

Summary
Aim
Insulin‐like growth factor I (IGF‐I) is a neuroprotective agent in animal models of ischemic stroke. The purpose of this study was to determine whether systemically injected IGF‐I exerts its neuroprotective action by binding to IGF‐I receptors in the brain after crossing the blood–brain barrier, or via peripheral effects.
Methods
To differentiate the central effects of IGF‐I from systemic effects, ischemic stroke was induced in conscious male Wistar Kyoto rats by the injection of endothelin‐1 adjacent to the middle cerebral artery in the right hemisphere, while either the IGF‐I receptor antagonist JB‐1 or vehicle was introduced into the right lateral ventricle.
Results
Intravenous injection of recombinant human (rh)IGF‐I resulted in 50% reduction in infarct size, which was counteracted by the central administration of JB‐1. Furthermore, rhIGF‐I was detected in both the ischemic and nonischemic hemisphere.
Conclusions
Systemically injected rhIGF‐I passes the blood–brain barrier and protects neurons via IGF‐I receptors in the brain in rats with an ischemic stroke.
Keywords: Blood–brain barrier, Cerebral stroke, Endothelins, Insulin‐like growth factor I, Middle cerebral artery, Neuroprotection
Introduction
Insulin‐like growth factor I (IGF‐I) exerts neuroprotective effects in different experimental models of ischemic stroke 1. In humans, higher circulating levels of IGF‐I measured early after stroke onset have been associated with better outcome, suggesting that systemic injection of IGF‐I could be a treatment option for stroke patients 2, 3, 4, 5. Several studies have suggested that neuroprotection by IGF‐I in preclinical stroke models may be mediated by the modulation of several events of the ischemic cascade including glial cell death, neuroinflammation, oxidative stress, and inhibition of excitotoxicity 6. Despite these data, it remains uncertain whether systemically injected IGF‐I exerts its neuroprotective effects through binding to the receptors in the brain, or through systemic effects. In the latter case, IGF‐I‐mediated modulation of adhesion molecules on endothelial cells or circulating leukocytes could affect the infiltration of leukocytes (predominantly monocytes and neutrophils) in response to cerebral ischemia. Moreover, hyperglycemia exacerbates poor outcome after stroke 7 and IGF‐I exerts hypoglycemic effects 8. Therefore, it is possible that IGF‐I positively influences the outcome after ischemic stroke via the restoration of glucose levels.
In the current study, we assessed, for the first time, the transport to the brain of systemically injected IGF‐I and the putative central effects of IGF‐I on neuroprotection in a preclinical stroke model. To dissect the central action component from the systemic component, we combined systemic IGF‐I administration with central injection of the IGF‐I receptor antagonist JB‐1 9. We found that systemically injected IGF‐I reduced infarct size through central actions via its receptor in the brain.
Materials and Methods
Surgery
Protocols for animal experiments were designed according to the European Guidelines on Animal Experimentation and approved by the Ethical Committee for Animal Experimentation of the Vrije Universiteit Brussel (VUB). Male albino Wistar Kyoto rats (WKYR: Charles River Laboratories, L'Arbresle Cedex, France) were housed in groups of four and allowed to recover from transport and to habituate to their new environment in the animal house for 1 week having free access to tap water and standard laboratory chow.
Surgery for the induction of ischemic stroke was performed on 10‐ to 12‐week‐old male rats weighing 275–300 g as described earlier 10. The rats were anaesthetized by intraperitoneal injection of 75 mg/kg ketamine and 3.5 mg/kg diazepam. The stereotactic coordinates for the injection of endothelin‐1 (Et‐1) close to the middle cerebral artery (MCA) in the piriform cortex were determined using the Paxinos and Watson rat brain atlas 11 (coordinates relative to bregma: anterior/posterior +0.9 mm, lateral +5.0 mm, and ventral +2.8 mm). A guide was implanted using a stereotactic frame. In this model, the core of the infarct is located in the striatum, whereas the penumbra is present in the cortex and striatum. Postoperative pain treatment consisted of an intraperitoneal injection of 5 mg/kg ketoprofen.
To address the role of central IGF‐I receptors, a second guide was implanted for the infusion of JB‐1 into the right lateral ventricle (coordinates relative to bregma; anterior/posterior −0.9, lateral +1.4, and ventral +3.5) for intracerebroventricular administration of JB‐1. Furthermore, an indwelling catheter filled with 0.9% NaCl was implanted in the left femoral vena and subcutaneously tunneled to the back of the neck and exteriorized 12. The catheter was made from 20‐cm pyrogen‐free polyethylene tubing (internal diameter 0.58 mm; outer diameter 0.96 mm) (Portex Limited, Hyte, Kent, UK).
Induction of Ischemic Stroke
Twenty‐four hours after surgery, the guide positioned close to the MCA in the piriform cortex was replaced by a cannula, and 6 μL of 200 pmol Et‐1 (Sigma, St Louis, MO, USA) in iso‐osmotic Ringer's solution was infused at a rate of 1 μL/min into freely moving animals.
IGF‐I Treatment and Administration of JB‐1
The rats were randomly assigned to the treatment groups. IGF‐I was a gift from Ipsen NV (Merelbeke, Belgium). Doses of 300 μg were prepared in 0.4 mL vehicle solution (0.9% NaCl) and injected intravenously at 30 min after the insult via the catheter. Placebo rats were injected with vehicle solution alone. The guide in the right lateral ventricle was replaced by a cannula for two injections of JB‐1 at 30 min before and again at 15 min after Et‐1 administration. For each injection, 10 μg JB‐1 (Bachem, Bubendorf, Switzerland) in 3 μL 0.9% NaCl was infused at a rate of 1 μL/min.
Histology and Immunohistochemistry
Infarct sizes and the presence of neurons were determined at 24 h after the insult. The rats received an intraperitoneal injection of sodium pentobarbital and were subsequently transcardially perfused for 5 min with 0.9% NaCl followed by perfusion for 5 min with a 4% phosphate‐buffered paraformaldehyde solution (pH 7.42). Subsequently, the brains were postfixed in paraformaldehyde and the brain slices of 50 μm were made using a vibratome (Leica VTS1000, Bensheim, Germany). The slices were kept at 4°C in PBS containing 0.01% sodium azide.
Infarct size was assessed using a series of equidistant 50‐μm sections, comprising every 4th section, from 4.70 to −1.80 mm from bregma. The sections were mounted onto gelatin‐coated microscope slides and stained with cresyl violet. Infarct sizes were calculated using Image J software (NIH, version 1.43, http://www.ncbi.nlm.nih.gov). The Cavalieri principle for the estimation of volumes 13 was used to estimate the infarct size (v) using the following formula: v = d × Σa, where d is the distance between the upper (rostral) surfaces of two consecutive analyzed sections and a is the surface area of a section. Edema corrections were made according to the following equation: infarct volume * (volume contralateral side/volume ipsilateral side).
To detect the neurons, 50‐μm coronal brain slices were mounted on 3‐aminopropyltriethoxylane (APES)‐coated slides and incubated with a mouse monoclonal anti‐NeuN antibody (1:1.000 in normal goat serum/PBS, Millipore, Temecula, CA, USA, catalog number: MAB377) and a peroxidase‐conjugated sheep anti‐mouse IgG as a second antibody (1:100 in normal goat serum/PBS, GE Healthcare UK Limited, Little Chalfont, Buckinghamshire, UK, catalog number: NA931V) 14.
Micrographs were taken using a Canon Powershot G5 camera (5.0‐M pixel CCD) attached to a Zeiss Axioskop 40 microscope (Oberkochen, Germany) with a plan‐Neofluar lens (10 × 0.30). Micrographs were processed using Adobe Photoshop CS3 for conversion to black and white and to correct for vignetting and to optimize contrast and image sharpening.
Assessment of IGF‐I Transport to the Brain
Solutions containing 300 μg recombinant human rhIGF‐I were prepared as described above and injected subcutaneously at 30 min after the injection of Et‐1 (stroke) or Ringer's solution (sham). The rats were sacrificed 90 min after the insult, perfused with saline, followed by prelevation of the brain. Left and right brain hemispheres were then snap‐frozen and stored at −80°C. Frozen hemispheres were homogenized in HEPES buffer (20 mM HEPES pH 7.4 with phosphoric acid, NaOH, and 1.5 mM EDTA) with protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Homogenates were then sonicated for 60 s at high pulse rate and centrifuged at 12,000 × g for 30 min at 4°C. The supernatant was stored at −20°C. The concentration of rhIGF‐I in the supernatant was determined using a human IGF‐I‐specific ELISA (ELISA Quantikine® kit, R&D systems Bio‐rad laboratories, München, Germany). Total protein concentrations in the homogenates were assessed using the Bradford method 15.
Determination of Blood rhIGF‐I and Glucose Levels
Blood samples were obtained by nicking the lateral tail vein with a scalpel and the glucose levels were determined using the Accu Chek (Aviva, Roche Diagnostics, Vilvoorde, Belgium) glucometer. After the preparation of serum, the levels of rhIGF‐I were measured as described above.
Statistics
The mortality rate in the experiments with the JB‐1 (Figure 1) was 27.5%. For this study, 40 rats were used, five animals died during surgery, and six rats died within one hour after the induction of stroke. In addition, one animal removed the guide, six animals were excluded after the histological assessment because of an incorrect placement of one of the guides, and four rats were excluded due to the damage of brain slices. In the study on IGF‐I transport (Figure 2B), 46 rats were used, two died during surgery, and two died directly after the induction of stroke.
Figure 1.
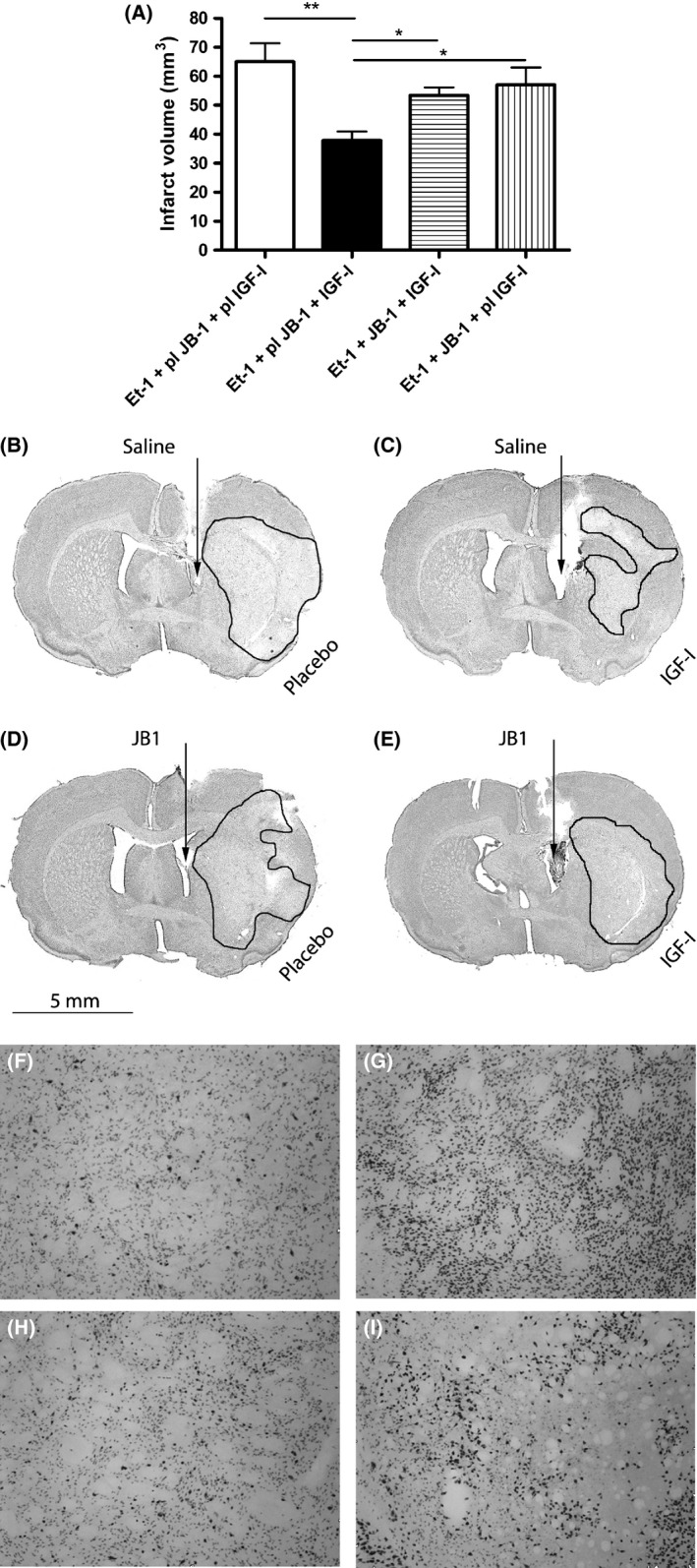
Central administration of JB‐1 blocks the neuroprotective effect of systemic IGF‐I injection. The effects of JB‐1 on the infarct volume are shown in Figure 1(A). Ischemic stroke was induced in all groups by the administration of endothelin‐1 (Et‐1). Control experiments consisted of the administration of vehicle solutions for JB‐1 (saline) or IGF‐I (placebo). Representative micrographs of the cresyl violet staining are shown in micrographs 1(B–E). Representative micrographs of NeuN immunohistochemistry are demonstrated in panels (F–I). (B+F): rats that received saline and placebo (n = 4); (C+G): rats that received saline and 300 μg IGF‐I (n = 6); (D+I): rats that received JB‐1 and placebo (n = 3); (E+H): rats that received JB‐1 and IGF‐I (n = 5). A statistical significance was assessed by an one‐way ANOVA followed by the Newman–Keuls post hoc tests. *Significant difference between the groups (P < 0.05). **Significant difference between the groups (P < 0.01).
Figure 2.
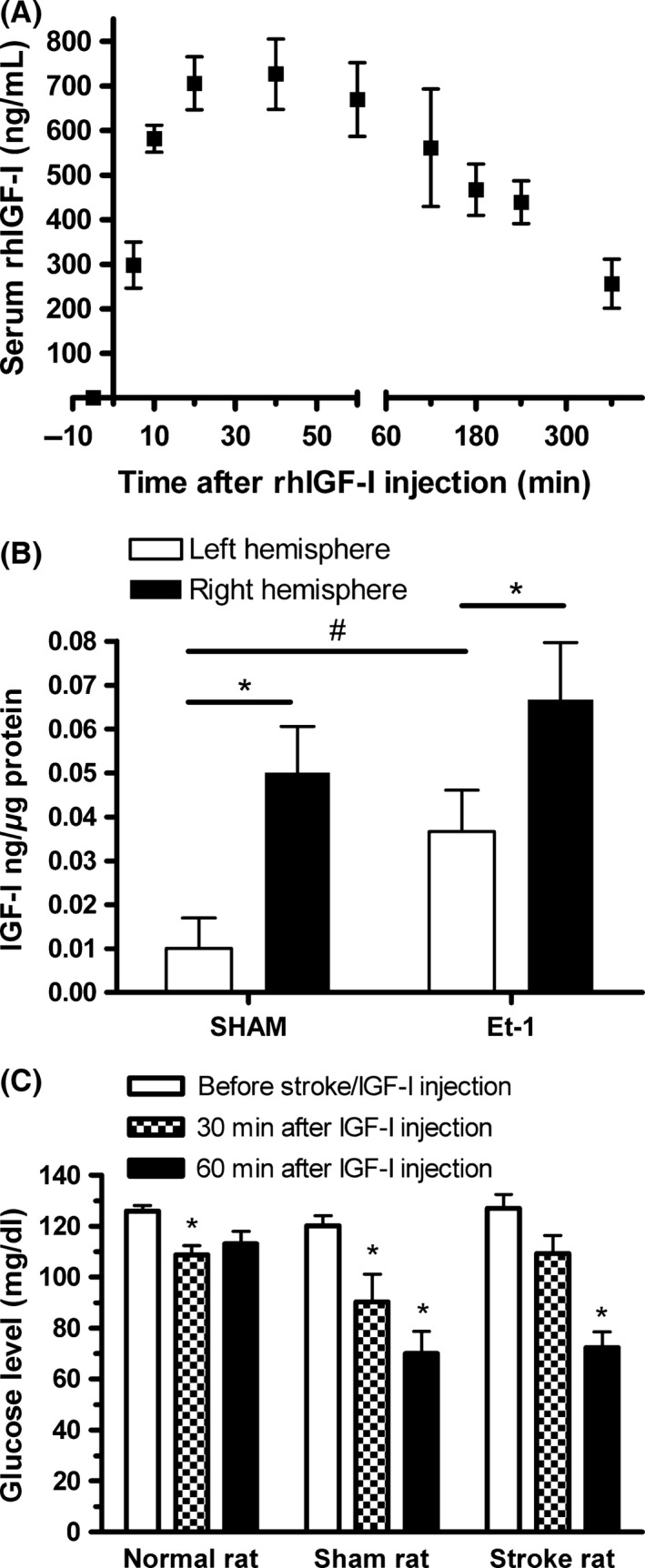
(A) Determination of serum rhIGF‐I levels after subcutaneous injection of 300 ug rhIGF‐I. A human IGF‐I‐specific ELISA was used to quantify rhIGF‐I. No IGF‐I was detected in the serum before the injection of rhIGF‐I (see time point ‐5 min). Values represent mean values ± SEM of four rats. (B) Transport to the brain of systemically injected rhIGF‐I. Levels of rhIGF‐I were measured in the ipsi‐ and contralateral hemisphere of sham‐operated animals and animals with an ischemic stroke in the right hemisphere induced by the injection of endothelin‐1. #Statistically significant (P < 0.05) as assessed by the unpaired Student's t‐test. *Statistically significant (P < 0.05) as assessed by the paired Student's t‐test. Each of the four groups consisted of 9–12 rats. (C) Measurements of blood glucose levels in normal (n = 14), sham‐operated (n = 9), and stroke rats (n = 7). Normal rats did not undergo surgery or any other treatment than the injection of rhIGF‐I. Blood was collected from the tail vein 15 min before the insult and 30 and 60 min after the injection of rhIGF‐I. IGF‐I was injected 30 min after the induction of ischemic stroke. In normal rats, the first measurement was taken 45 min before IGF‐I injection. * (P < 0.05) as assessed by the two‐way ANOVA.
Statistical analysis was performed using GraphPad Prism (version 4.03, GraphPad Software, San Diego, California, USA). Statistical significance (P < 0.05) between two groups was assessed by the paired or unpaired Student's t‐test. Statistical difference between more than two groups was evaluated via the one‐way ANOVA followed by the Newman–Keuls multiple comparison test or a two‐way ANOVA followed by the Bonferroni posttest. All data represent mean values ± SEM.
Results
Although IGF‐I has been shown to pass the blood–brain barrier (BBB) within 20 min after intravenous injection 16, it has never been investigated whether the systemic administration of IGF‐I indeed leads to the interaction with its receptors in the brain or whether the systemic effects are involved. To address the possible direct effects of IGF‐I on its receptors in the brain, we studied the effects of the selective IGF‐I receptor antagonist JB‐1. This antagonist was injected into the right lateral ventricle at 60 and 15 min before IGF‐I injection. Figure 1 reveals that IGF‐I effectively reduced the infarct volumes and blocking the cerebral IGF‐I receptor by intraventricular administration of JB‐1 significantly (P < 0.05) reduced the neuroprotective effects of IGF‐I. Representative micrographs of the infarcts are shown in Figure 1B–E. In addition, immunohistochemical staining with NeuN, a marker for neurons, shows that IGF‐I partially reverses the reduction in NeuN expression at the insult site and that this effect is also attenuated by JB‐1 (micrographs in Figure 1F–I). According to the literature, the latter effect may reflect the stimulation of nuclear factor kappa B (NFκB) by IGF‐I, mostly acting as a survival factor in neurons 17. Figure 1A also shows that central injection of JB‐1 in the absence of IGF‐I administration did not affect the infarct volume, ruling out the possible effects of JB‐1 through the inhibition of endogenous IGF‐I. Taken together, these results suggest that systemically administered IGF‐I acts on the brain. Direct evidence for IGF‐I transport to the brain in a rat model for ischemic stroke was obtained by measuring the amount of rhIGF‐I in the brain using a human IGF‐I‐specific ELISA after the systemic injection of rhIGF‐I. We first measured rhIGF‐I levels in the serum in normal rats and found that rhIGF‐I was detectable 5 min after the injection and that peak levels of ca 700 ng/mL were obtained 20 min after injection (Figure 2A). One hour after rhIGF‐I injection in stroke rats and sham‐operated rats, we perfused the brain with PBS and made tissue extracts of the ipsi‐ and contralateral hemispheres. No IGF‐I was detected after the injection of the vehicle, confirming that the ELISA does not recognize rat IGF‐I (data not shown). Human IGF‐I was detected in both hemispheres of rhIGF‐I‐treated rats with ischemic stroke; however, the amount of IGF‐I in the ischemic hemisphere was significantly higher than in the nonischemic hemisphere (Figure 2B). Remarkably, IGF‐I was also clearly detected in the ischemic hemisphere of sham‐operated rats. An explanation for this could be that part of the transport to the brain is due to the disruption of the BBB by the placement of the guide. It should be noted, however, that the induction of stroke significantly increases the amount of IGF‐I in the nonischemic hemisphere, suggesting that ischemic stroke augments IGF‐I transport to the brain via a bona fide transport mechanism. This idea is supported by the absence of BBB disruption in the contralateral side as established by staining with Evan's blue (data not shown).
Both anesthesia by the injection of ketamine and the induction of ischemic stroke may lead to acute hyperglycemia in rats 18, 19, which on its turn can trigger the disruption of the blood–brain barrier 20, 21. Conversely, severe hypoglycemia may also induce BBB damage 22. Therefore, we assessed the blood glucose levels in normal, stroke, and sham‐operated rats that were treated with rhIGF‐I. It appeared that prestroke glucose levels were normal and that rhIGF‐I injection lowered glucose levels, but did not induce severe hypoglycemia (Figure 2C). A reduction in glucose levels by IGF‐I administration has been shown before in diabetic rats with ischemic stroke 23, and this could contribute to neuroprotection in these animals.
Discussion
IGF‐I has been tested as a treatment for focal cerebral ischemia in animal models. It reduced infarct volumes and improved neurological outcome when administered topically on the cerebral cortex or intracerebroventricularly 24. The application of intranasal IGF‐I administration was effective, especially when administered early after the insult 25. Three studies revealed that systemic injection of IGF‐I also leads to neuroprotection 23, 24, 26. The current study proves for the first time that intravenously administered IGF‐I exerts neuroprotective actions via the direct interaction with its receptors in the brain. Furthermore, systemically administered IGF‐I is indeed transported to the brain in our model for ischemic stroke. These observations imply that therapeutic interventions aiming to increase the transport of IGF‐I to the brain may enhance the efficacy of IGF‐I treatment. The neuroprotective effect of IGF‐I is demonstrated by the finding that IGF‐I attenuates the loss of NeuN‐positive cells. Because the implantation of the catheter severely affected the neurological deficit score, the functional outcome has not been assessed. However, it has been demonstrated that even a smaller reduction in infarct size coincides with an amelioration of the neurological deficit score 26.
It has been shown before that IGF‐I can pass the BBB via a genuine saturable transport system 27, probably involving IGF‐I receptors 28 and transcytosis across the capillary endothelium 29. It has also been suggested that IGF‐I transport to the brain is regulated by neuronal activity 30. IGF‐I can also enter the brain through the transport across the choroid plexus, where IGF‐I binds to its membrane receptor, which interacts with the multicargo membrane protein and transporter megaline/LRP2. The perimembrane domain of megaline is instrumental to the internalization of IGF‐I, and interestingly, the C‐terminal region associates with a glycogen synthase kinase‐3 (GSK‐3), which acts as a key regulator of IGF‐I transport. Mutations of the regulatory site of GSK‐3 modulate IGF‐I internalization, and pharmacological inhibition of GSK‐3 at the choroid plexus increases the internalization of IGF‐I 30, 31, 32. It would be interesting to test whether GSK‐3 inhibitors can stimulate the transport of systemically injected IGF‐I across the BBB and increase the treatment efficacy of IGF‐I. The IGF‐I receptor and LRP1, a variant of LRP2, are expressed in brain microvessels 33, 34 and could be involved in this process. Indeed, LRP1 has already been associated with endothelial transcytosis 35. Upregulation of LRP1 expression in endothelial cells in response to hypoxia 36 could play a role in the augmentation of IGF‐I transport into the ischemic hemisphere. Alternatively, the release of proinflammatory mediators triggered by ischemic stroke can also enhance the expression of LRP1 37. Interestingly, insulin transport across the BBB also involves a saturable mechanism, possibly via insulin receptors, and this kind of transport can be modulated through proinflammatory mediators such as nitric oxide 38. Production of these factors in the ischemic hemisphere could explain the enhancement of IGF‐I transport in the nonischemic hemisphere. Alternatively, the inhibition of GSK‐3 or upregulation of IGF‐I receptor levels may also lead to an enhancement of IGF‐I transport. Upregulation of IGF‐I receptor levels in the rat models of ischemic stroke has been shown before 39, but an early assessment of modulation of IGF‐I receptor expression in the BBB has not been performed yet.
In order to evaluate the clinical applicability of IGF‐I treatment, the outcome also has to be determined at later time points. It has been shown in the literature that neuroprotection by IGF‐I can persist for at least a week. A daily subcutaneous injection of 200 ug IGF‐I resulted in a decrease in infarct size and an amelioration of the functional outcome at 7 days after the insult 24. The current study focused on the basic mechanism of IGF‐I on early events of the ischemic cascade occurring within the first 24 h. This is relevant for stroke studies, because neuronal death predominantly occurs within 1 day. In addition, we demonstrated that the window for IGF‐I treatment is not longer than 4 h 26. Hence, the stimulation of IGF‐I transport shortly after the insult could be very effective. It remains to be established, however, whether putative effects of IGF‐I at later time points, targeting later events of the ischemic cascade, also depend on the transport across the BBB.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We thank Ipsen (Ipsen Nv, Merelbeke, Belgium) for supplying rhIGF‐I; Mrs. Peggy Verdood, Laura Punak, and Julie Verdood for their practical help; and Dr. Cathy Jensen for proofreading and correcting the language. Financial support was obtained from the Research Foundation‐Flanders and from the Wetenschappelijk Fonds Willy Gepts of the Universitair Ziekenhuis Brussel. Deborah De Geyter and Wendy Stoop are PhD students of the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen. Ann De Smedt is a PhD Fellow of the Research Foundation‐Flanders.
The first two authors contributed equally to this work.
References
- 1. Kooijman R, Sarre S, Michotte Y, De Keyser J. Insulin‐like growth factor I: a potential neuroprotective compound for the treatment of acute ischemic stroke? Stroke 2009;40:e83–e88. [DOI] [PubMed] [Google Scholar]
- 2. De Smedt A, Brouns R, Uyttenboogaart M, et al. Insulin‐like growth factor I serum levels influence ischemic stroke outcome. Stroke 2011;42:2180–2185. [DOI] [PubMed] [Google Scholar]
- 3. Aberg D, Jood K, Blomstrand C, et al. Serum IGF‐I levels correlate to improvement of functional outcome after ischemic stroke. J Clin Endocrinol Metab 2011;96:E1055–E1064. [DOI] [PubMed] [Google Scholar]
- 4. Tang JH, Ma LL, Yu TX, et al. Insulin‐like growth factor‐1 as a prognostic marker in patients with acute ischemic stroke. PLoS ONE 2014;9:e99186. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. van Rijn MJ, Slooter AJ, Bos MJ, et al. Insulin‐like growth factor I promoter polymorphism, risk of stroke, and survival after stroke: the Rotterdam study. J Neurol Neurosurg Psychiatry 2006;77:24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin‐like growth factor system and its pleiotropic functions in brain. Endocr Rev 2005;26:916–943. [DOI] [PubMed] [Google Scholar]
- 7. Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke 1989;20:1627–1642. [DOI] [PubMed] [Google Scholar]
- 8. Jacob R, Barrett E, Plewe G, Fagin KD, Sherwin RS. Acute effects of insulin‐like growth factor I on glucose and amino acid metabolism in the awake fasted rat. Comparison with insulin. J Clin Invest 1989;83:1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pietrzkowski Z, Wernicke D, Porcu P, Jameson BA, Baserga R. Inhibition of cellular proliferation by peptide analogues of insulin‐like growth factor 1. Cancer Res 1992;52:6447–6451. [PubMed] [Google Scholar]
- 10. Van Hemelrijck A, Vermijlen D, Hachimi‐Idrissi S, Sarre S, Ebinger G, Michotte Y. Effect of resuscitative mild hypothermia on glutamate and dopamine release, apoptosis and ischaemic brain damage in the endothelin‐1 rat model for focal cerebral ischaemia. J Neurochem 2003;87:66–75. [DOI] [PubMed] [Google Scholar]
- 11. Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Elsevier, 2006. [DOI] [PubMed] [Google Scholar]
- 12. Clinckers R, Smolders I, Michotte Y, et al. Impact of efflux transporters and of seizures on the pharmacokinetics of oxcarbazepine metabolite in the rat brain. Br J Pharmacol 2008;155:1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Avendano C, Roda JM, Carceller F, Diez‐Tejedor E. Morphometric study of focal cerebral ischemia in rats: a stereological evaluation. Brain Res 1995;673:83–92. [DOI] [PubMed] [Google Scholar]
- 14. De Geyter D, Stoop W, Zgavc T, et al. Spontaneously hypertensive rats display reduced microglial activation in response to ischemic stroke and lipopolysaccharide. J Neuroinflammation 2012;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 16. Pan W, Kastin AJ. Interactions of IGF‐1 with the blood‐brain barrier in vivo and in situ. Neuroendocrinology 2000;72:171–178. [DOI] [PubMed] [Google Scholar]
- 17. Heck S, Lezoualc'h F, Engert S, Behl C. Insulin‐like growth factor‐1‐mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem 1999;274(14):9828–35. [DOI] [PubMed] [Google Scholar]
- 18. Saha JK, Xia J, Grondin JM, Engle SK, Jakubowski JA. Acute hyperglycemia induced by ketamine/xylazine anesthesia in rats: mechanisms and implications for preclinical models. Exp Biol Med 2005;230:777–784. [DOI] [PubMed] [Google Scholar]
- 19. Wang YY, Chen CJ, Lin SY, Chuang YH, Sheu WH, Tung KC. Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Mol Cell Endocrinol 2013;367:50–56. [DOI] [PubMed] [Google Scholar]
- 20. Dietrich WD, Alonso O, Busto R. Moderate hyperglycemia worsens acute blood‐brain barrier injury after forebrain ischemia in rats. Stroke 1993;24:111–116. [DOI] [PubMed] [Google Scholar]
- 21. Huang J, Liu B, Yang C, Chen H, Eunice D, Yuan Z. Acute hyperglycemia worsens ischemic stroke‐induced brain damage via high mobility group box‐1 in rats. Brain Res 2013;1535:148–155. [DOI] [PubMed] [Google Scholar]
- 22. Oztaş B, Küçük M, Sandalci U. Effect of insulin‐induced hypoglycemia on blood‐brain barrier permeability. Exp Neurol 1985;87:129–136. [DOI] [PubMed] [Google Scholar]
- 23. Rizk NN, Myatt‐Jones J, Rafols J, Dunbar JC. Insulin like growth factor‐1 (IGF‐1) decreases ischemia‐reperfusion induced apoptosis and necrosis in diabetic rats. Endocrine 2007;31:66–71. [DOI] [PubMed] [Google Scholar]
- 24. Schabitz WR, Hoffmann TT, Heiland S, et al. Delayed neuroprotective effect of insulin‐like growth factor‐i after experimental transient focal cerebral ischemia monitored with mri. Stroke 2001;32:1226–1233. [DOI] [PubMed] [Google Scholar]
- 25. Liu XF, Fawcett JR, Hanson LR, Frey WH 2nd. The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin‐like growth factor‐I in rats. J Stroke Cerebrovasc Dis 2004;13:16–23. [DOI] [PubMed] [Google Scholar]
- 26. De Geyter D, Stoop W, Sarre S, De Keyser J, Kooijman R. Neuroprotective efficacy of subcutaneous insulin‐like growth factor‐I administration in normotensive and hypertensive rats with an ischemic stroke. Neuroscience 2013;250:253–262. [DOI] [PubMed] [Google Scholar]
- 27. Pan W, Kastin AJ. Penetration of neurotrophins and cytokines across the blood‐brain/blood‐spinal cord barrier. Adv Drug Deliv Rev 1999;36:291–298. [DOI] [PubMed] [Google Scholar]
- 28. Carro E, Torres‐Aleman I. Serum insulin‐like growth factor I in brain function. Keio J Med 2006;55:59–63. [DOI] [PubMed] [Google Scholar]
- 29. Reinhardt RR, Bondy CA. Insulin‐like growth factors cross the blood‐brain barrier. Endocrinology 1994;135:1753–1761. [DOI] [PubMed] [Google Scholar]
- 30. Nishijima T, Piriz J, Duflot S, et al. Neuronal activity drives localized blood‐brain‐barrier transport of serum insulin‐like growth factor‐I into the CNS. Neuron 2010;67:834–846. [DOI] [PubMed] [Google Scholar]
- 31. McKinnie SM, Rodriguez‐Lopez EM, Vederas JC, et al. Differential response of orthologous L, L‐diaminopimelate aminotransferases (DapL) to enzyme inhibitory antibiotic lead compounds. Bioorg Med Chem 2014;22:523–530. [DOI] [PubMed] [Google Scholar]
- 32. Bolos M, Fernandez S, Torres‐Aleman I. Oral administration of a GSK3 inhibitor increases brain insulin‐like growth factor I levels. J Biol Chem 2010;285:17693–17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frank HJ, Pardridge WM, Morris WL, Rosenfeld RG, Choi TB. Binding and internalization of insulin and insulin‐like growth factors by isolated brain microvessels. Diabetes 1986;35:654–661. [DOI] [PubMed] [Google Scholar]
- 34. Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer's amyloid‐ss(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 2000;106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tian X, Nyberg S, Sharp P, et al. LRP‐1‐mediated intracellular antibody delivery to the central nervous system. Sci Rep 2015;5:11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xi Li XP. LRP1 is a novel receptor of Bmper and required for pathological angiogenesis. Circulation 2014;130:A15833. [Google Scholar]
- 37. Kovac A, Erickson MA, Banks WA. Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP‐1 expression in response to lipopolysaccharide. J Neuroinflammation 2011;8:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Banks WA, Dohgu S, Lynch JL, et al. Nitric oxide isoenzymes regulate lipopolysaccharide‐enhanced insulin transport across the blood‐brain barrier. Endocrinology 2008;149:1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J, Li Y, Chen J, Yang M, et al. Expression of insulin‐like growth factor 1 and receptor in ischemic rats treated with human marrow stromal cells. Brain Res 2004;1030:19–27. [DOI] [PubMed] [Google Scholar]