterstained with Hoechst 33342 (1 μg/mL). The dead or apoptotic cells were labeled green with the TUNEL staining kit. Images were acquired under a fluorescent microscope (Olympus, Tokyo, Japan) with 12.8 M pixel recording digital color cooled camera (Olympus). The ratio of TUNEL stained cells to those labeled by Hoechst is defined as death or apoptosis rate.
Statistical Analysis
Data are expressed as mean ± standard deviation. For experiments involving only two groups, data were analyzed with t‐test or t'‐test. For experiments involving more than two groups, data were analyzed with anova followed by Bonferroni test or Games‐Howell test. P < 0.05 was considered statistically significant. Analyses were performed using SPSS 21.0 (SPSS, Chicago, IL, USA).
Results
Sodium Sulfide Protects Against Acute Cerebral Ischemia
Sodium sulfide decreased infarct size of rats dose‐dependently compared with control group, achieving the nadir at 20 mg/kg (24.5% vs. 40.5%; P < 0.001; Figure 1A). The neurological deficit score was decreased concomitantly at 20 mg/kg by 30.8% (Figure 1B). The efficacy of sodium sulfide against acute cerebral ischemia was stronger than nimodipine, although it had weaker potency than nimodipine (Figure 1C). Sodium sulfide (10 mg/kg) was effective when given within 2 h after MCAO (Figure 1D).
Figure 1.
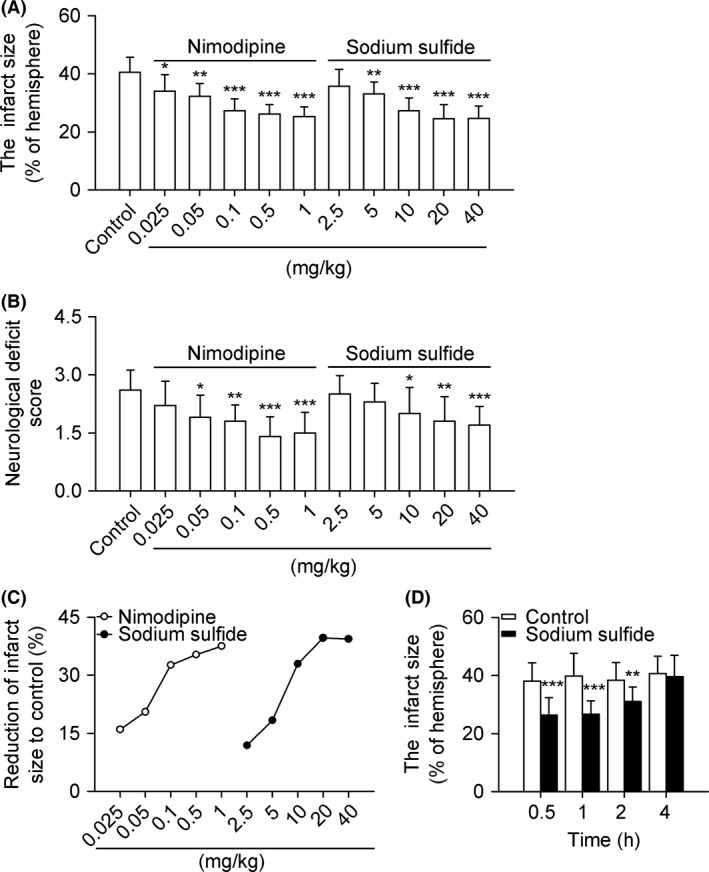
Sodium sulfide protects against acute cerebral ischemia in rats. SD rats received nimodipine (0.025–1 mg/kg, i.v.), sodium sulfide (2.5–40 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 20 mL/kg, i.v.). Both nimodipine and sodium sulfide decreased infarct size dose‐dependently (A). The neurological deficit scores were decreased concomitantly both in nimodipine and sodium sulfide groups (B). The efficacy and potency of sodium sulfide and nimodipine on reduction of infarct size were compared (C). Time window for sodium sulfide (10 mg/kg, i.v.) effective on infarct size was tested within 4 h after MCAO (D). n = 10 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Sodium Sulfide Protects Against Cerebral Global Ischemia
Sodium sulfide decreased recovery time of EEG of rats dose‐dependently compared with control group (Figure 2A). Lac and ATP in brain tissue were increased by sodium sulfide dose‐dependently (Figure 2C,D). Sodium sulfide had no effect on water content of wet brain tissue and CP in brain tissue (Figure 2B,E). Immunohistochemistry demonstrated that sodium sulfide did not decrease necrosis of neuronal cells, and had no effect on brain edema (Figure 2F).
Figure 2.
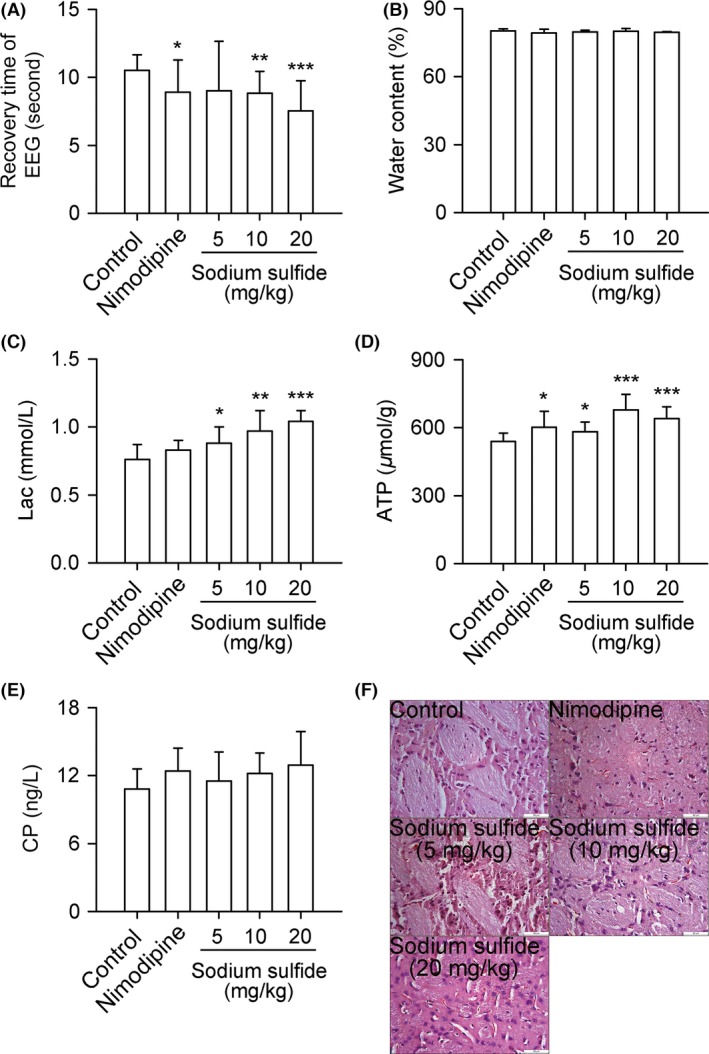
Sodium sulfide protects against cerebral global ischemia in rats. SD rats received nimodipine (0.1 mg/kg, i.v.), sodium sulfide (5, 10, 20 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 20 mL/kg, i.v.). Sodium sulfide decreased recovery time of EEG dose‐dependently (A). Lac and ATP in brain tissue were increased by sodium sulfide dose‐dependently (C, D). Sodium sulfide had no effect on water content of wet brain tissue and CP in brain tissue (B, E). Representative images of brain tissue immunohistochemistry (F). Scale bars = 50 μm. n = 10 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Sodium Sulfide Improves CBF of Dogs and Inhibits CVC
Sodium sulfide increased CBF and decreased CVR of dogs at a dose of 6 mg/kg from1 h to 2 h after administration (Figure 3C,D), with no effect on MBP and HR (Figure 3A,B). Sodium sulfide inhibited CVC dose‐dependently, with an efficacy 70.7% at 10 mmol/L, which was larger than that of nimodipine, although sodium sulfide had weaker potency than nimodipine. IC50 for sodium sulfide to inhibit CVC is 407 μmol/L (Figure 3E), and IC50 for sodium sulfide to inhibit VPMC is 4269 μmol/L (Figure 3F).
Figure 3.
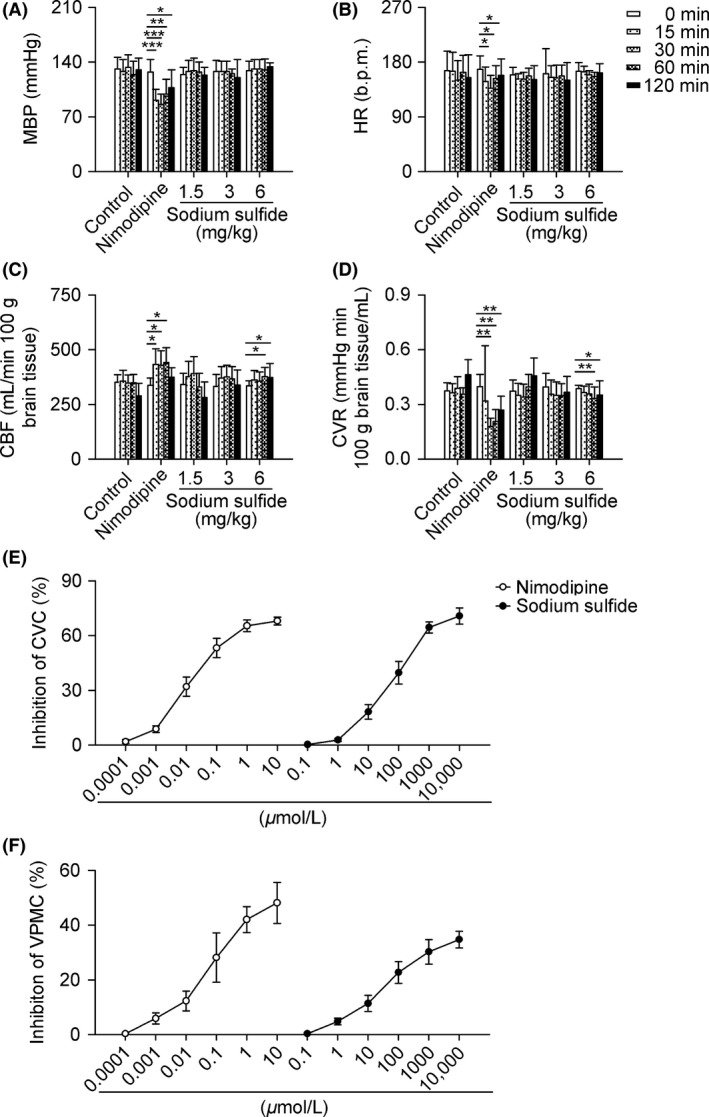
Sodium sulfide improves CBF of dogs and inhibits CVC. Beagle dogs received nimodipine (0.03 mg/kg, i.v.), sodium sulfide (1.5, 3, 6 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 1 mL/kg, i.v.). Sodium sulfide increased CBF and decreased CVR at a dose of 6 mg/kg from1 h to 2 h after administration (C, D), with no effect on MBP and HR (A, B). In another set of experiments, basilar arterial rings and ventricular papillary muscles from dog were treated with nimodipine (0.0001–10 μmol/L), sodium sulfide (0.1–10000 μmol/L), or injection water as control. Sodium sulfide inhibited CVC and VPMC dose‐dependently (E, F). n = 6 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus 0 min.
Sodium Sulfide Inhibits PAG and Decreases Blood Viscosity
Sodium sulfide inhibited PAG of rabbits at a dose of 10 mg/kg with an inhibition rate of 24.4% compared with control group (Figure 4A). The blood viscosity was also decreased at 10 mg/kg (Figure 4B,C), especially at a shear rate of 10/second by 15.9% (P < 0.05).
Figure 4.
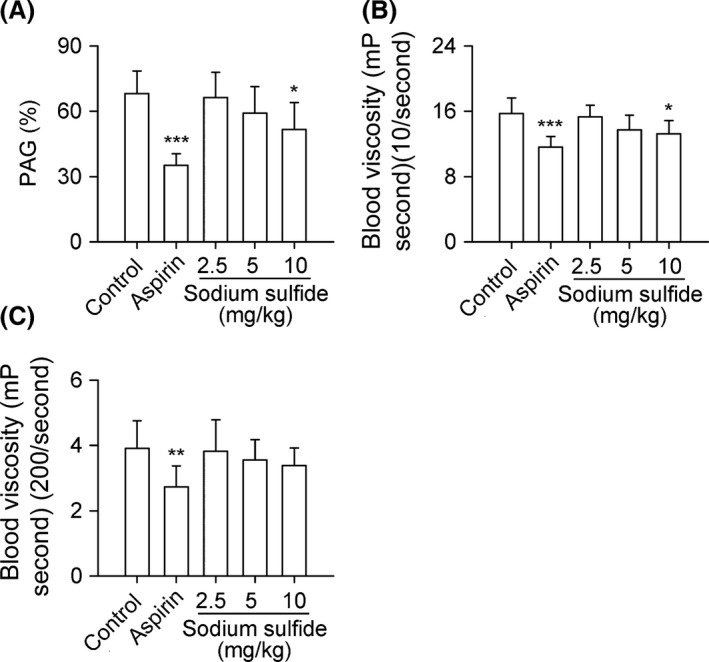
Sodium sulfide inhibits PAG and decreases blood viscosity in rabbits. New Zealand white rabbits received aspirin (10 mg/kg, i.v.), sodium sulfide (2.5, 5, 10 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 10 mL/kg, i.v.) for 7 days. Sodium sulfide inhibited PAG at a dose of 10 mg/kg (A). The blood viscosity was also decreased by sodium sulfide at 10 mg/kg at a shear rate of both 10 and 200/second (B, C). n = 8 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Sodium Sulfide Protects Against Acute Cerebral Anoxia
Sodium sulfide prolonged survival time dose‐dependently both in hypobaric anoxic mice and hypoxic anoxic rats compared with control groups (Figure 5,B).
Figure 5.
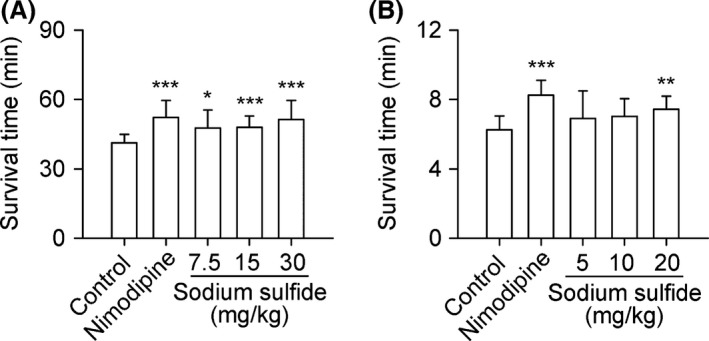
Sodium sulfide protects against acute cerebral anoxia. C57BL/6 mice received nimodipine (0.15 mg/kg, i.v.), sodium sulfide (7.5, 15, 30 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 10 mL/kg, i.v.). In another set of experiments, SD rats received nimodipine (0.1 mg/kg, i.v.), sodium sulfide (5, 10, 20 mg/kg, i.v.), or injection water (20 mL/kg, i.v.) as control. Sodium sulfide prolonged survival time dose‐dependently both in hypobaric anoxic mice (A, n = 12) and hypoxic anoxic rats (B, n = 10). *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Sodium Sulfide Inhibits Thrombogenesis and Decreases CVP
Sodium sulfide decreased thrombus weight and EB content of rats dose‐dependently compared with control group (Figure 6A,B), reflecting less thrombogenesis and lower CVP, respectively.
Figure 6.
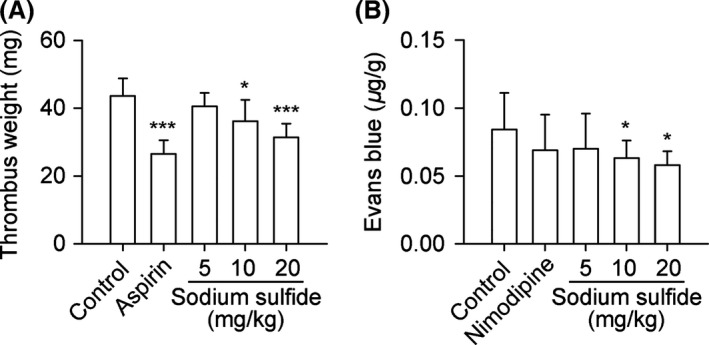
Sodium sulfide inhibits thrombogenesis and decreases CVP in rats. SD rats received aspirin (20 mg/kg, i.v.), sodium sulfide (5, 10, 20 mg/kg, i.v.), or 0.3% citric acid solution (vehicle control, 20 mL/kg, i.v.). In another set of experiments, SD rats received nimodipine (0.1 mg/kg, i.v.), sodium sulfide (5, 10, 20 mg/kg, i.v.), or injection water (20 mL/kg, i.v.) as control. Sodium sulfide inhibited thrombogenesis (A) and decreased CVP dose‐dependently (B). n = 10 per group. *P < 0.05, ***P < 0.001 versus control.
Sodium Sulfide Protects Neuron from OGD
Sodium sulfide increased cell viability and decreased cell apoptosis of primarily cultured neurons under OGD (Figure 7A,B), but had no effect on TNF‐α and IL‐6 secreted by primarily cultured neurons under OGD (Figure 7C,D).
Figure 7.
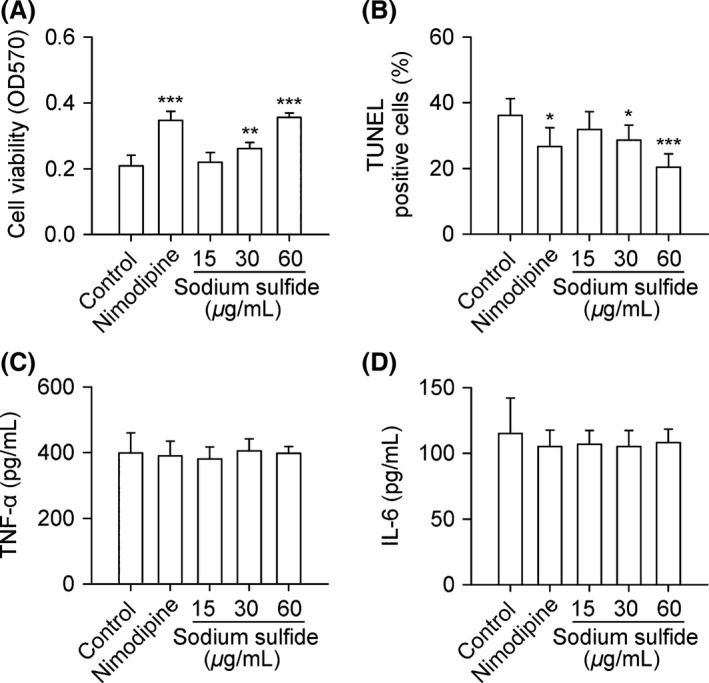
Sodium sulfide protects neuron from OGD. Primary neurons from SD rats were treated with nimodipine (0.3 μg/mL), sodium sulfide (15, 30, 60 μg/mL), or 0.3% citric acid solution (vehicle control). Sodium sulfide increased cell viability and decreased cell apoptosis under OGD (A, B), but had no effect on TNF‐α and IL‐6 secreted by primary neurons under OGD (C, D). n = 6 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Discussion
The major findings of the present study include: (1) sodium sulfide protects against acute cerebral ischemia and acute cerebral anoxia; (2) sodium sulfide has a stronger efficacy against acute cerebral ischemia than nimodipine, without influence on MBP and HR; (3) sodium sulfide protects against neuron apoptosis in vitro.
H2S is an endogenously produced gaseous signaling molecule and is maintained at physiologic concentrations in mammalian systems 27, 28. Sodium sulfide, a H2S donor, has been used in the study of cytoprotective effects of H2S 10, 29. Nevertheless, the role of H2S in cerebral ischemia remains controversial 10, 11, 12, 13, 14, 15, 30, 31. In the present study, sodium sulfide protected against acute cerebral ischemia in rats with MCAO, manifested as decreased infarct size and neurological deficit score. Such effects were confirmed by comparison with nimodipine which exerted definite protection on acute cerebral ischemia. Our results showed that the efficacy of sodium sulfide against acute cerebral ischemia was slightly stronger than nimodipine, and the effective treatment window was limited to 2 h after MCAO. The protective effects of sodium sulfide was further tested in rats with cerebral global ischemia, and proved by shortened recovery time of EEG and improved cerebral energy metabolism. Meanwhile, sodium sulfide prolonged survival time of rodents with acute cerebral anoxia.
Severity of cerebral ischemia depends on lots of physiological indexes. Consistent with the above results, we observed increased CBF and decreased CVR, CVP, and thrombogenesis in animals treated with sodium sulfide. Inspiringly, different from nimodipine, sodium sulfide had no influence on MBP and HR at therapeutic doses. PAG and blood viscosity were also decreased by sodium sulfide, with aspirin as positive control. Sodium sulfide exerted a selective inhibition of CVC compared with VPMC. Furthermore, results from in vitro study demonstrated that sodium sulfide increased neuron viability and decreased neuron apoptosis. However, brain edema and inflammation were not alleviated by sodium sulfide, which were similar in nimodipine groups.
It has been reported that H2S may exert neuroprotective effects at low concentrations and destructive effects at high concentrations in cerebral ischemia 9, and the protective effects of H2S are mediated by thiosulfate 10. In the present study, sodium sulfide may emit H2S at physiological concentrations, and protect against acute cerebral ischemia through thiosulfate. Additional studies are needed to clarify the mechanisms of the neuroprotective effects of sodium sulfide in cerebral ischemia.
In conclusion, results from the current study indicated that sodium sulfide, a H2S releasing molecule, could protect against acute cerebral ischemia, and deserved further investigation for clinical application.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (81102453).
The first three authors contributed equally to this work.
References
- 1. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet 2008;371:1612–1623. [DOI] [PubMed] [Google Scholar]
- 2. Tang YH, Ma YY, Zhang ZJ, Wang YT, Yang GY. Opportunities and challenges: stem cell‐based therapy for the treatment of ischemic stroke. CNS Neurosci Ther 2015;21:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koronowski KB, Dave KR, Saul I, et al. Resveratrol preconditioning induces a novel extended window of ischemic tolerance in the mouse brain. Stroke 2015;46:2293–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun Y, Zhang L, Chen Y, Zhan L, Gao Z. Therapeutic targets for cerebral ischemia based on the signaling pathways of the GluN2B C terminus. Stroke 2015;46:2347–2353. [DOI] [PubMed] [Google Scholar]
- 5. Zuloaga KL, Zhang W, Roese NE, Alkayed NJ. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front Pharmacol 2014;5:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei HJ, Li X, Tang XQ. Therapeutic benefits of H(2)S in Alzheimer's disease. J Clin Neurosci 2014;21:1665–1669. [DOI] [PubMed] [Google Scholar]
- 7. Salloum FN. Hydrogen sulfide and cardioprotection–Mechanistic insights and clinical translatability. Pharmacol Ther 2015;152:11–17. [DOI] [PubMed] [Google Scholar]
- 8. Motta JP, Flannigan KL, Agbor TA, et al. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm Bowel Dis 2015;21:1006–1017. [DOI] [PubMed] [Google Scholar]
- 9. Hadadha M, Vakili A, Bandegi AR. Effect of the inhibition of hydrogen sulfide synthesis on ischemic injury and oxidative stress biomarkers in a transient model of focal cerebral ischemia in rats. J Stroke Cerebrovasc Dis 2015;24:2676–2684. [DOI] [PubMed] [Google Scholar]
- 10. Marutani E, Yamada M, Ida T, et al. Thiosulfate mediates cytoprotective effects of hydrogen sulfide against neuronal ischemia. J Am Heart Assoc 2015;4:e002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei X, Zhang B, Cheng L, et al. Hydrogen sulfide induces neuroprotection against experimental stroke in rats by down‐regulation of AQP4 via activating PKC. Brain Res 2015;1622:292–299. [DOI] [PubMed] [Google Scholar]
- 12. Geng Y, Li E, Mu Q, et al. Hydrogen sulfide inhalation decreases early blood‐brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J Cereb Blood Flow Metab 2015;35:494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 2004;18:1165–1167. [DOI] [PubMed] [Google Scholar]
- 14. Wong PT, Qu K, Chimon GN, et al. High plasma cyst(e)ine level may indicate poor clinical outcome in patients with acute stroke: possible involvement of hydrogen sulfide. J Neuropathol Exp Neurol 2006;65:109–115. [DOI] [PubMed] [Google Scholar]
- 15. Qu K, Chen CP, Halliwell B, Moore PK, Wong PT. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006;37:889–893. [DOI] [PubMed] [Google Scholar]
- 16. Dabus G, Nogueira RG. Current options for the management of aneurysmal subarachnoid hemorrhage‐induced cerebral vasospasm: a comprehensive review of the literature. Interv Neurol 2013;2:30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth 2012;109:315–329. [DOI] [PubMed] [Google Scholar]
- 18. Li Y, Xu XL, Zhao D, et al. TLR3 ligand poly IC attenuates reactive astrogliosis and improves recovery of rats after focal cerebral ischemia. CNS Neurosci Ther 2015;21:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu JH, Feng D, Zhang YF, et al. Chloral hydrate preconditioning protects against ischemic stroke via upregulating annexin A1. CNS Neurosci Ther 2015;21:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen HS, Chen XM, Feng JH, Liu KJ, Qi SH, Shen JG. Peroxynitrite decomposition catalyst reduces delayed thrombolysis‐induced hemorrhagic transformation in ischemia‐reperfused rat brains. CNS Neurosci Ther 2015;21:585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han J, Chen ZW, He GW. Acetylcholine‐ and sodium hydrosulfide‐induced endothelium‐dependent relaxation and hyperpolarization in cerebral vessels of global cerebral ischemia‐reperfusion rat. J Pharmacol Sci 2013;121:318–326. [DOI] [PubMed] [Google Scholar]
- 22. Zheng JL, Li GZ, Chen SZ, et al. Angiotensin converting enzyme 2/Ang‐(1‐7)/mas axis protects brain from ischemic injury with a tendency of age‐dependence. CNS Neurosci Ther 2014;20:452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y, Liu XZ, Tian WW, Guan YF, Wang P, Miao CY. Extracellular visfatin has nicotinamide phosphoribosyltransferase enzymatic activity and is neuroprotective against ischemic injury. CNS Neurosci Ther 2014;20:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sinagra T, Merlo S, Spampinato SF, Pasquale RD, Sortino MA. High mobility group box 1 contributes to wound healing induced by inhibition of dipeptidylpeptidase 4 in cultured keratinocytes. Front Pharmacol 2015;6:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoppstadter J, Seif M, Dembek A, et al. M2 polarization enhances silica nanoparticle uptake by macrophages. Front Pharmacol 2015;6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abo Elnazar SY, Ghazy AA, Ghoneim HE, Taha AR, Abouelella AM. Effect of ultra violet irradiation on the interplay between Th1 and Th2 lymphocytes. Front Pharmacol 2015;6:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kimura H, Shibuya N, Kimura Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal 2012;17:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimura H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide 2014;41:4–10. [DOI] [PubMed] [Google Scholar]
- 29. Balaban CL, Rodriguez JV, Tiribelli C, Guibert EE. The effect of a hydrogen sulfide releasing molecule (Na2S) on the cold storage of livers from cardiac dead donor rats. A study in an ex vivo model. Cryobiology 2015;71:24–32. [DOI] [PubMed] [Google Scholar]
- 30. Cheung NS, Peng ZF, Chen MJ, Moore PK, Whiteman M. Hydrogen sulfide induced neuronal death occurs via glutamate receptor and is associated with calpain activation and lysosomal rupture in mouse primary cortical neurons. Neuropharmacology 2007;53:505–514. [DOI] [PubMed] [Google Scholar]
- 31. Chen MJ, Peng ZF, Manikandan J, et al. Gene profiling reveals hydrogen sulphide recruits death signaling via the N‐methyl‐D‐aspartate receptor identifying commonalities with excitotoxicity. J Cell Physiol 2011;226:1308–1322. [DOI] [PubMed] [Google Scholar]