

Summary
Background and Purpose
Sprawling (Swl) is a radiation‐induced mutation which has been identified to have a nine base pair deletion in dynein heavy chain 1 (DYNC1H1: encoded by a single gene Dync1h1). This study is to investigate the phenotype and the underlying mechanism of the Dync1h1 mutant.
Methods and Results
To display the phenotype of Swl mutant mice, we examined the embryos of homozygous (Swl/Swl) and heterozygous (Swl/+) mice and their postnatal dorsal root ganglion (DRG) of surviving Swl/+ mice. The Swl/+ mice could survive for a normal life span, while Swl/Swl could only survive till embryonic (E) 8.5 days. Excessive apoptosis of Swl/+ DRG neurons was revealed during E11.5–E15.5 days, and the peak rate was at E13.5 days. In vitro study of mutated DRG neurons showed impaired retrograde transport of dynein‐driven nerve growth factor (NGF). Mitochondria, another dynein‐driven cargo, demonstrated much slower retrograde transport velocity in Swl/+ neurons than in wild‐type (WT) neurons. Nevertheless, the Swl, Loa, and Cra mutations did not affect homodimerization of DYNC1H1.
Conclusion
The Swl/Swl mutation of Dync1h1 gene led to embryonic mal‐development and lethality, whereas the Swl/+ DRG neurons demonstrated deficient retrograde transport in dynein‐driven cargos and excessive apoptosis during mid‐ to late‐developmental stages. The underlying mechanism of the mutation may not be due to impaired homodimerization of DYNC1H1.
Keywords: Dynein heavy chain, Mouse mutant, Neuron loss, Retrograde transport, Sprawling mutation
Introduction
Axonal transport plays important roles on neurons survival, function, and morphogenesis. The long‐distance axonal transport in neurons mainly involves microtubules and their associated molecular motors, cytoplasmic dynein and kinesins, two major motor molecules 1, 2. Cytoplasmic dynein, one of the microtubule‐associated proteins transporting cargoes to the minus end of microtubule, predominantly contributes to the retrograde axonal transport of organelles, proteins, and vesicles in neurons 3, 4. Cytoplasmic dynein is one of the key molecules transporting membrane‐bound organelles back to the cell body. It consists of two homodimerized heavy chains, and several intermediate chains, light intermediate chains, and light chains, which assemble into different dynein subcomplexes 3, 5.
Dysfunction of dynein would cause severe disruptions in long‐distance retrograde axonal transport, and decrease the survival of long‐axon neurons, including sensory and motor neurons. Hafezparast et al. reported that two independent mouse strains generated by N‐ethyl‐N‐nitrosourea (ENU)‐induced mutagenesis, Legs at odd angles (Loa) and Cramping 1 (Cra1, further referred to as Cra), harbored dominantly inherited missense point mutations in the cargo‐binding domain of DYNC1H1. Heterozygous mice from both lines showed an age‐related, progressive motor neuron disease, with cramping of hind limbs, gait abnormalities, and paralysis in late stage of life 6. A nine base pair deletion in the cargo‐binding domain of DYNC1H1, close to the Cra mutation, was identified in mice heterozygous as the radiation‐induced mutation Sprawling (Swl) 7. These Swl/+ mice displayed an early‐onset proprioceptive sensory neuronopathy characterized by predominantly lumbar proprioceptive DRG neuron loss along with muscle spindle degeneration, while locomotion and motor neuron numbers appeared normal in these animals 7. Swl/+ mice have a moderate sensory neuropathy that does not affect the nociceptive modality or motor function 7.
Neuronal system defects caused by cytoplasmic DYNC1H1 mutations have been found in developmental and neurodegenerative diseases in mice as well as in humans. In 2010, the first human DYNC1H1 mutation (DYNC1H1 H3822P) was found in an individual with developmental delay, hypotonia, and brain malformations 8. Michael et al. reported a mutation in a large dominant pedigree, c.917A>G in DYNC1H1, which led to a missense substitute, p.His306Arg at highly conserved residue within the homodimerization domain, leads to an autosomal dominant Charcot–Marie–Tooth disease 9. Many other DYNC1H1 mutations, inducing quite different clinical phenotypes, have been recently reported 10, 11, 12. These studies highly strengthened the importance of dynein and retrograde axonal transport in neuronal function of humans.
Although many mutations related to DYNC1H1 have been revealed, the underlying mechanisms for the different phenotypes of each mutation still remain largely unknown. In this study, we tried to investigate the DYNC1H1 mutation especially in Swl/Swl embryos and Swl/+ mice and also explore the possible mechanisms underlying neuronal degeneration related to dynein in vitro. We found an excessive apoptosis event of Swl/+ DRG neurons during E11.5–E15.5 days, and the peak rate was at E13.5 days. Furthermore, the retrograde transport of dynein‐driven NGF and mitochondria was affected in Swl/+ DRG neurons. Nevertheless, in vitro study showed evidence which Swl, Loa and Cra mutations did not affect homodimerization of dynein heavy chains. This study offers new insights into the underlying mechanism for the specific phenotype of Swl mutation and also points out a new direction for further studies.
Materials and Methods
Mice
Swl/+ mutant mice were obtained as described previously 7. The mice were genotyped by polymerase chain reaction (PCR) analysis of tail biopsies. All mice were singly housed in Plexiglas cages in a controlled environment at a constant temperature of 24°C and humidity of 50 ± 10% with food and water available ad libitum. For developmental studies, the day on which a plug was detected in the morning was considered as embryonic (E) 0.5 day. All experiments were carried out in accordance with the guidelines laid down by the National Institutes of Health regarding the care and use of animals for experimental procedures. Additionally, all experimental procedures which involve in animals and animal cares were approved by Medical Experimental Animal Administrative Committee of Fudan University.
Tissue Processing
The mouse embryos at E11.5, E13.5, E15.5, E17.5 days and the postnatal (P) 5‐day‐old mice were collected for tissue processing. The embryos of wild‐type (WT) and mutant mice were dissected out from the uteri and immersed in the fixation solution (4% paraformaldehyde in 0.1 M Millonig's buffer, pH 7.4) for 2 h at room temperature. Under the light microscopy, without staining, the embryos were displayed in grayscale pattern and were examined for the gross anatomy, including three germ layers (endoderm, mesoderm, ectoderm layer) and two sac (amniotic sac, yolk sac) structures, and potential developmental defects for the comparison of Swl/+, Swl/Swl, and WT mouse embryos. Meanwhile, the postnatal dorsal root ganglia (DRGs) were embedded in paraffin and sectioned into 7‐μm serial paraffin sections.
Cell Culture and Transfection
Dorsal root ganglion neurons were isolated and cultured as described in the previous study 13 from newborn mice (within 24 h after birth). Generally, freshly prepared cells were plated onto Petri dishes (Falcon) for 1 h at 37°C in 5% CO2 to allow attachment of nonneuronal cells. The suspended DRG neurons were then seeded onto sterile glass‐bottomed culture dishes (WillCo Wells BV, Amsterdam, the Netherlands) coated with 10 μg/mL poly‐L‐Lysin and 12 μg/mL laminin (Sigma, St. Louis, MO, USA). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (Gibco, Waltham, MA, USA) and 2 mM l‐glutamine (Sigma). After 2‐h incubation, we changed the medium to D/F‐12 medium (Gibco) with 2 mM l‐glutamine, 2% B27 (Gibco), and 50 ng/mL NGF (Invitrogen, Waltham, MA, USA). Neurons were used for the following experiments within 3 days of culture. Explant culture of DRG was established from DRGs obtained from the E12.5 WT and Swl/+ mouse. Briefly, DRGs were dissected and digested with 0.25% trypsin/EDTA (Invitrogen) for 5 min. The ganglia were placed into 24‐well plates coated with collagen (4 mg/mL) and then maintained in neuron culture medium for observation. HEK 293T cells were maintained in DMEM with 10% fetal bovine serum (Gibco) and transfected using Lipofectamine 2000 (Invitrogen). All cells were maintained in a humidified atmosphere containing 5% CO2.
TUNEL and Immunohistochemistry
For apoptosis analysis, every fourth section in the series from the cervical (C) 4 or lumbar (L) 4 was selected. Apoptosis was quantified using a commercially available fluorescent terminal deoxynucleotidyl transferase nick‐end labeling (TUNEL) kit, in accordance with the manufacturer's protocol (ApopTag kit, Chemicon, Billerica, MA, USA, S7100). Meanwhile, the sections were stained by DAPI for cell nuclear detection. Slides were visualized at a magnification of ×20 under Axiovert‐25 microscope with digital imaging from Carl Zeiss MicroImaging (Gottingen, Germany). The number of TUNEL positive cells (green) was counted in 15 separate fields for each slide of three independent experiments.
Campenot Chamber Experiment
The compartmentalized Campenot chambers were prepared as described previously 14. DRG neurons were plated into the inner cell body compartment (CB) of the chambers, and axons were allowed to grow into the outer distal axon compartment (DA) (Figure 3B). DRGs from three Swl/+ mice and three WT littermates at E13.5 (three independent experiments) were dissected and plated at 1 × 105 cells per culture in the inner chamber. To eliminate nonneuronal cells, 0.3 μM Ara‐C was added for 24 h. DRG cells were cultured in DMEM/10% FBS/1×P/S/100 ng/mL NGF for 15 days, allowing the neurites to sufficiently cross the barrier. Then, NGF was withdrawn from the cells in different compartments for 2 days. At DIV18, cells were fixed and stained with phosphorylated neurofilament antibody (SMI‐31, Covance, Princeton, NJ, USA) to identify neurons and axons and Hoechst 33258 (Sigma) to identify nuclei.
Construction of Plasmids
Total RNA was extracted from brains of WT and Swl/+ mutant mice using the TRIZOL reagent (Invitrogen). Dync1h1 cDNA was reverse‐transcribed by a specific primer (GAATTCTGTCATGTTTGA) using First Stand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA). DYNC1H1 cDNA encoding amino acids 230‐1110 was amplified by PCR with the forward primer: 5′‐GATCGCTAGCGGTGATAAGGTTGAAGAC and the reverse primer: 5′‐AATTGCGGCCGCTTAGACCTTGCCGTAATCTAT. The cDNA fragment was then inserted into the mammalian expression vector pcDNA3 (Invitrogen). To express flag‐tagged dynein heavy chain protein fragments, we subcloned the cDNA fragments into pUHD30F, a vector containing a flag tag 15, further replaced the flag tag with GFP. Finally, eight plasmids were constructed (Table 1): pUHD30F‐flag‐WT, pUHD30F‐flag‐Swl, pUHD30F‐flag‐Cra, pUHD30F‐flag‐Loa, pUHD30F‐GFP‐WT, pUHD30F‐GFP‐Swl, pUHD30F‐GFP‐Cra, and pUHD30F‐GFP‐Loa. Firefly luciferase cDNA was amplified by PCR from pGL3‐Basic vector (Promega, Madison, WI, USA) and inserted into pCMV to express FLAG‐Luc as a blank control. The confirmatory DNA sequencing was performed by ABI310 genetic analyzer (Applied Biosystems, Foster City, CA, USA). All plasmids were amplified using mini preparation kit (Qiagen, Hilden, Germany) for cell transfection.
Table 1.
The transfection combination of GFP‐tagged and flag‐tagged plasmids with their abbreviations in the article
| Transfection combination of plasmid | Abbreviations in text |
|---|---|
| pUHD30F‐flag‐wild type + pUHD30F‐GFP‐wild type | F‐WT/G‐WT |
| pUHD30F‐flag‐Swl + pUHD30F‐GFP‐wild type | F‐Swl/G‐WT |
| pUHD30F‐flag‐Cra + pUHD30F‐GFP‐wild type | F‐Cra/G‐WT |
| pUHD30F‐flag‐Loa + pUHD30F‐GFP‐wild type | F‐Loa/G‐WT |
| pUHD30F‐flag‐Swl + pUHD30F‐GFP‐Cra | F‐Swl/G‐Cra |
|
pUHD30F‐flag‐Swl + pUHD30F‐GFP‐Loa pUHD30F‐flag‐Swl + pUHD30F‐GFP‐Swl |
F‐Swl/G‐Loa F‐Swl/G‐Swl |
| Luci‐flag + pUHD30F‐GFP‐wild type | F‐Luci/G‐WT |
Co‐immunoprecipitation and Immunoblotting
After transfection for 48 h, HEK293T cells were harvested using lysis buffer. Co‐immunoprecipitation (co‐IP) using anti‐flag M2 affinity resin (Sigma) was performed as described previously 16. Briefly, after centrifugation of the lysate, the supernatant was incubated with anti‐flag beads for 2 h. The beads were washed extensively six times with wash buffer, and the proteins bound to beads were then eluted with 0.8 mg of flag peptide/mL in a total volume of 30 μL.
Protein samples were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). After protein transfer, the membrane was incubated in a blocking buffer (5% nonfat milk in TBST) for 30 min and then with rabbit anti‐GFP antibody (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C, then processed using an HRP‐conjugated anti‐rabbit antibody (Millipore, Billerica, MA, USA) followed by ECL detection (Thermo Scientific, Waltham, MA, USA). The same membrane was stripped by stripping buffer for 30 min in a 55°C water bath. After washing, the membrane was incubated with rabbit anti‐flag antibody (Sigma) overnight and then incubated with HRP‐antibody as the procedure of anti‐GFP labeling.
Time‐lapse Microscopy
Time‐lapse microscopy was performed as described previously 13. To label mitochondria, neurons were treated with 100 nM Mitotracker Red (Molecular Probes) which was diluted in growth medium without serum. Neurons were incubated at 37°C for 1 min and then recover for 2 h in fresh media before time‐lapse microscopy recording. DRG cells from E12.5 WT mice and Swl/+ mice were scanned at 37°C in an atmosphere of 5% CO2, using Leica AS MDW system with a motorized stage and a HCX PL APO 63/1.30 GLYC CORR 37°C objective. Images were captured with a CCD camera (CoolSNAP HQ, Roper Scientific, Tucson, AZ, USA). Vesicle movement was recorded every 2 seconds. Images were then integrated for the continuous description or graph of vesicle movement or transport in the axons. The final kymographs were generated from time‐lapse images using ImageJ software (NIH, Bethesda, MD, USA).
Statistics
All quantified data were analyzed by one‐way ANOVA with a Tukey's posttest. Error bars represent standard deviation, and P < 0.05 was considered statistically significant.
Results
Homozygous Swl/Swl Mutation Leads to Embryonic Lethality and Maldevelopment of Mouse Embryos
In the procedure of mouse breeding, we noticed that all homozygous (Swl/Swl or −/−) embryos demised at E7.5–E8.5 days, whereas the heterozygous (Swl/+ or −/+) embryos could survive through the pregnancy and the postnatal 1‐week‐old mice were evidently distinguishable from the WT mice due to the unsteady gait and wobbly locomotion (Figure 1A), which is consistent with our previous study 7. To detect any morphological changes in mutated embryos, we took out the embryos at E7.5 days. The Swl/Swl mice had fetal defects in the intrauterous development, based on their size, the mobility, and the morphological abnormality. The size of the Swl/Swl embryo body was much smaller than Swl/+ or WT embryo (Figure 1B–D). The morphological defects of Swl/Swl embryo were displayed as lack of amniotic sac, smaller size and unstructured anatomy of somatic body (Figure 1D). These changes indicate that Swl mutation influences the development of mouse embryos, and dynein protein is essential for the survival of mouse embryos.
Figure 1.
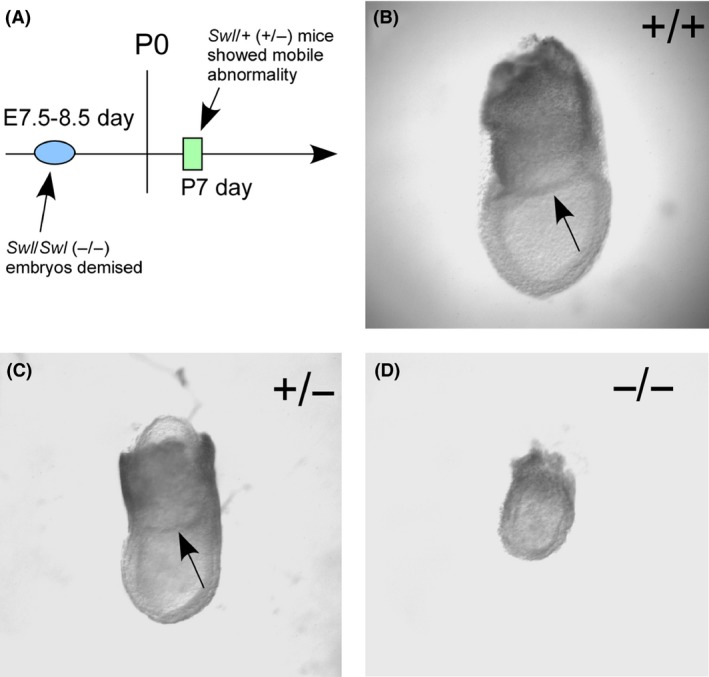
Morphological analysis of WT, Swl/+, Swl/Swl mouse embryos. To investigate the influence of the Swl mutation on the development of mouse embryos, we compared mutated and WT mouse embryos at E7.5 days. (A) The time line of observed defects occurred in homozygous (−/−) and heterozygous (+/−) mutated mice. (B–D) The morphology of WT (B), heterozygous (C), and homozygous (D) embryos showed that the size of homozygous(−/−) embryos was much smaller than heterozygous(+/−) or WT embryos, and the amniotic sac (pointed out by arrowhead) was absent in homozygous (−/−) embryos.
DRG Neurons of Swl/+ Mice Exhibit Excessive Apoptosis During Mid‐ to Late‐ Developmental Stages
Based on a previous study 7, the number of Swl/+ DRGs neurons was already decreased at E15.5 days. At birth (P0), the number of Swl/+ DRGs neurons was decreased by 70%. To determine when the changes of neuron number in Swl/+ mouse embryos occurred, we analyzed the DRG neuronal apoptosis at different time points (E11.5, E13.5, E15.5, E17.5, and P0.5) by TUNEL assay. The apoptosis rate of WT DRG neurons was 7.89 ± 2.18% at E11.5 days, 7.67 ± 2.56% at E13.5 days, 5.01 ± 0.62% at E15.5 days, 0.51 ± 0.11% at E17.5 days, and 0.15 ± 0.18% at P0.5 days (Figure 2A). The apoptosis rate of Swl/+ DRG neurons at those days was 6.73 ± 1.08% at E11.5 days, 18.91 ± 1.17% at E13.5 days, 6.51 ± 1.02% at E15.5 days, 2.48 ± 0.41% at E17.5 days, and 2.21 ± 0.50% at P0.5 days (Figure 2A). The statistical analysis between two groups (Swl/+ and WT) showed that the apoptosis rate of Swl/+ was significantly higher than that of WT at the same embryonic age, noted by an apparent apoptotic peak (18.91 ± 1.17%) at E13.5 in Swl/+(As shown in Figure 2B). This finding demonstrated that an excessive apoptosis event occurred between E11.5 and E15.5 selective to DRG sensory neurons.
Figure 2.
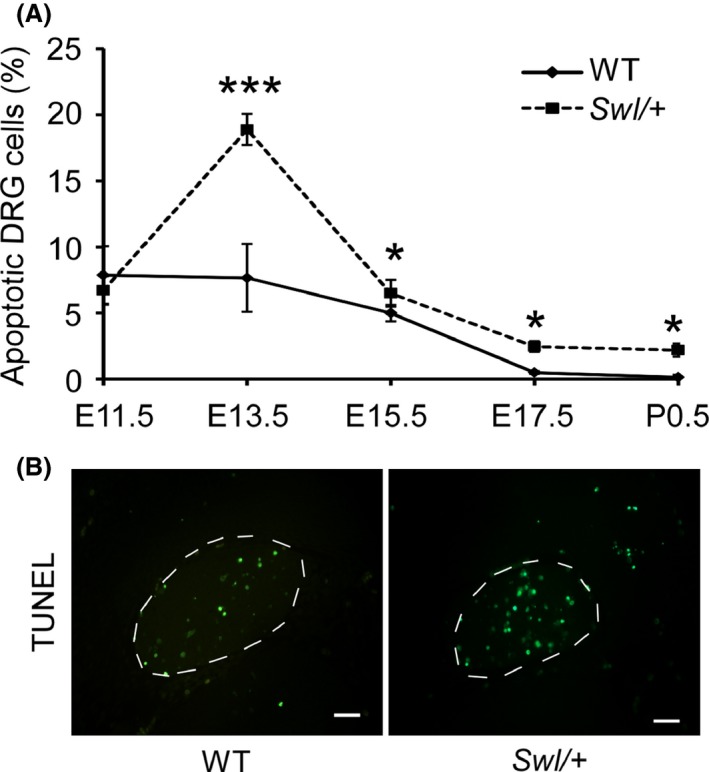
Dorsal root ganglion (DRG) neurons of Swl/+ mice exhibit selective apoptosis at different developmental stages. To explore the changes of neurons in mutated mouse embryos, we analyzed the DRG neuronal apoptosis at different time points (E11.5, E13.5, E15.5, E17.5, and P0.5) by TUNEL assay. (A) Apoptotic cell numbers in Swl/+ mice were significantly increased at E11.5 to P0.5 days, compared to that of WT, whereas the peak apoptotic rate appeared at E13.5 days. (B) Representative figures of TUNEL staining in Swl/+ mice and WT mice. At least eight sections from each time points were stained and analyzed. Scale bar 100 μm. Data represent mean ± SD *P < 0.05; ***P < 0.001.
Impairment of NGF Retrograde Transport Increase Neuron Apoptosis in Developmental Swl/+ DRG Neurons
To explore the underlying mechanism for the selective apoptosis of sensory neurons in Swl/+ mice, we did primary neuron culture and explant culture of DRGs from WT and Swl/+ mice. DRG neurons from E12.5 Swl/+ mice showed significant decrease in cell number and neurite outgrowth (Figure 3A). To explore whether Swl mutation influences the retrograde axonal transport of dynein‐driven NGF, we cultured E12.5 DRG neurons from WT and Swl/+ mice in a Campenot chambers system with or without NGF deprivation in either cell body (CB) or distal axon (DA) compartment (Figure 3B). The condition of neuronal growth was assessed by the apoptotic cell number and the visible axonal growth in Campenot chambers. With complete NGF deprivation for both the neuronal body and distal axon (None), the WT and Swl/+ neurons showed high apoptosis rates, 10.4±2.83% and 11.7±2.7%, respectively, which did not differ significantly. When NGF was withdrawn from the distal axons medium but not from the neuronal body medium (CB), the neuronal apoptosis rate was much lower and similar both in WT and Swl/+ neurons, 1.2±0.3% and 1.6±0.58%, respectively. When NGF was withdrawn from the neuronal body medium while the distal axon medium still contained NGF (DA), the neuronal apoptosis rate in the WT group was low, 1.0±0.5% compared to a significantly higher apoptosis rate in the Swl/+ group 6.1±1.64% (P < 0.05) (Figure 3C). The survival of DRG neurons needs endocytosed NGF retrogradely transported from nerve terminals to cell bodies. Thus, this chamber culture data strongly supported the notion that disrupted retrograde axonal transport of some critical endosomes such as NGF probably leads to DRG neuron apoptosis in Swl/+ mice.
Figure 3.
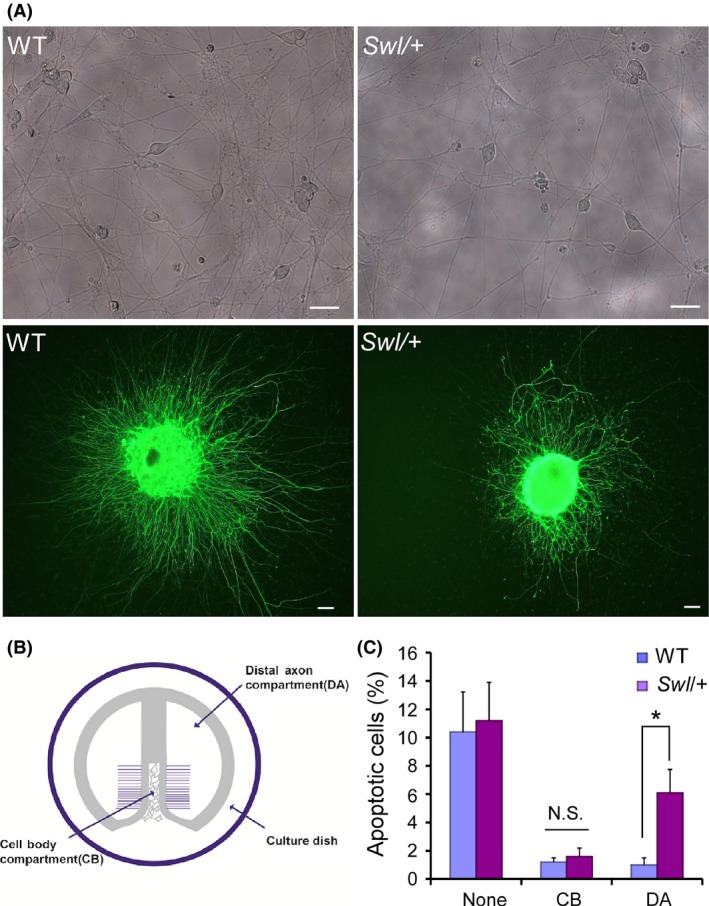
Impaired retrograde transport of nerve growth factor (NGF) results in excessive apoptosis of Swl/+ DRG neurons. To explore the underlying mechanism for the selective apoptosis of sensory neurons in Swl/+ mice, we cultured dorsal root ganglion (DRG) neurons from WT and Swl/+ mice in Campenot chambers system with or without NGF deprivation in two compartments. (A) Both primary neuron culture and explant culture of E12.5 DRG neurons showed significant decrease in cell number and neurite outgrowth in Swl/+ mice. (B) The schematic diagram of Campenot chambers system. In the laminin‐coated 35‐mm tissue culture dish, a specialized Teflon divider separates the compartment into two parts, the distal axonal (DA) compartment and the cell body (CB) compartment; the growing axons of neurons penetrate the high melting point gel underneath the divider into the DA compartments. (C) DRG neuron apoptosis in different groups with or without NGF deprivation was analyzed by TUNEL and quantified statistically. Data represent mean ± SD from three independent experiments. *P < 0.05; N.S. not significant.
Retrograde Transport of Dynein‐Driven Membrane‐Bound Mitochondria was Impaired in Developmental Swl/+ DRG Neurons
Dynein retrogradely transports membrane‐bound organelles including mitochondria. We quantified the velocity of mitochondria transport to evaluate whether the Dync1h1 Swl/+ mutation affects the retrograde transport functionality of membrane‐bound cargo. Using time‐lapse microscopy, we examined the transport velocity of mitochondria by MitoTracker Red labeling in the E12.5 Swl/+ and WT DRG axons. When neurons were monitored for 224‐second at 2‐second intervals, 12 motile vesicles in average were observed per 100 μm axon. Fifteen neurons/axons from each group were observed and analyzed. The average velocity of mitochondria anterograde transport was 0.452 μm/min in Swl/+ axons and 0.418 μm/min in WT axons, respectively (P > 0.05). Nevertheless, the velocity of retrograde transport of Swl/+ axons was 0.357 μm/min, significantly slower than 0.604 μm/min in WT axons (P < 0.05) (Figure 4B). These data indicated a significant deficit of retrograde transport functionality of dynein‐driven mitochondria in Swl/+ axons.
Figure 4.
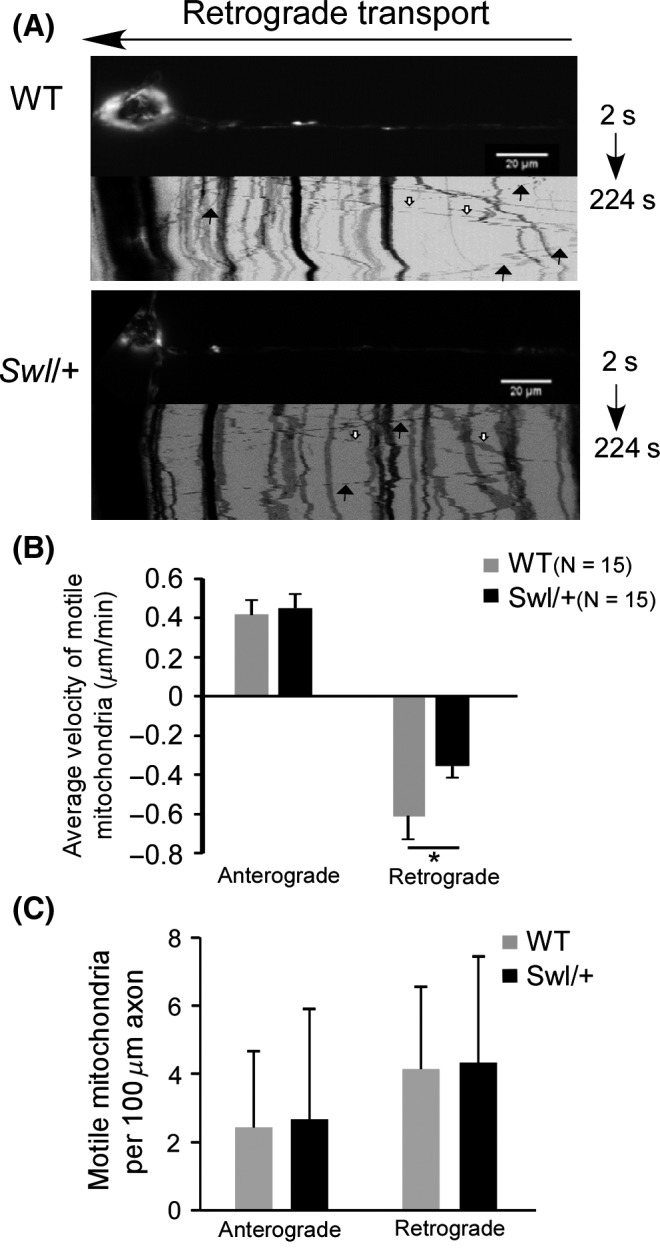
Retrograde axonal transport of mitochondria was significantly impaired in Swl/+ DRG neurons. To quantify the axonal transport in Swl/+ DRG neurons, we employed time‐lapse microscopy to examine the transport rate of mitochondria labeled by Mitotracker Red in the axons of Swl/+ and WT DRG neurons. (A) Kymographs of retrograde transport of motile mitochondria in dorsal root ganglion (DRG) neurons from E12.5 Swl/+ and WT mice. Solid or hollow arrows indicate typical retrograde or anterograde motile vesicles, respectively. (B) The mean velocity of anterograde and retrograde transport of mitochondria in DRG neurons was analyzed statistically. (C) The mean motile mitochondria counts analyzed in each directions. For the figures shown in B and C, 15 neurons from each group were observed and analyzed. Data represent mean ± SD from three independent experiments. *P < 0.05.
Furthermore, the initial mitochondria movement, indicated by the mean motile mitochondria counts, was analyzed in both directions. The mean anterograde motile mitochondrial counts were 2.43 in WT axons and 2.67 in Swl/+ axons (P > 0.05). The mean retrograde motile mitochondria counts were 4.14 in WT axons and 4.33 in Swl/+ axons (P > 0.05). This result implicated that Swl/+ mutation did not affect the initial procedure of retrograde mitochondria movement (Figure 4C).
Swl, Loa and Cra Mutations in DYNC1H1 Do not Influence Heavy Chain Dimerization
Our data of embryonic development, neural cell apoptosis, and retrograde transport velocity, could lead to the notion that the Dync1h1 Swl/+ mutation causes dysfunction of the dynein protein. As the Swl, Loa, and Cra mutations lie within the binding domain of DYNC1H1 and the completion of dynein functionality needs homodimerization of two heavy chains of DYNC1H1, we then asked whether these Dync1h1 mutations influenced the homodimerization of dynein heavy chains in Swl/+ mice. Using co‐IP and immunoblotting, we evaluated the dimerization of three mutated dynein heavy chains fragments (Swl, Loa, and Cra synthetic peptides by mutagenesis and cloning in vitro, Figure 5A). First, we detected the dimerization between mutant fragments and WT control (Figure 5B). Our results did not show a significant difference in the ratios of the GFP‐labeled and flag‐labeled protein between each group (Figure 5C), indicating that the labeled two chains had similar levels of pull‐down and that these mutations did not influence the dimerization between mutants fragments and WT controls. The Swl mutant fragment was then evaluated specifically for their binding ability to Loa or Cra mutant fragments (Figure 6A), which also did not show any significant difference (Figure 6B). The co‐IP findings demonstrated that the Swl mutated DYNC1H1 fragment has the same binding ability to the other mutated DYNC1H1 fragments, in other word, that the dimerization of dynein heavy chain was not impaired by the mutations of DYNC1H1.
Figure 5.
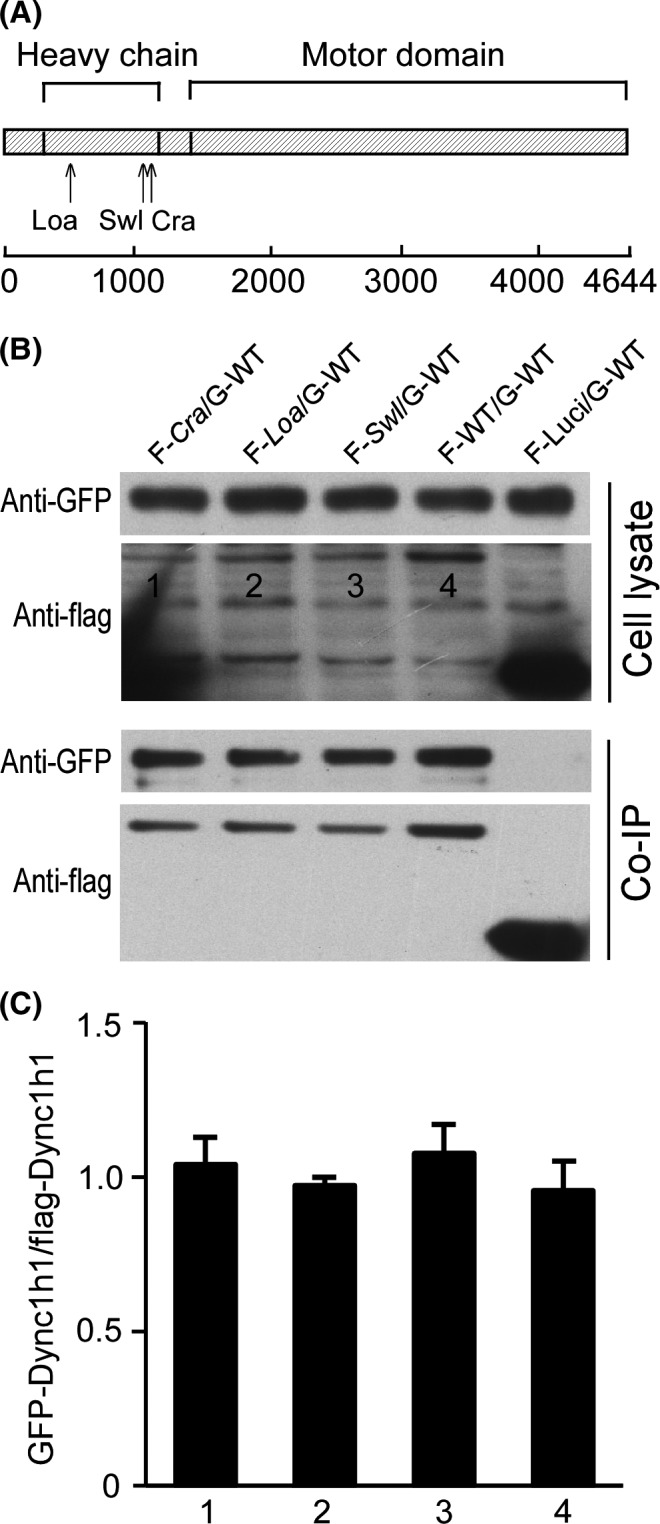
The Swl, Loa, and Cra mutations do not influence the dimerization of DYNC1H1 fragments with WT DYNC1H1. To explore the impact of the DYNC1H1 mutations on the structure of the dynein complex, we measured the dimerization between mutated DYNC1H1 fragments and WT. Different combinations of plasmids with different mutated DYNC1H1 fragments were co‐transfected into HEK 293T cells using co‐IP and immunoblots for further analysis. (A) Domain diagram of the DYNC1H1 protein showing the position of the known mouse mutants (Loa, Swl, Cra). (B) Immunoblot images of GFP‐labeled and flag‐labeled proteins (GFP‐WT set) in cell lysate and co‐IP liquid. (C) Quantification of the band density ratio. FLAG‐Luci as a blank control. Numbers “1, 2, 3, 4” represent “F‐Cra/G‐WT, F‐Loa/G‐WT, F‐Swl/G‐WT, F‐WT/G‐WT,” respectively. Data represent mean ± SD from three independent experiments.
Figure 6.
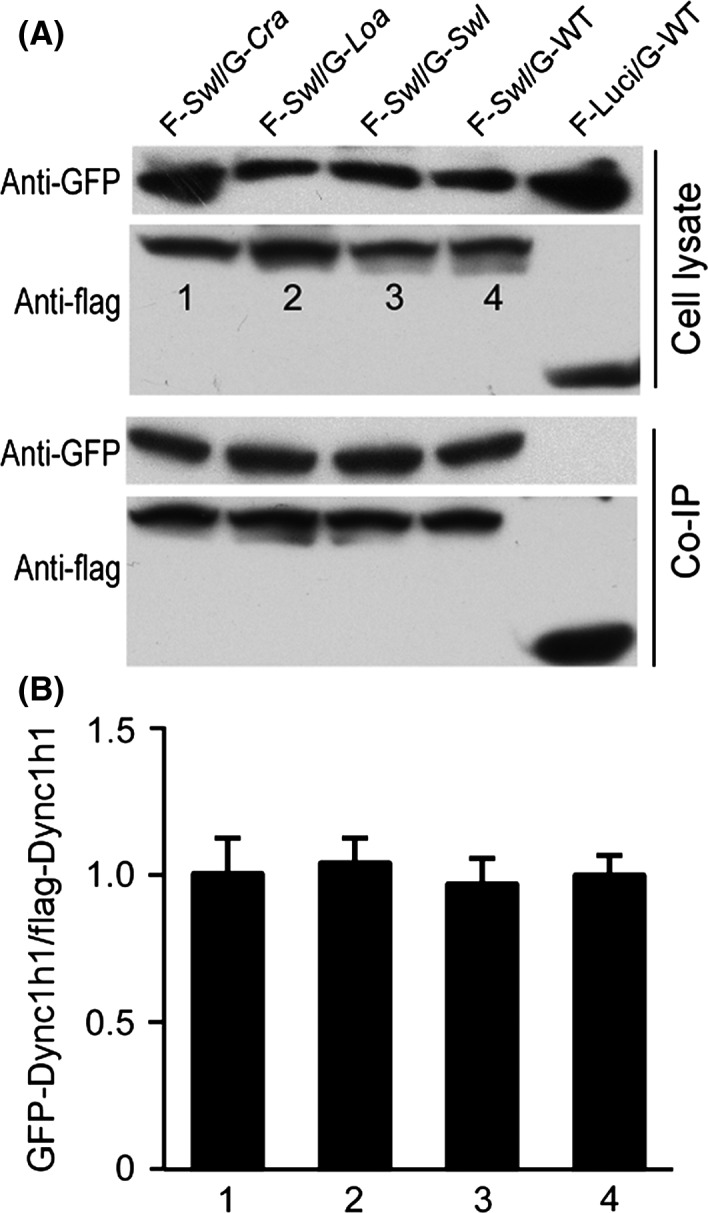
Dimerization between the Swl DYNC1H1 fragment and other mutated DYNC1H1 fragments was not influenced by the mutations. The dimerization between Swl DYNC1H1 and other mutated DYNC1H1 fragments was analyzed using above‐mentioned co‐IP assay. (A) Immunoblot images of (flag‐Swl set) in cell lysate and co‐IP liquid. (B) Quantification of the bands density ratio. FLAG‐Luci as a blank control. Numbers “1, 2, 3, 4” represent “F‐Swl/G‐Cra, F‐Swl/G‐Loa, F‐Swl/G‐Swl, F‐Swl/G‐WT,” respectively. Data represent mean ± SD from three independent experiments.
Discussion
In this study, we further characterized the phenotypes of the Swl mutation in DYNC1H1 on morphology of embryos and DRG neurons, the sensory neuron survival, retrograde transport functionality of dynein‐driven membrane‐bound vesicles in neuronal axons. The Sprawling (Swl) mouse, firstly described in 1967, had been generated from an X‐irradiation experiment 17. However, the causative mutation was not identified until 2007, Chen et al. 7 determined that the defective sensory neuronal phenotype arose from a nine base pair in‐frame deletion in exon 12 of Dync1h1 that changed four amino acids (GIVT) to a single alanine (A) residue in a cargo‐binding domain. Heterozygous mice for Swl mutation have been reported displaying an early‐onset sensory neuronopathy with muscle spindle deficiency 18, 19.
The dynein complex has many critical housekeeping functions in all cells, including orientation of the mitotic spindle, nuclear positioning, Golgi maintenance, and endosomal dynamics 20, 21. In the nervous system, the dynein complex takes on additional roles specific to neurons. Dynein is involved in multiple neuron‐specific processes, such as neuronal migration 22, synapse formation 23, growth and development of neurites 24, as well as axonal transport of microtubules, neurofilaments 25, and organelles 26. Homozygosity for null mutations of the gene causes early embryonic lethality in both Drosophila and mice 27, 28. Among three Dync1h1 mutations, homozygous Loa and Cra mice die by one day after birth, whereas Swl homozygotes die at the late implantation to early gastrulation stage 6, 7. In this study, we found that homozygous Swl/Swl embryos aborted at or around E7.5 days, in agreement with previous finding and again indicative of the importance of dynein in embryonic development.
Chen et al. 7 observed a significant reduction in the number of Swl/+ lumbar DRG proprioceptive sensory neurons compared to that of WT mice at P0.5. In this study, we further detected an excessive DRG neuronal apoptosis occurring between E11.5 and E15.5 days, with a peak at E13.5 days. Lines of evidence have shown that larger motor and proprioceptive sensory neurons with longer axons are more vulnerable to the diseased conditions than small‐sized neurons 29. Proprioceptive sensory neurons possess a high energy demand and require significant trafficking capacity and hence may be particularly more sensitive to vesicular and axonal transport defects. It was reported that there is a change of neurotrophins on DRG neurons during development. In vitro studies have suggested that sensory neurons switching from dependence on brain‐derived neurotrophic factor (BDNF) or NT‐3, to dependence on NGF at approximately E13 days 30. We have observed an excess of apoptosis event between E11.5 and E15.5 and a marked neuron loss at P0.5 in the Swl/+ lumbar DRGs. This time frame of DRG neuron loss in Swl/+ mice is consistent with the actions of neurotrophins on DRG neurons during development, suggesting that disrupted neurotrophin signaling may be responsible for sensory neuron loss in Swl/+ mice. Dynein has been found to play an important role in neurotrophic factor/Trk complexes transport 31, 32. It is possible that the degeneration of proprioceptive sensory neurons in Swl/+ mice is one of consequences of defective dynein‐based axonal transport. Results from our chamber experiments revealed that the retrograde transport of NGF is impaired, which could be one of the causes for the DRG neuron loss in Swl/+ mice.
Among these Dync1h1 mutant mouse strains, Loa/+, Cra/+ mice have been well studied and gave us many insights into the possible mechanisms of defects caused by Dync1h1 mutations. A study by Deng et al. 33 reported that the Loa mutation in Dync1h1 led to increased affinity of this subunit of cytoplasmic dynein to light intermediate and a population of intermediate chains, and a suppressed association of dynactin to dynein. It was also reported that there is a marked inhibition of motor run‐length in vitro and in vivo, and significantly altered motor domain coordination in the dynein from Loa/+ mice 34. The study on Loa/+ mice have also shown that the Dync1h1 mutation changed the conformation of dynein heavy chain, modifying its interaction with dynein intermediate chain (DIC), dynein light chain (DLIC1), and dynactin 33, 35. It seems that the mutations in DYNC1H1 may influence the coordination of subunits in dynein complex. The 9‐bp Swl deletion is outside the DIC binding region but within the putative homodimerization domain 36. To explore the underlying mechanism further, we asked whether the Swl mutation could influence the formation of the dynein complex. However, this study showed that the mutated DYNC1H1 fragments did not impair the dimerization of two homogenous heavy chain fragments. Further studies warrant expanding knowledge on the interaction between different subunits in dynein complexes, such as DIC, DLIC1, dynactin, and even the cargos transported by the dynein complexes.
With respect to neurodegeneration, defects in retrograde axonal transport, mainly driven by cytoplasmic dynein, have already been reported in several neurodegenerative diseases 3, 37. An interesting phenomenon has been noticed in the studies of the dynein‐related diseases. Two independent studies have shown that interactions between mutant SOD1 G93A and the dynein Loa/+ and Cra/+ mutations with a significant involvement of motor neurons can markedly attenuate motor neuron degeneration in double‐heterozygote mice 38, 39. Double mutants had an increased rate of retrograde axonal transport in embryonic motor neuron cultures compared with their single‐mutant and WT littermates 38. No significant increase in life span was observed when the Loa/+ mouse was crossed to two other SOD1‐ALS models: SOD1 G37R and SOD1 G85R transgenic mice 40. Similarly, no evidence supports a significant delay of average survival time, body weight loss or motor dysfunction in Swl/SOD1 G93A mice 7. Despite intense research in this field, the molecular basis for these DYNC1H1 functional discrepancies remains largely unknown at present. More and more mutations identified in human DYNC1H1 are enriching the picture and clearly show pivotal roles for this retrograde axonal transport motor complex in neuronal function. The known mutations have overlapping phenotypes but remain distinct from each other. This could be partly explained by potential disruption of binding by different cargoes and adaptors 41. Many intriguing questions still remain, which may be the key point to understand the functions of dynein and the underlying mechanisms of selective neuronal dysfunction in dynein‐related neurodegenerative diseases (so called “dyneinopathies”).
In conclusion, the Dync1h1 Swl/+ mutation led to maldevelopment of embryos, sensory neuron loss, and impaired mitochondria transport in DRG neurons. Those deficits, coupling with the failure of endocytosed NGF retrograde transportation in Swl/+ mice, could likely contribute to the excessive apoptosis of DRG neurons during the development stages. All of these findings could enrich our understanding of the pathogenesis underlying Swl/+ mice and shed new insights onto the functions of dynein.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work is funded by the National Natural Sciences Foundation of China (grant numbers 30870873 and 81171187) to XJ Chen.
References
- 1. Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998;279:519–526. [DOI] [PubMed] [Google Scholar]
- 2. Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 2005;6:201–214. [DOI] [PubMed] [Google Scholar]
- 3. Schiavo G, Greensmith L, Hafezparast M, Fisher EM. Cytoplasmic dynein heavy chain: The servant of many masters. Trends Neurosci 2013;36:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldstein LS, Yang Z. Microtubule‐based transport systems in neurons: The roles of kinesins and dyneins. Annu Rev Neurosci 2000;23:39–71. [DOI] [PubMed] [Google Scholar]
- 5. Pfister KK, Fisher EM, Gibbons IR, et al. Cytoplasmic dynein nomenclature. J Cell Biol 2005;171:411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003;300:808–812. [DOI] [PubMed] [Google Scholar]
- 7. Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci 2007;27:14515–14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet 2010;42:1109–1112. [DOI] [PubMed] [Google Scholar]
- 9. Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a dync1h1 mutation in a large pedigree with dominant axonal charcot‐marie‐tooth disease. Am J Hum Genet 2011;89:308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsurusaki Y, Saitoh S, Tomizawa K, et al. A dync1h1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics 2012;13:327–332. [DOI] [PubMed] [Google Scholar]
- 11. Willemsen MH, Vissers LE, Willemsen MA, et al. Mutations in dync1h1 cause severe intellectual disability with neuronal migration defects. J Med Genet 2012;49:179–183. [DOI] [PubMed] [Google Scholar]
- 12. Poirier K, Lebrun N, Broix L, et al. Mutations in tubg1, dync1h1, kif5c and kif2a cause malformations of cortical development and microcephaly. Nat Genet 2013;45:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Q, Wang F, Cao J, et al. Nudel promotes axonal lysosome clearance and endo‐lysosome formation via dynein‐mediated transport. Traffic 2009;10:1337–1349. [DOI] [PubMed] [Google Scholar]
- 14. Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci USA 1977;74:4516–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu X. Structural requirements and dynamics of mitosin‐kinetochore interaction in m phase. Mol Cell Biol 1999;19:1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan X, Li F, Liang Y, et al. Human nudel and nude as regulators of cytoplasmic dynein in poleward protein transport along the mitotic spindle. Mol Cell Biol 2003;23:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duchen LW. A dominant hereditary sensory disorder in the mouse with deficiency of muscle spindles: The mutant sprawling. J Physiol 1974;237:10P–11P. [PubMed] [Google Scholar]
- 18. Duchen LW, Scaravilli F. The structure and composition of peripheral nerves and nerve roots in the sprawling mouse. J Anat 1977;123:763–775. [PMC free article] [PubMed] [Google Scholar]
- 19. Brook GA, Duchen LW. End‐plates, transmission and contractile characteristics of muscles without spindles in the hereditary sensory neuropathy of the sprawling mouse. Brain 1990;113:867–891. [DOI] [PubMed] [Google Scholar]
- 20. Bader JR, Vaughan KT. Dynein at the kinetochore: Timing, interactions and functions. Semin Cell Dev Biol 2010;21:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vallee RB, Seale GE, Tsai JW. Emerging roles for myosin ii and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol 2009;19:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sasaki S, Shionoya A, Ishida M, et al. A lis1/nudel/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 2000;28:681–696. [DOI] [PubMed] [Google Scholar]
- 23. Cheng HH, Liu SH, Lee HC, et al. Heavy chain of cytoplasmic dynein is a major component of the postsynaptic density fraction. J Neurosci Res 2006;84:244–254. [DOI] [PubMed] [Google Scholar]
- 24. Barakat‐Walter I, Riederer BM. Triiodothyronine and nerve growth factor are required to induce cytoplasmic dynein expression in rat dorsal root ganglion cultures. Brain Res Dev Brain Res 1996;96:109–119. [PubMed] [Google Scholar]
- 25. He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol 2005;168:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA 1989;86:1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gepner J, Li M, Ludmann S, et al. Cytoplasmic dynein function is essential in drosophila melanogaster. Genetics 1996;142:865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol 1998;141:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McIlwain DL. Nuclear and cell body size in spinal motor neurons. Adv Neurol 1991;56:67–74. [PubMed] [Google Scholar]
- 30. Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development 1993;118:989–1001. [DOI] [PubMed] [Google Scholar]
- 31. Yano H, Lee FS, Kong H, et al. Association of trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci 2001;21:RC125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated trks to promote survival of target‐dependent neurons. Nat Neurosci 2004;7:596–604. [DOI] [PubMed] [Google Scholar]
- 33. Deng W, Garrett C, Dombert B, et al. Neurodegenerative mutation in cytoplasmic dynein alters its organization and dynein‐dynactin and dynein‐kinesin interactions. J Biol Chem 2010;285:39922–39934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ori‐McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol 2010;12:1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eyckerman S, Verhee A, der Heyden JV, et al. Design and application of a cytokine‐receptor‐based interaction trap. Nat Cell Biol 2001;3:1114–1119. [DOI] [PubMed] [Google Scholar]
- 36. Banks GT, Fisher EM. Cytoplasmic dynein could be key to understanding neurodegeneration. Genome Biol 2008;9:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu XA, Rizzo V, Puthanveettil SV. Pathologies of axonal transport in neurodegenerative diseases. Transl Neurosci 2012;3:355–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kieran D, Hafezparast M, Bohnert S, et al. A mutation in dynein rescues axonal transport defects and extends the life span of als mice. J Cell Biol 2005;169:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teuchert M, Fischer D, Schwalenstoecker B, Habisch HJ, Bockers TM, Ludolph AC. A dynein mutation attenuates motor neuron degeneration in sod1(g93a) mice. Exp Neurol 2006;198:271–274. [DOI] [PubMed] [Google Scholar]
- 40. Ilieva HS, Yamanaka K, Malkmus S, et al. Mutant dynein (loa) triggers proprioceptive axon loss that extends survival only in the sod1 als model with highest motor neuron death. Proc Natl Acad Sci USA 2008;105:12599–12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Franker MA, Hoogenraad CC. Microtubule‐based transport – basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 2013;126:2319–2329. [DOI] [PubMed] [Google Scholar]