

SUMMARY
Activation of the TLR4 signaling pathway by lipopoly-saccharide (LPS) leads to induction of both inflammatory and interferon-stimulated genes, but the mechanisms through which these coordinately activated transcriptional programs are balanced to promote an optimal innate immune response remain poorly understood. In a genome-wide small interfering RNA (siRNA) screen of the LPS-induced tumor necrosis factor α (TNF-α) response in macrophages, we identify the interferon-stimulated protein IFIT1 as a negative regulator of the inflammatory gene program. Transcriptional profiling further identifies a positive regulatory role for IFIT1 in type I interferon expression, implicating IFIT1 as a reciprocal modulator of LPS-induced gene classes. We demonstrate that these effects of IFIT1 are mediated through modulation of a Sin3A-HDAC2 transcriptional regulatory complex at LPS-induced gene loci. Beyond the well-studied role of cytosolic IFIT1 in restricting viral replication, our data demonstrate a function for nuclear IFIT1 in differential transcriptional regulation of separate branches of the LPS-induced gene program.
Graphical Abstract
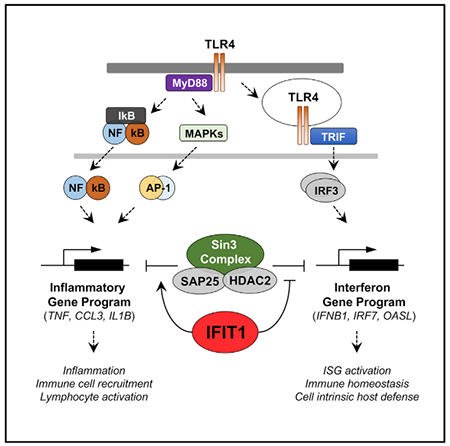
In Brief
John et al. describe a function for IFIT1 in the innate immune response. Previously considered an antiviral protein, IFIT1 is identified as a reciprocal modulator of bacterially induced pro-inflammatory and interferon genes and shown to associate with chromatin regulators to modulate transcription and the host response to bacterial infection.
INTRODUCTION
Toll-like receptors (TLRs) play a critical role in the innate immune response through their recognition of pathogen-associated molecular patterns (PAMPs) (Akira et al., 2006). Pathogen-derived macromolecular components such as lipids, lipopeptides, and nucleic acids serve as the primary ligands for TLRs (Akira et al., 2006; Gay and Gangloff, 2007; Takeda et al., 2003), and receptor engagement initiates a complex signaling and transcriptional response orchestrated primarily by members of the nuclear factor κB (NF-κB), AP-1, and interferon regulatory factor (IRF) transcription factor families (Schwabe et al., 2006; Seki and Brenner, 2008; Zhang and Lu, 2015). There are two major branches of the TLR signaling pathway characterized by the receptor-recruited adaptor proteins myeloid differentiation primary response protein 88 (MyD88) and TIR-domain-containing adaptor protein inducing interferon (TRIF), and among the TLR-activating PAMPs, lipopolysaccharide (LPS) is unique in its ability to induce both adaptor pathways through TLR4. The MyD88-dependent pathway more strongly induces pro-inflammatory cytokine genes such as TNF and IL6, primarily through the activation of NF-κB and API (Schwabe et al., 2006; Zhang and Lu, 2015), while activation of IRF3 through the TRIF-dependent pathway induces the IFNB1 gene and a broad range of interferon-stimulated genes (ISGs) (Schwabe et al., 2006; Seki and Brenner, 2008). LPS thus induces a more complex gene program than other TLR ligands through both the direct activation of each adaptor pathway and a poorly characterized interplay between the MyD88 and TRIF signaling branches (Bagchi et al., 2007; Liu et al., 2015; Napolitani et al., 2005; Suet Ting Tan et al., 2013). Prior studies in TLR-activated dendritic cells have suggested that opposing and interconnected transcriptional regulatory networks may coordinately control these inflammatory and antiviral gene programs (Amit et al., 2009), but the mechanisms of this control remain poorly defined.
Numerous transcriptional regulators have been associated with the expression of inflammatory and antiviral genes. Notably, HDAC complexes have been implicated both in positively regulating the expression of IFNB1 (Chen etal., 2012) and in negatively regulating the expression of inflammatory cytokine genes such as TNF and IL1B in macrophage cells (Hu et al., 2017; Lohman et al., 2016). In complex with the transcriptional co-repressor Sin3A, HDAC family members have been shown to regulate eukaryotic gene transcription through a complex interaction with chromatin, where depending upon the nature of cofactor binding, Sin3-HDAC complexes can either repress or activate transcription (Kadamb et al., 2013). Transcriptional repression of genes by Sin3-HDAC complexes is generally achieved through the deacetylation of histones H3 and H4 (Bansal et al., 2011; Das et al., 2013; van Oevelen et al., 2010), while the mechanism for gene activation is less well established (Wang et al., 2009). Although the role of individual HDAC-Sin3 complexes is not well studied in innate immune cells, HDAC3 was recently shown to be essential to support the LPS-induced gene program in mouse primary macrophages (Chen etal., 2012). However, it has yet to be established whether these chromatin regulatory factors influence the balance between the MyD88 and TRIF-dependent transcriptional programs induced by LPS challenge.
Genome-wide gene perturbation screens, either through RNAi or CRISPR-Cas9 gene editing, are useful tools to characterize molecular pathways and cellular regulatory networks in an unbiased manner (Aregger etal., 2015; Chia et al., 2012; Parnas et al., 2015). In a genome-wide small interfering RNA (siRNA) screen for genes regulating LPS-induced TNF activation in human macrophages, we discovered a previously unappreciated negative regulatory role for the interferon-induced protein with tetratrico-peptide repeats-1 (IFIT1). IFITs are an IRF-inducible family of genes with antiviral activity against several RNA viruses (Fensterl and Sen, 2015), and while numerous antiviral functions for IFIT proteins have been described (Diamond and Farzan, 2013; Guo et al., 2000; Li et al., 2009), they have not been implicated in regulation of the TLR-activated gene program. Although the most common stimuli for induction of IFIT1 are interferons, the JAK-signal transducer and activator of transcription (STAT) pathway is dispensable for induction (Bandyopadhyay et al., 1995), and IFIT1 can be induced directly by LPS in macrophages through TRIF-dependent IRF3 activation (Chevrier et al., 2011; Tong et al., 2016). We find that IFIT1 negatively regulates the expression of TNF and numerous additional inflammatory cytokine genes in LPS-treated macrophages while simultaneously promoting the expression of IFNB1 and the subsequent interferon gene program. The positive regulatory role of IFIT1 in IFNB1 induction is observed for a broad range of interferon-inducing stimuli and occurs downstream of IRF3 activation. We propose a previously unappreciated role for a nuclear pool of IFIT1 in facilitating the removal of Sin3A-HDAC complexes from ISG loci, which permits effective recruitment of the IRF3 transcription factor. We further demonstrate that IFIT1-dependent IFNB1 induction is necessary for effective macrophage control of Gram-negative bacterial infection.
RESULTS
A Genome-wide siRNA Screen Identifies IFIT1 as a Negative Regulator of LPS-Induced TNF-α Induction
Using a recently developed macrophage cell system for large-scale perturbation screening (Sun et al., 2016; Li et al., 2015) (Figure 1A), we conducted a genome-wide RNAi screen to identify regulators of the LPS-induced TNF transcriptional response in differentiated THP1 human macrophage-like cells (Sun et al., 2017). Among the putative negative regulators of TNF induction was the host viral restriction factor IFIT1 (Figure 1B), a gene induced early in response to LPS treatment in differentiated macrophage cells (Tong et al., 2016). We observed increased tumor necrosis factor α (TNF-α) reporter expression upon knockdown of IFIT1, both with a pool of 4 siRNAs in the primary siRNA screen (Figure 1B) and with additional independent siRNA sequences in a secondary screen (Figure S1). We then tested whether this apparent negative regulatory function for IFIT1 was also observed in siRNA-transfected human primary macrophages. We achieved sustained perturbation of IFIT1 expression in primary human cells (Figure 1C), and consistent with our THP1 siRNA screen data, we observed elevated TNF mRNA expression (Figure 1D) and TNF-α protein secretion (Figure 1E) in response to LPS.
Figure 1. IFIT1 Depletion in Human Macrophages Leads to an Enhanced LPS-Induced TNF-α Response.
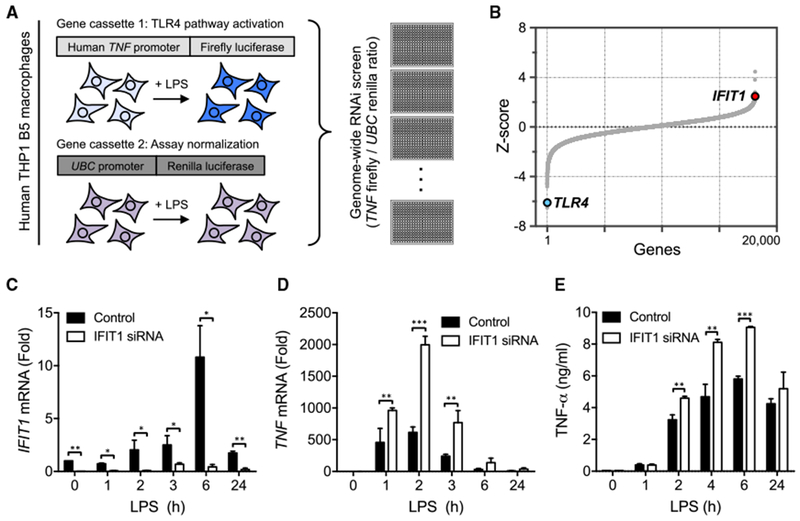
(A) Schematic for the TNF-α reporter assay used in a genome-wide screen of the LPS response in human THP1 cells (Sun et al., 2017). (B) Screen hit distribution showing TLR4 as a validating positive regulator hit and identification of IFIT1 as a negative regulator of LPS-induced TNF-α expression. (C-E) Primary human macrophages transfected with control or IFIT1 siRNA were stimulated with 100 ng/mL LPS for the indicated times, IFIT1 (C) or TNF (D) mRNA was measured by qPCR, and secreted TNF-α (E) was measured by ELISA. Data are representative of four independent experiments and expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA followed by Sidak’s multiple comparison test). See also Figure S1.
Transcriptomic Analysis Identifies a Role for IFIT1 as a Reciprocal Regulator of the Pro-inflammatory and Antiviral Gene Programs
TLR4 activation leads to the induction of numerous genes involved in the innate immune response. To further investigate the function of IFIT1 in regulating TLR-activated macrophages, we generated a THP1 cell line stably expressing an IFIT1 small hairpin RNA (shRNA) (shIFIT1), which exhibited substantial and sustained perturbation of IFIT1 expression (Figures S2A and S2B). To assess the genome-scale consequences of IFIT1 depletion, we conducted microarray analysis in LPS-stimulated wild-type (WT) and shIFIT1 THP1 cells (Data S1; STAR Methods). To more efficiently suppress IFIT1 expression in response to the LPS stimulus, we also transfected the IFIT1 shRNA cells with an IFIT1 siRNA, which led to a sustained attenuation of IFIT1 induction after LPS stimulation (Figure S2C). Microarray analysis across a comprehensive time course confirmed the enhanced expression of TNF-α in IFIT1-depleted cells, in addition to a substantial number of further inflammatory cytokines whose LPS-induced expression was also enhanced (Figure 2A, group 1 genes). Ingenuity Pathway Analysis (IPA) of the upregulated genes identified substantial known functional connections among this gene set, with a strong enrichment for genes linked to the inflammatory response and to recruitment of leukocytes and myeloid cells to sites of infection (Figure 2B). Further IPA analysis of the most strongly enriched pathways among this gene set implicated numerous pro-inflammatory cytokine pathways driven by interleukin-17 (IL-17) and IL-6 and enrichments for TLR, mitogen-activated protein kinase (MAPK), and PPAR signaling (Figure S2D).
Figure 2. IFIT1 Depletion Differentially Affects LPS-Induced Gene Programs.
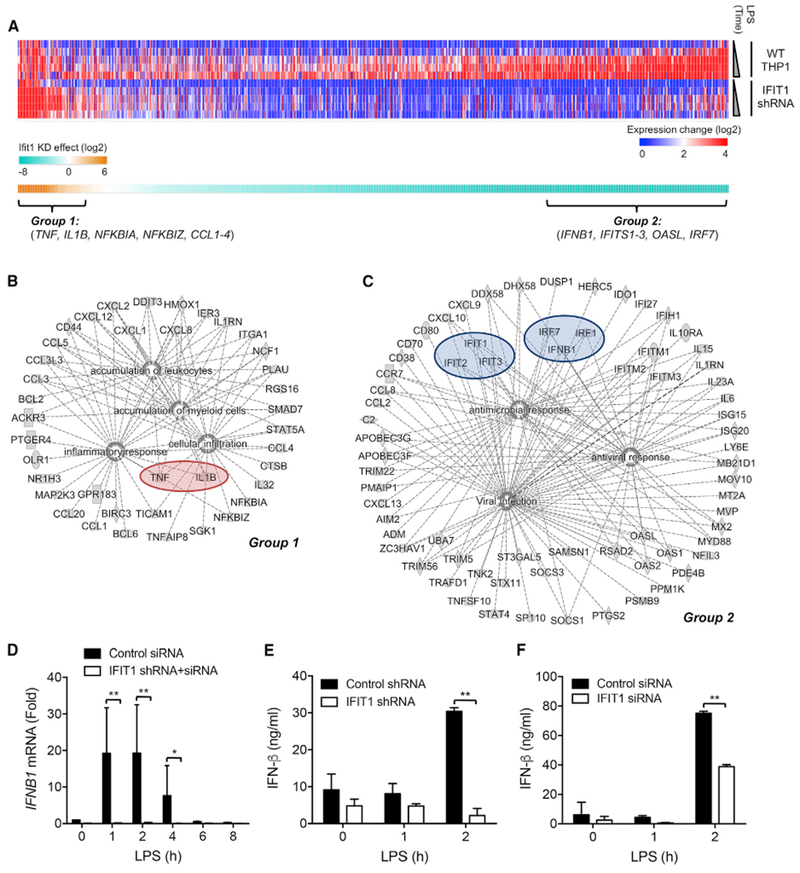
(A) Control and IFIT1-depleted THP1 cells were stimulated with 100 ng/mL LPS for 1, 2, 4, 6, and 24 hr, and transcriptional responses were measured by microarray. The upper heatmap shows genes with substantial expression increases in WT cells (STAR Methods) compared to their expression level in IFIT1-depleted cells. The lower heatmap shows the sum of the expression difference between WT and IFIT1-depleted cells across the time course (Data S1).
(B and C) The network connections of genes that showed (B) enhanced expression (group 1; Data S2) or (C) reduced expression (group 2; Data S3) in IFIT1-depleted cells, as determined by IPA.
(D) Transfection of siRNA into IFIT1 shRNA-expressing cells sustains a stable suppression of IFIT1 induction in response to 100 ng/mL LPS.
(E and F) Control and IFIT1-depleted THP1 cells (E) or human primary macrophages (F) were stimulated with 100 ng/mL LPS, and secreted IFN-β protein was measured by ELISA.
Data are representative of two (A) or three (D–F) independent experiments. Data in (D)–(F) are expressed as mean SD; *p < 0.05, **p < 0.01 (two-way ANOVA followed by Sidak’s multiple comparison test). See also Figure S2 and Data S1, S2, and S3.
While the identification of an enriched set of enhanced pro-inflammatory genes in IFIT1-depleted cells validated the IFIT1 phenotype from our siRNA screen, we also observed an unexpected pattern of gene suppression in our microarray dataset, where a substantial group of genes showed a significant attenuation in their LPS-induced expression (Figure 2A, group 2 genes). We noted that this gene set included a large number of ISGs, and consistent with this, IPA analysis showed a strong enrichment for the antiviral gene program and the interferon pathway (Figures 2C and S2E). This effect of IFIT1 perturbation could be explained by a notable attenuation of the principal type I interferon gene induced by LPS, IFNB1 (Figure 2C). While IFIT1 is a well-established ISG, it has not been previously implicated as a modulator of the expression of IFNB1 itself. This effect of IFIT1 perturbation on IFNB1 induction was confirmed at both the mRNA (Figure 2D) and protein secretion levels (Figure 2E). Similar reduction in the induction of IFNB1 was also seen in IFIT1-depleted human primary macrophages (Figure 2F). The opposite effect of IFIT1 perturbation in enhancing the induction of TNF-α was also confirmed at both the protein (Figure S2F) and mRNA levels (Figure S2G). These results show a previously unappreciated dual role for IFIT1 in LPS-stimulated macrophages in attenuating the degree of inflammatory cytokine induction while also promoting the expression of IFNB1 and the subsequent induction of a broad range of ISGs.
Opposing Effects of IFIT1 Perturbation on LPS-Induced Gene Sets Are Reflected in Signaling Responses and Conserved in Mouse Macrophages
To determine whether the enhanced TNF-α induction in IFIT1-perturbed cells correlates with alterations in LPS-induced signaling responses known to drive TNF expression (Zhang et al., 2015), we measured phosphorylation of the NF-κB component p65 and the MAPK p38 and the nuclear localization of NF-κB p50 in shIFIT1 cells. We observed stronger and more sustained activation of p65, p38, and p50 (Figure 3A), which may contribute to the elevated TNF expression seen with IFIT1 depletion.
Figure 3. Signaling Responses in IFIT1-Depleted Cells.
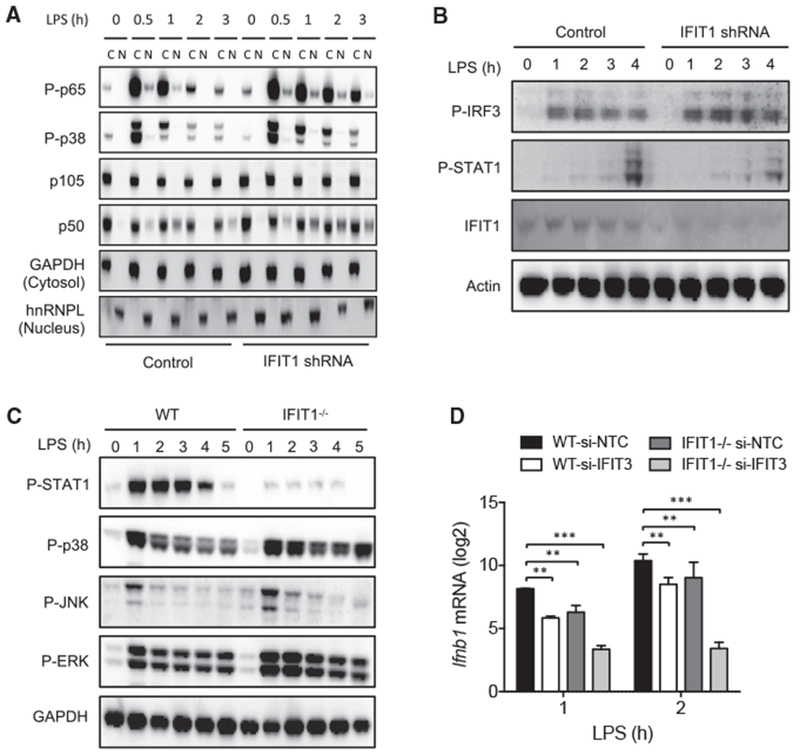
(A) Control and IFIT1 shRNA-expressing THP1 cells were stimulated with 100 ng/mL LPS for the indicated times, and cytosolic and nuclear fractions were subjected to western blot analysis for phos-pho-p65/NF-κB (Ser536), phospho p38 and MAPK (Thr180/Tyr182), p105/NF-κB, and p50/NF-κB. GAPDH and heterogenous nuclear ribonucleoprotein L (hnRNPL) were detected as positive controls for cytosolic and nuclear fractions.
(B) Control and IFIT1 shRNA-expressing THP1 cells were stimulated with 100 ng/mL LPS for the indicated times, and whole-cell lysates were subjected to western blot analysis for phospho-IRF3 (S386), phospho-STAT1 (Tyr701), IFIT1, and actin (loading control).
(C) WT and Ifit1−/− BMDM cells were stimulated with 100 ng/mL LPS for the indicated times, and whole-cell lysates were subjected to western blot analysis for phospho-STAT1 (Tyr701), phospho-p38 and MAPK (Thr180/Tyr182), phospho-JNK (Thr183/Tyr185), phospho-ERK (Thr202/Tyr204), and GAPDH (loading control).
(D) WT and Ifit1−/− mouse BMDM transfected with non-targeting control (NTC) or IFIT3 siRNA were stimulated with 100 ng/mL LPS and Ifnb1 mRNA was measured by qPCR.
Data are representative of three independent experiments. Data in (D) are expressed as mean ± SD; **p < 0.01, ***p < 0.001 (two-way ANOVA followed by Sidak’s multiple comparison test). See also Figure S3.
To further assess whether the reduction in IFNB1 expression also correlated with the activation state of the primary signaling pathway responsible for IFNB1 induction, we assessed the LPS-induced phosphorylation of IRF3 in IFIT1-perturbed cells (Fitzgerald et al., 2003; Sin et al., 2012). We observed no attenuation of IRF3 activation in shIFIT1 cells, with a slight increase in phosphorylation evident in response to LPS (Figure 3B). STAT1 phosphorylation, which is induced by delayed autocrine type I IFN signaling in LPS-treated macrophages, was reduced in the shIFIT1 cells, consistent with reduced IFNB1 production (Figure 4B). This suggests that the positive regulatory effect of IFIT1 on LPS-induced IFNB1 occurs downstream of IRF3 activation.
Figure 4. IFNB1 Induction Is Attenuated in IFIT1-Depleted Cells in Response to Multiple Different Stimuli.
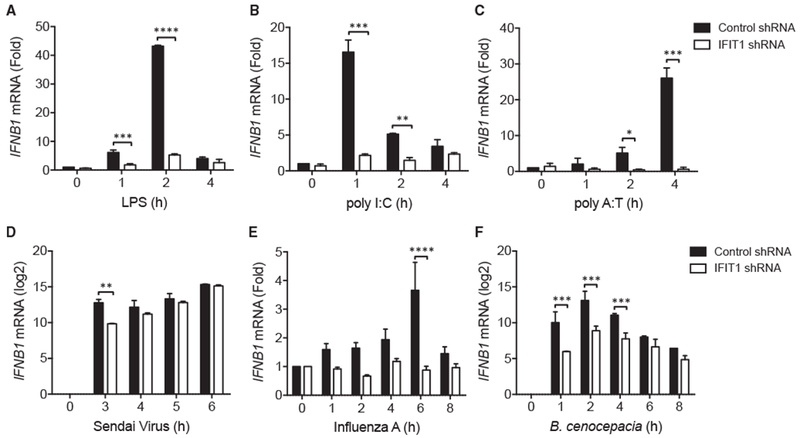
(A–F) Control and IFIT1 shRNA-expressing THP1 cells were stimulated with (A) 100 ng/mL LPS, (B) 10 μg/mL poly(I:C), (C) 10 μg/mL poly(dA:dT), (D) Sendai virus (Cantell) MOI 5, (E) influenza A virus (Texas/36/91) MOI 5, and (F) B. cenocepacia MOI 5, and IFNB1 mRNA was measured by qPCR. Data are representative of four (A) or three (B–F) independent experiments and are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-way ANOVA followed by Sidak’s multiple comparison test).
To compare the effects of IFIT1 depletion in mouse macrophages, we measured signaling responses in bone-marrow-derived macrophage cells from WT and Ifit1−/− mice (Szretter et al., 2012). Consistent with the effects seen in human macrophages, LPS-induced STAT1 phosphorylation was strongly reduced in Ifit1−/− bone marrow-derived macrophage cells (BMDMs) (Figure 3C), reflecting reduced Ifnb1 induction in response to LPS (Figure 3D). Activation of the mouse macrophage MAPK pathways was increased and sustained (Figure 3C), similar to human cells and consistent with a negative role for IFIT1 in the degree and duration of activation of the inflammatory program.
We noted that the relative defect in early induction of Ifnb1 in Ifit1−/− mouse macrophages was less than that observed in human macrophages. This may be related to accumulating evidence suggesting that the functions of individual gene products in the innate immune response can show considerable differences between these species (Seok et al., 2013; Sun et al., 2016), and it has also been shown that human and mouse IFIT proteins are not well conserved at the amino acid level and that they may not be directly orthologous (Daugherty et al., 2016). Since mouse macrophages also express high levels of the related Ifit3 protein, we tested the effect of Ifit3 knockdown in mouse BMDMs. Both Ifit1 and Ifit3 knockdown led to defective Ifnbl mRNA induction (Figure 3D). Notably, siRNA-based knockdown of Ifit3 in the Ifit1−/− background (Figure S3) caused a more substantial defect in Ifnbl mRNA induction (Figure 3D), suggesting possible redundancy between mouse Ifit1 and Ifit3 in their contribution to LPS-induced Ifnbl expression.
IFN-β Expression Is Attenuated in IFIT1-Depleted Cells in Response to Multiple IFN-Inducing Stimuli
To evaluate whether the effect of IFIT1 in supporting IFNB1 expression is observed with other interferon (IFN)-inducing stimuli, we challenged cells with poly(I:C) or poly(dA:dT), mimicking double-stranded RNA and DNA, respectively. Analysis of expression of IFNB1 showed reduced induction in shIFIT1 cells compared with control cells, consistent with that observed for LPS stimulation (Figures 4A–4C).
We then used live pathogens that induce IFNB1 through different PRR pathways to test whether the modulatory role of IFIT1 broadly affects IFNB1 induction by multiple signaling routes. We infected shIFIT1 cells and control cells with two viral pathogens and one bacterial pathogen. Infection with Sendai virus (Cantell) (Figure 4D) and influenza A (TX91) (Figure 4E) showed reduced early induction of IFNB1 in shIFIT1 macrophages. Similarly, the Gram-negative bacterium Burkholderia cenocepacia elicited reduced induction of IFNB1 in shIFIT1 cells (Figure 4F). Our results suggest a broad role for IFIT1 in the transcriptional induction of IFNB1 downstream of several different pathogenic stimuli.
IFIT1 Modulates the Association of an LPS-Regulated HDAC2-SAP25 Protein Complex
Taken together, our data suggest a role for IFIT1 in regulating the balance and transition between early pro-inflammatory cytokine transcription and the subsequent type I IFN gene program induced by LPS. In this context, IFIT1 appears to limit the strength and duration of the primarily NF-κB and MAPK-activated pro-inflammatory gene program typified by TNF while also playing a key role in supporting the expression of type I IFN and the consequent induction of a broad range of ISGs. Previous reports have suggested a reciprocal counterbalance between these gene programs (Guarda et al., 2011; Quartier et al., 2011), and studies in LPS-stimulated dendritic cells suggest control by interacting inflammatory and antiviral transcriptional regulatory networks (Amit et al., 2009). The Sin3-associated protein (SAP) family is among the transcriptional regulators proposed to regulate this balance (Amit et al., 2009); furthermore, SAP25 in particular has been identified as an interactor of both IFIT1 (Shiio et al., 2006) and the histone deacetylase HDAC2 (BioGRID; Shiio et al., 2006). The involvement of HDAC complexes in the positive regulation of IFN expression in macrophages has been previously reported (Chen et al., 2012), while through inhibitor analysis and gene perturbation, HDAC complexes have also been shown to attenuate the production of pro-inflammatory cytokines such as TNF and IL1B in macrophages (Chen et al., 2012; Hu et al., 2017; Lohman et al., 2016). Since this reciprocal role for HDACs in the regulation of the IFN and inflammatory gene programs in macrophages resembles our findings with IFIT1, we first tested whether a SAP25-HDAC2-IFIT1-containing protein complex exists in macrophages. Using immunoprecipitation (IP), we observed an LPS-induced increase in co-precipitation of SAP25 and HDAC2, with a weaker but detectable co-precipitation of IFIT1 (Figure 5A). In IFIT1-depleted cells, SAP25/HDAC2 co-precipitation was substantially reduced and failed to sustain the levels observed in WT cells. Reduced association of HDAC2 and SAP25 in IFIT1 knockdown cells suggests that IFIT1 may stabilize an HDAC2-SAP25 complex.
Figure 5. IFIT1 Modulates the Association of an LPS-Regulated HDAC2-SAP25 Protein Complex.
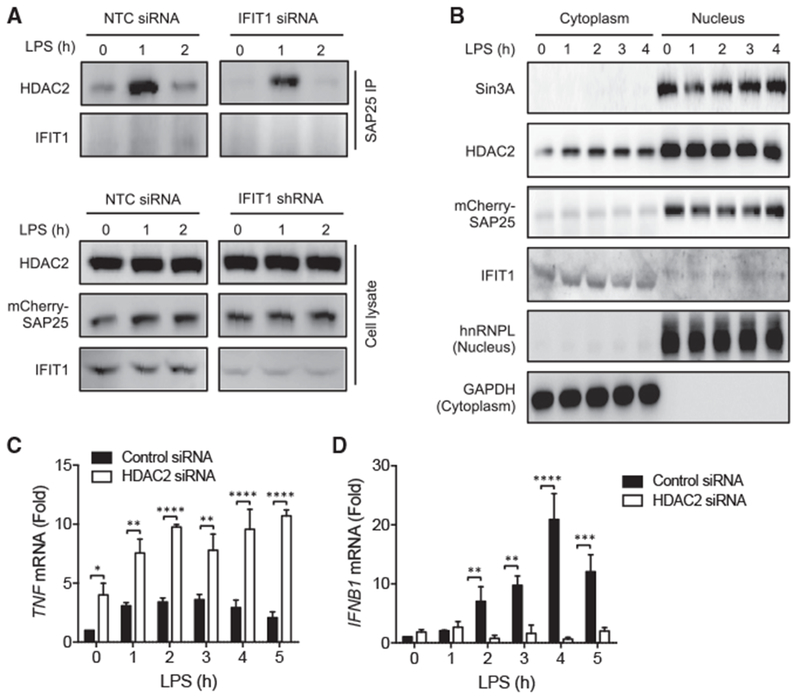
(A) THP1 cells stably expressing a SAP25-mCherry fusion protein were transfected with non-targeting control (NTC) or IFIT1 siRNA and stimulated with 100 ng/mL LPS for 0, 1, or 2 hr. The indicated proteins were detected by western blot in either a SAP25-mCherry immunoprecipitate (top) or in whole-cell lysates (bottom).
(B) Cytosolic and nuclear fractions were isolated from 100 ng/ml LPS treated mCherry-SAP25 expressing cells and subjected to western blot analysis for Sin3A, HDAC2, mCherry, hnRNPL (nuclear marker) and GAPDH (cytosolic marker).
(C and D) THP1 cells transfected with non-targeting control or HDAC2 siRNA were stimulated with 100 ng/mL LPS for the indicated times and mRNA levels of (C) TNF and (D) IFNB1 were measured by qPCR.
Data are representative of three independent experiments. Data in (C) and (D) are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-way ANOVA followed by Sidak’s multiple comparison test). See also Figure S4.
Previous reports have shown that IFIT1 is localized primarily to the cytoplasm (Fensterl and Sen, 2011; Pichlmair et al., 2011). Since cytoplasmic localization of IFIT1 is not consistent with a role in the nuclear regulation of gene expression, we analyzed the subcellular localization of IFIT1 in differentiated macrophage cells by both subcellular fractionation and confocal microscopy. While we found the majority of IFIT1 localized in the cytosol, as previously reported, we also consistently detected an IFIT1 immunoreactive band in nuclear lysates of slightly higher molecular weight than the cytosolic protein (Figure 5B). Presence of IFIT1 in the nucleus was further supported by confocal microscopy (Figure S4A), with ~10%–15% of endogenous IFIT1 detected in the nucleus (Figure S4B). HDAC2, SAP25, and the SAP-family associated transcriptional regulator Sin3A were also detected in the macrophage nuclear lysates (Figure 5B). To further investigate IFIT1 nuclear localization, we expressed FLAG-tagged IFIT1 in THP1 cells and compared nuclear levels with and without the nuclear export inhibitor leptomycin B (Kudo et al., 1998). We observed a significant increase in the nuclear FLAG immunoreactive signal in leptomycin-B-treated cells (Figures S4C and S4D), suggesting that IFIT1 is actively shuttled between the cytoplasm and nucleus. We then sought to determine whether increasing IFIT1 nuclear localization with an exogenous nuclear localization sequence (NLS) would influence IFIT1 regulation of the SAP25-HDAC2 complex. We first overexpressed IFIT1 with and without an NLS in HEK293 cells and observed a substantial increase in nuclear localization of the NLS-tagged IFIT1 protein (Figures S5A and S5B). We also observed a marked increase in SAP25-HDAC2 interaction in cells overexpressing NLS-IFIT1 (Figure S5C), supporting a role for nuclear IFIT1 in regulation of this interaction. To test whether targeting IFIT1 to the nucleus in macrophages would influence IFNB1 transcription, we stably expressed IFIT1 with or without an NLS sequence in U937 cells (Figures S5D and S5E) and observed a significant increase in IFNB1 expression that was further enhanced by NLS-driven nuclear localization of IFIT1 (Figure S5E). These data suggest that a fraction of IFIT1 is localized in the macrophage nucleus and that this pool of IFIT1 could be involved in the modulation of LPS-induced gene expression through association with an HDAC2-SAP25 complex.
Since expression of LPS induced genes has been shown to be differentially regulated by HDAC-Sin3 complexes in macrophages (Chen et al., 2012; Hu et al., 2017; Lohman et al., 2016), we tested the effect of HDAC2 knockdown in LPS-treated THP1 cells (Figure S6). Consistent with previous reports of the differential effect of HDACs on IFNB1 (Chen et al., 2012) and TNF (Hu et al., 2017; Lohman et al., 2016) induction, we found that HDAC2 knockdown leads to upregulation of TNF (Figure 5C) and downregulation of IFNB1 (Figure 5D), similar to the effects observed with IFIT1 knockdown (Figures 1D, 1E, and 2D–2F). These results suggest a related role for HDAC complexes and IFIT1 in regulating the inflammatory and antiviral gene programs.
IFIT1 Facilitates Sin3A Corepressor Removal and IRF3 Recruitment to Multiple ISG Loci
Sin3-HDAC complexes are most often considered to act as transcriptional co-repressors (Bansal et al., 2011; Buttar et al., 2010; Muñoz et al., 2012). Interestingly, analysis of regulatory motifs among the IFIT1-dependent ISG-dominated gene set showed enrichment for the Sin3A-associated REST transcriptional repressor (Grimes et al., 2000), along with the expected enrichment for IFN-sensitive response element (ISRE), IRF, and STAT motifs (Figure S7A). Since the interaction of HDAC2 and SAP25, key components in the Sin3A repressor complex, is diminished in IFIT1-depleted cells, we speculated that IFIT1 may be required to regulate binding of a Sin3A-containing repressor complex at the IFNB1 locus during gene induction. Chromatin immunoprecipitation (ChIP) PCR analysis of Sin3A at the IFNB1 locus showed that LPS activation leads to a transient reduction in Sin3A corepressor binding, which could be required for effective IFNB1 induction (Figure 6A). Furthermore, we observed substantial dysregulation of Sin3A removal from the IFNB1 locus in IFIT1-perturbed cells (Figure 6A). Consistent with opposing effects of IFIT1 on regulation of genes in the IFN and inflammatory gene programs, we observed reduced basal state binding of Sin3A at the promoters of the inflammatory genes TNFand CCL3 in IFIT1-depleted cells (Figures S7B and S7C), which may contribute to their enhanced expression in cells with diminished IFIT1 expression.
Figure 6. IFIT1 Facilitates Sin3A Corepressor Removal and IRF3 Recruitment to Multiple ISG Loci.

(A–C) Control and IFIT1 shRNA-expressing THP1 cells were stimulated with 100 ng/mL LPS for the indicated times, and Sin3A binding was measured by ChIP-qPCR at the (A) IFNB1, (B) IRF7, and (C) IRF8 promoters.
(D–F) Control and IFIT1 shRNA-expressing THP1 cells were stimulated with 100 ng/mL LPS for the indicated times, and IRF3 binding was measured by ChIP-qPCR at the (D) IFNB1, (E) IRF7, and (F) IRF8 promoters.
Data are representative of three independent experiments and are expressed as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-way ANOVA followed by Sidak’s multiple comparison test). See also Figure S5.
It has been shown that type I IFN gene induction involves an initial phase dependent on IRF3 recruitment and a secondary phase that is supported by IRF7 and IRF8 activity (Ikushima et al., 2013; Honda et al., 2004; Kawai et al., 2004; Lazear et al., 2013; Li et al., 2011). This secondary phase requires IRF7 and IRF8 gene induction, and we find that both LPS and Sendai-virus-induced expression of IRF7 and IRF8 is significantly attenuated in IFIT1-depleted cells (Data S1; Figures S7D and S7E), which likely contributes to the IFNB1 expression defect. Interestingly, the human IRF7 promoter also includes evidence of both REST and Sin3A binding from Encyclopedia of DNA Elements (ENCODE) data (Figure S7F). We used ChIP-PCR to determine whether the IRF7 and IRF8 gene loci in macrophages bound Sin3A, and we observed a pattern similar to the IFNB1 locus, with LPS-induced removal of the Sin3A corepressor from the IRF7 and IRF8 loci in control macrophages, and dysregulated Sin3A removal in IFIT1-depleted cells (Figures 6B and 6C). These data suggest a model where the interaction of SAP25-HDAC2-Sin3A, facilitated by IFIT1, could be required for the efficient removal of the repressor complex from the promoters of IFNB1, IRF7, and IRF8 during gene induction.
To further test whether such corepressor removal is required to permit recruitment of activating transcription factors, we assessed IRF3 binding at these genes. We observe a robust LPS-induced recruitment of IRF3 to all three gene loci, which was strongly perturbed in IFIT1-depleted cells (Figures 6D–6F). These data demonstrate a critical role for nuclear IFIT1 to facilitate the appropriate regulation of a Sin3A-HDAC repressor complex both at the IFNB1 locus and at the IRF7 and IRF8 loci that are required to support sustained expression of the IFN gene program.
IFIT1 Is Required for Regulation of Intracellular Bacterial Infection in Macrophages
Finally, to test the impact of reduced LPS-stimulated IFNB1 induction in the context of a Gram-negative bacterial challenge, we infected IFIT1-depleted and control macrophages with the bacterium B. cenocepacia. Using a previously established imaging assay for intracellular bacterial replication (Miller etal., 2015), we observed substantially increased B. cenocepacia replication in IFIT1-depleted cells (Figures 7A and 7B). Prior treatment of cells with recombinant IFN-β protein reduced bacterial replication in both WT and IFIT1-depleted cells (Figures 7A and 7B). Furthermore, pretreatment of IFIT1-depleted cells with IFN-β reduced replication of B. cenocepacia to the levels observed in untreated WT cells. These data imply that the type I IFN induced by LPS has an important host-protective role during Gram-negative bacterial infection (Assani et al., 2014; Schmeisser et al., 2014) and further suggest an important role for IFIT1 in supporting a host-protective type I IFN response.
Figure 7. IFIT1 Is Required for IFN-β-Mediated Regulation of Intracellular Bacterial Infection in Macrophages.
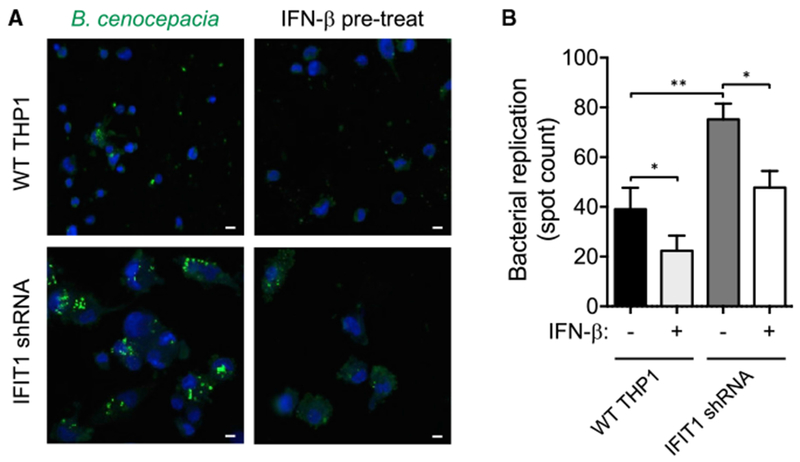
Control and IFIT1 shRNA-expressing THP1 cells with or without 3,000 IU/mL IFN-β pretreatment were infected with B. cenocepacia at MOI 1 for 22 hr.
(A) Representative images from WT and IFIT1 shRNA-expressing THP1 cells. Scale bars, 20 μm.
(B) Quantification of bacterial replication by spot count (STAR Methods). Data are representative of four independent experiments and are expressed as mean ± SD; *p < 0.05, **p < 0.01 (paired t test).
DISCUSSION
Systematic studies with knockdown of transcriptional regulators have suggested the existence of two opposing gene regulatory networks for the inflammatory and antiviral gene programs induced in TLR-activated dendritic cells (Amit et al., 2009). Such cross regulation of type I IFN and inflammatory cytokines has been implicated in earlier studies and also observed in clinical outcomes of arthritis patients treated using TNF blockade (Cantaert et al., 2010; Cuchacovich et al., 2008), where unexpected onset of psoriasis-like symptoms in TNF-blocked patients were attributed to increased type I IFN levels (Cuchacovich et al., 2008). Further clinical evidence of cross-regulation includes IFN therapy reducing IL-1β levels in multiple sclerosis patients (Guarda et al., 2011) and IL-1 receptor antagonism inducing an IFN signature in juvenile idiopathic arthritis (Quartier et al., 2011). It is also becoming clear that different phenotypic states of macrophages are often controlled at the level of chromatin regulation (Piccolo et al., 2017).
Here, we have provided considerable insight to mechanisms contributing to reciprocal regulation of the inflammatory and IFN gene programs. We first identified IFIT1 as a negative regulator of the pro-inflammatory cytokine gene TNF in a genomewide siRNA screen and subsequently identified an opposing positive regulatory role for IFIT1 in the induction of the IFNB1 gene and the antiviral IFN gene program. IFIT proteins have been heavily studied as important viral restriction factors with a range of established antiviral functions (Diamond and Farzan, 2013; Fensterl and Sen, 2015; Guo et al., 2000; Li et al., 2009). However, while they are well-known ISGs themselves, IFIT proteins have not previously been considered as positive regulators of IFN levels, and IFIT1 depletion can lead to a markedly increased viral load and consequent ISG response (Li et al., 2009). However, overexpression of IFIT1 has been shown to increase endogenous IFNB1 levels during Alpha virus infection (Reynaud et al., 2015), suggesting a supporting role for IFIT1 in IFN gene expression.
Prior studies have implicated both HDACs (Chen et al., 2012; Hu et al., 2017; Lohman et al., 2016) and SAPs (Amit et al., 2009) in the differential regulation of IFNB1 and TNF. Our data suggest that a nuclear pool of IFIT1 can stabilize an LPS-induced association of HDAC2 and SAP25, as this association is substantially diminished in IFIT1-depleted cells and enhanced in cells expressing a nuclear targeted IFIT1 protein. Diminished HDAC2-SAP25 interaction may contribute to the inefficient removal of Sin3A from the promoters of IFNB1 and the transcription factors (IRF7 and IRF8) that support the induction of IFNB1 upon ISG stimulation. Lack of efficient removal of Sin3A, and possibly other transcriptional repressors associated with this complex, hinders the timely binding of activated IRF3 at the promoters of these genes.
IRF7 has been shown to play a key role in the induction of IFN by TLR ligands (Honda et al., 2004; Ikushima et al., 2013; Kawai et al., 2004), and we find that IRF7 expression is reduced both at the basal state and stimulated state in IFIT1-depleted cells. Notably, we also found that the expression of IRF8, which also promotes IFN induction (Tailor et al., 2007), is significantly attenuated in IFIT1 knockdown cells. IRF8 and IRF3 have been shown to cooperatively regulate rapid induction of IFNB1 in human monocytes (Li et al., 2011). Although we find that IRF3 is expressed and activated normally in IFIT1-depleted cells, the attenuated recruitment of IRF3 to ISG promoters, and the consequent reduced expression of IRF7 and IRF8, could together contribute to the impaired induction of IFNB1 in response to various stimuli.
Multiple mechanisms may contribute to the negative regulation of TNF and the inflammatory gene program inIFIT1-depleted cells. It has been shown that these genes are more strongly induced by Myd88-dependent activation of the NF-κB and MAPK pathways in macrophages (Schwabe et al., 2006; Zhang and Lu, 2015), and we find that the activation levels of NF-κB and MAPK components are higher and more sustained when IFIT1 expression is attenuated. Furthermore, the expression of inflammatory cytokine genes like TNF are often inhibited by HDAC complexes (Hu et al., 2017; Lohman et al., 2016), so our observation of reduced HDAC2-SAP25 association in IFIT1-depleted cells suggests that this reduced association may also impair the negative regulation of TNF transcription. This is also supported by the increased TNF transcription observed when we knocked down HDAC2 expression. In addition, downregulation of IFNB1 may increase inflammatory cytokines through reduced negative feedback as a consequence of reduced regulatory inhibitors such as SOCS proteins (Shuai and Liu, 2003).
Gene transcription is a complex process coordinated by numerous transcription factors and co-regulators. A recent high-lytime resolved transcriptional analysis ofthe LPS-induced gene program also concluded that genes could be classified on the basis of inflammatorytranscription factor binding strength, motif content, and nucleosome profile (Tong et al., 2016). We find that in this context, IFNB1 is a member of a specific class among LPS-induced genes that exhibits early transcription factor (TF) binding, strong motif strength and dynamic nucleosome alterations, while TNF is a member of a separate class with a markedly different profile. Thus, association of unique TFs and co-regulators in the induction of the two gene classes could mediate the opposing consequences of IFIT1 depletion for the inflammatory and IFN gene classes.
While the role of type IIFN has been extensively characterized in the context of viral infection, the role for this immune mediator in the host response to bacterial infections is less clear. Since IFNB1 is strongly induced by LPS activation of the TRIF pathway during Gram-negative infection, it seems unlikely that there is not a host benefit for the LPS-induced IFN program. The effect of IFIT1 depletion, in favoring the transcriptional balance toward higher inflammatory gene transcription and lower IFN induction, allowed us to test the consequences of this bias in the context of infection with the bacterium B. cenocepacia, which escapes the macrophage endolysosomal pathway to replicate in the cytosol (Al-Khodor et al., 2014; Ganesan and Sajjan, 2012; Vergunst et al., 2010). We find that B. cenocepacia intracellular replication levels were substantially higher in IFIT1-depleted cells, suggesting that modulation of gene expression by IFIT1 is critical in maintaining the transcriptional balance for host cell resistance to infection by pathogens. Moreover, this increased replication could be reversed with addition of recombinant IFN-β, suggesting a likely role for IFN in limiting bacterial evasion of the endolysosomal pathway.
In summary, through an unbiased genome-wide siRNA screen, we have identified a role for IFIT1 in reciprocal regulation of the balance between the inflammatory and antiviral gene programs in LPS-activated macrophages. While the antiviral functions of IFIT1 through cellular regulation of protein synthesis and immune activation through mitochondrial antiviral-signaling protein (MAVS) have been extensively reported (Li et al., 2009), a role for IFIT1 in regulating the response to bacteria has not been demonstrated, despite its strong induction by bacterial stimuli (Nau et al., 2003). We show that a previously unrecognized nuclear pool of IFIT1 stabilizes the interaction of HDAC2 with the Sin3A-corepressor-associated protein SAP25, promoting efficient removal of the Sin3A corepressor and consequent IRF3 recruitment at ISG loci. We propose that IFIT1 regulates an important balance between inflammatory and IFN gene programs to facilitate an optimal innate immune transcriptional response to microbial challenge.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Iain D.C. Fraser (fraseri@niaid.nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and stimulation
Human cells
THP1 monocytes (ATCC), U937 cells (ATCC), THP1 B5 reporter cells (Li etal., 2015) and Human blood derived monocytes were propagated in RPMI media with 10% FBS, 10 mM HEPES, and Betamercaptoethanol in 5% CO2 at 37°C. Human peripheral blood monocyte samples from screened, healthy donors were obtained under the NIH Clinical Center IRB-approved protocol 99-CC-0168 from the NIH Department of Transfusion Medicine. THP1 cells and were differentiated into a macrophage-like state with 10 ng/ml PMA (Sigma) for three days. Human primary monocytes were differentiated with 10 ng/ml GM-CSF (R&D) for 7 days. HEK293T cells were propagated in DMEM media with 10% FBS.
Mouse cells
Mice
All mice were bred and maintained under pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care accredited animal facility at the NIAID and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by the NIAID Animal Care and Use Committee. Ifit1−/− mice were a gift from Dr. Michael Diamond (Szretter et al., 2012). All mice used were between 6 and 13 weeks of age. Mouse BMDM were prepared by differentiation from bone marrow for 6 days in DMEM, 10%FBS, 20mM HEPES, 100U/ml Penicillin-Streptomycin solution (CellGrow) and 2 mM glutamine containing 60 ng/ml M-CSF (R&D).
TLR ligands
LPS derived from Salmonella was from ENZO Life Sciences, Minnesota R595 TLRgrade, ALX-581-008-L002; poly I:C was from Enzo Life Sciences, ALX-746-021-M005, poly dAdT was from Invivogen, tlrn-patn. Cells were plated and differentiated in 24-well plate for ELISA, qPCR and microarray experiments. Ligands were diluted in culture media and added onto differentiated cells at the respective time points.
Viral and bacterial infection of cells
Sendai virus (Cantell strain, obtained from Dr. Kathryn Zoon, (Bedsaul etal., 2016)) and Influenza A virus (Texas/36/91 strain, obtained from Dr. Ronald Germain, (Brandes et al., 2013)) were diluted in RPMI media and added onto 3×105 THP1 cells differentiated for three days with PMA in 1 mL volume at an MOI of 5 and infected for the indicated time period in a 24-well plate. Media was removed after the indicated time, cells were washed with PBS and lyzed in 350 μL RLT buffer (QIAGEN) for isolating RNA. Infection of THP1 cells with B. cenocepacia at an MOI of 1 was conducted using the infection and imaging methods previously described (Al-Khodor et al., 2014; Miller et al., 2015). Recombinant IFN-β was obtained from R&D systems (Cat# 11415-1) and used at a final concentration of 3000 IU/ml.
METHOD DETAILS
siRNA knockdown
THP1 cells (3×105 cells) were cultured and differentiated in 24-well plates, and siRNA transfection was performed using Hiperfect (QIAGEN) reagent for human THP1 cells and RNAi MAX (Thermofisher) for mouse BMDMs with 50 nM siRNA, following a previously established protocol (Li et al., 2015). Human primary macrophages were electroporated with 1 μg siRNA per 7×105 cells using the VPA-1008 Amaxa electroporation kit (Lonza) following the manufacturer’s recommended protocol. The following siRNA were used; human HDAC2: Dharmacon, D-003495-05, mouse Ifit3: Dharmacon, D-040963-03, human IFIT1: Ambion, Cat# s7150 (siRNA1), s7151 (siRNA2), s7152 (siRNA3); non-targeting (NT) siRNA: Dharmacon, Cat# D-001210-05-05, human TLR4: Dharmacon M-008088-01-0003.
Dual luciferase assay of THP1 B5 human macrophage cells
The human THP1 B5 cell clone (Li etal., 2015) was differentiated into a macrophage-like state, transfected with siRNA and stimulated with LPS as described above. Firefly and renilla luciferase activity in the cell lysates was determined using the Dual-Luciferase Reporter Assay System (Promega, E1960), following the manufacturers protocol, and the ratio of firefly luminescence to renilla luminescence was used to reflect the cell response to LPS stimulation.
Quantitative PCR
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN). cDNA was reverse transcribed from 1 μg RNA using the Biorad Reverse Transcription kit. For qPCR, 200 ng RNA equivalent of cDNA was used per reaction with gene specific primers and FAM conjugated probes (IDT DNA) and qPCR Solaris mix (Dharmacon/Life Technologies). PCR reactions were performed in an Eppendorf PCR thermocycler with the following thermal cycles 95°C for 15 min, (95°C for 15 s, 60°C for 60 s) × 40 cycles. The Ct values were analyzed with Eppendorf Realplex 1.5 software.
ELISA
THP1 cells or differentiated primary macrophage cells were cultured in 24-well plates at a density of 3×105 cells/well. After ligand treatment, the supernatants were collected and the concentration of TNF-α or IFN-β was quantitated by ELISA. ELISA kits were from BD bioscience OptEIA (TNF-α) and PBL assay science (IFN-β).
Generation of IFIT1 shRNA-expressing THP1 cell line
shRNAs targeting human IFIT1 were designed using the Hannon Lab online tool (http://cancan.cshl.edu/RNAi_central/RNAi.cgi?type=shRNA) and cloned into the pEN_miRc2 vector ((Shin et al., 2006)). The knockdown efficiency was tested in 293T cells by transient co-expression of shRNAs with an YFP-IFIT1 fusion protein. The two most efficient shRNAs (Figure S2A, targeting the following sequences in human IFIT1; shRNA#1; 5′-CCAGACAATGGATATATTAAG-3′ and shRNA#4; 5′-GCCCTGAAGCTTCAGGATGAA-3) were subcloned to the pDS_FBneo plasmid and production of retrovirus and generation of stable cell lines were carried out as described previously (Zhu et al., 2007). Stable knockdown of IFIT1 was confirmed using qPCR (Figure S2B), and cells expressing shRNA#4 were used in subsequent experiments.
Generation of IFIT1-expressing cell lines
HA-tagged IFIT1 was amplified from human cDNA utilizing the primers:
HA-IFIT1-L:GTGTGGTGGAATTCTGCAGATACCATGGGATCCACCGGTTACCCATACGATGTTCCAGATACGCTCTCGAGATGCCTGATTTAGAAAACAGAGTCTTGG and
IFIT1-R:CCAGAGGTTGATTGTCGAGCGGCCGCCACTGTGCTGGATCTAAGGACCTTGTCTCACAGAGTTCTCAAAG and was cloned into the pLenti-Puro DEST gateway vector after gateway cassette excision by EcoRV followed by Gibson Assembly. The c-Myc unipartate NLS sequence was introduced through PCR amplification of HA-IFIT1 constructs with the primers:
NLS-HA-F:ACCGACTCTAGTCCAGTGTGGTGGAATTCTGCAGATACCATGGGATCCCCTGCAGCAAAACGTGTGAAACTCGACACCGGTTACCCATACGATGTTCCAG and
IFIT1-R:CCAGAGGTTGATTGTCGAGCGGCCGCCACTGTGCTGGATCTAAGGACCTTGTCTCACAGAGTTCTCAAAG followed by Gibson Assembly into the EcoRV sites of the pLenti-Puro DEST Gateway vector. THP1 cells stably expressing IFIT1 N-terminally tagged with FLAG were created using retroviral transduction as previously described (Zhu et al., 2007).
Lentivirus was produced by plating 3×105 293F/T cells/well in 6-well plates which were then transfected with an 8:4:1:1 ratio of Lenti Target Construct:pDeltaR(8.91):VSV-G:RSV-Rev. Cells were allowed to produce virus for 2 days in complete medium containing 2 mM Butyrate. Supernatants were collected, pooled, and concentrated with LentiX Concentrator (Takara) according to manufacturer protocols. Viral pellets were then suspended in 500 μL of complete medium and 100 μL was added to 105 freshly plated log-phase U937 or 293F/T cells while the rest was stored at −80°C. At day 2 post-transduction, we replaced the viral medium with selection medium containing 1 μg/ml Puromycin and cultured for two weeks prior to assay.
Microarray analysis
Wild-type THP1 and IFIT1 shRNA cells (3.5×105 per well) were transfected with NT siRNAand IFIT1 siRNA respectively as described above in 24-well plates, and differentiated with 10 ng/ml PMAfor 72 hours. Cells were stimulated with 100 ng/ml LPS for 0h, 1h, 2h, 4h, 6h and 24h. RNA was isolated with an RNeasy Mini Kit (QIAGEN) following the manufacturer’s protocol and eluted in RNase free TE buffer. Each condition was represented by two biological replicates. cRNA amplification and labeling were performed using the Illumina TotalPrep RNA Amplification Kit (Ambion), microarray hybridization and scanning protocols followed standard Illumina protocols. Signal data was extracted from the image files with the Gene Expression module (v. 1.9.0) of the GenomeStudio software (v. 2011.1), and Log2 signal intensity were determined. 925 genes were identified which showed a log2 change of > 1 from the untreated condition in at least 1 of the 5 treatment time points in WT THP1, or genes in which the sum of the log2 changes across all 5 time points in WT THP1 exceeded 3 (Data S1). To identify the effect of IFIT1 perturbation on this gene set, the difference in log2 signal change in the IFIT1 KD cells was summed across all time points (Data S1; ‘KD-WT SUM’). The microarray data from this study has been deposited with the Gene Expression Omnibus under GSE103829.
Pathway analysis
Genes which showed increased expression (106 genes; Data S2; Figures 2A and 2B ‘Group 1’) and decreased expression (185 genes; Data S3; Figures 2A and 2C ‘Group 2’) in IFIT1 depleted cells were subjected to Ingenuity Pathway Analysis (QIAGEN). Core analysis was carried out using the Ingenuity Knowledge based to identify network relationships within each gene group and enriched canonical pathways.
Western blotting
THP1 cells were lyzed in 1X SDS-PAGE loading buffer with protease and phosphatase inhibitor cocktails (Roche). Samples were resolved on a 4%−20% gradient SDS-PAGE gel. Following transfer of the proteins, the nitrocellulose membrane was blocked in 5% milk for 1h and probed with the following antibodies overnight at 4°C: goat anti-IFIT1 (Santa Cruz, 82946), Rabbit anti-phos-pho-p38 (Cell Signaling, 4511S), Rabbit anti-phospho-p65 (Cell Signaling, 3033S), Rabbit anti-p105 (Santa Cruz, sc293141), Mouse anti-GAPDH (Cell Signaling, 97166), Rabbit anti-hnRNPL (Santa Cruz, sc-32317), Rabbit anti-phospho-IRF3 (Abcam, ab76493), Rabbit anti-phospho-JNK (Cell Signaling, 9255), Rabbit anti-phospho-ERK (Cell Signaling, 4370), Rabbit anti-phospho-STAT1 (Cell Signaling, 9167S). Western blots were incubated with respective HRP-conjugated secondary antibodies and visualized using ECL reagents (Pierce).
Nuclear and cytoplasmic fractionation
Nuclear and cytosolic fractions were isolated from cells using NE-PER nuclear and cytosolic fractionation kit from Thermo Fisher (Cat# 78833). Control and IFIT1 shRNA-expressing THP1 cells (7×106 cells per sample) were PMA differentiated for three days and stimulated with 100 ng/ml LPS. For immunoprecipitation, 7×106 HEK293T cells expressing HA-IFIT1 and HA-NLS-IFIT1 were used for nuclear fractionation. Fractionation was conducted following the manufacturer’s protocol.
Immunoprecipitation
THP1 cells stably expressing SAP25 N-terminally tagged with mCherry were created using retroviral transduction as previously described (Zhu et al., 2007). After siRNA transfection and TLR ligand treatment, cells were lyzed in 1 mL CHAPSO buffer (50 mM Tris.HCL pH7.4, 150 mM NaCl, 0.5% CHAPSO (Sigma), Protease and Phosphatase inhibitors (Roche)). Lysates were passed through a 26G needle 5 times to degrade condensed chromatin and centrifuged at 10,000 g for 10 min to remove cell debris. Lysates were precleared with Protein A Dynabeads (Life Technologies) for 1 hr at 4°C. Anti-mCherry antibody (Clontech) 10 μl was used to bind Dyna Beads in CHAPSO buffer for 1h at RT. Beads were then washed with CHAPSO buffer and incubated with the lysate overnight. HEK293T cells stably expressing HA-IFIT1 and HA-NLS-IFIT1 were created using lentiviral transduction as described above. Nuclear fractions were diluted 5 times in CHAPSO buffer before incubating with FLAG M2 dynabeads (Sigma). After incubation, beads were washed 4 times in CHAPSO buffer and suspended in 30 μl elution buffer (50 mM Glycine pH 2.5) for 10 minutes at RT, then mixed with 30 μl of 2XSDS-PAGE loading buffer with BME and protease/phosphatase inhibitors. Beads were then heated at 95°C for 10 minutes and the eluted protein solution was separated from the Dyna Beads with a magnet. 10 μL of the eluted sample was loaded onto an SDS-PAGE gel for western blotting.
Confocal imaging
THP1 cells, HEK293T and U937 cells were cultured in 96-well plates at a density of 2×104 cells/well. THP1 and U937 cells were differentiated with PMA for three days. Where indicated, Leptomycin B (Abcam, cat# ab120501) was added to cells at a concentration of 1.25 ng/μl for 3h. Cells were fixed with 4% formaldehyde and blocked with 4% BSA for 1 hr. Cells were then incubated with an anti-IFIT1 antibody (Novus, cat# NBP2-33751), anti-FLAG antibody (Sigma, cat# F7425), or anti-HA antibody (Abcam, cat# ab130275) overnight at 4° Cat 1:500 dilution. Cells were washed in PBST three times and probed with secondary (anti-rabbit or anti-mouse) antibody coupled with Alexa 488 flourophor (Cell Signaling, Cat# 4412) for 1 hr at 1:1000 dilution, followed by washing with PBST and nuclear staining with Hoechst 33342 (Sigma, cat# 14533). Images were acquired on a Leica SP5 confocal microscope equipped with a 63X/1.4NA oil objective, an Argon 488nm laser and a 405nm diode laser, as well as HyD detectors. Nine to ten fields of view were collected for each sample with a voxel size of approximately 130nm. The fields collected encompassed more than 70 cells. Nuclear co-localization of FLAG, HA and IFIT1 was calculated based on the fluorescent intensity in the nucleus and cytoplasm using the software Imaris (Bitplane). Specificity of the IFIT1 antibody was confirmed by a lack of signal in cells transfected with IFIT1 siRNA.
Motif analysis
The gene group showing decreased expression in IFIT1 depleted cells (185 genes; Data S3; Figures 2A and 2C ‘Group 2’) was analyzed for transcriptional regulatory elements using DiRE (Gotea and Ovcharenko, 2008). Analysis was performed using default DiRE settings with a background gene set of 1280 LPS-induced genes from the microarray dataset.
Chromatin immunoprecipitation-PCR (ChIP-PCR)
WT or IFIT1 shRNA-expressing (shIFIT1) THP1 cells were differentiated for three days with PMA. After LPS stimulation, cells were washed in PBS and lysed in 1 mL Lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-HCl pH 8.1 and protease inhibitors (Sigma)). Lysate was then sonicated three times for 30 s (with 30 s breaks) at 4°C and centrifuged at 10,000×g for 10 min at 4°C to remove cell debris. A fraction of the lysate was used to quantitate chromatin DNA. Lysates were diluted in Dilution Buffer to 250ug chromatin/ml (0.01% SDS, 1.1% Triton x-100, 1.2mM EDTA, 16.7mM Tris-HCl pH 8.1, 167mM NaCl 1 and protease inhibitors). Lysates were precleared with Protein A Dynabeads (Life Technologies) for 1 hr at 4°C. 10 μL of antibodies, anti-Sin3A and anti-IRF3 (Santa Cruz, sc-136318X; sc-33641X) were used to bind Dyna Beads in Dilution Buffer for 1h at RT. Beads were then washed three times with 1 mL Dilution Buffer and incubated with the lysate overnight at 4°C. After incubation, beads were washed 2 times each in, 500 μL Low Salt Buffer (0.1% SDS, 1% Triton x-100, 2mM EDTA, 20mM Tris-HCl pH 8, 150mM NaCl), High Salt Buffer (0.1% SDS, 1% Triton x-100, 2mM EDTA, 20mM Tris-HCl pH 8.1, 500mM NaCl), LiCl Buffer (0.25M LiCl, 1% NP-40,1% deoxycholate, 1mM EDTA, 10mM Tris-HCl pH 8.1), and 1X TE Buffer (10mM Tris-HCl, 1mM EDTA, pH 8). The beads were then suspended in 400μl elution buffer (1% SDS, 10mM Tris-HCL pH 8., EDTA 5 mM, 300 mM NaCl) and incubated with 20 U Proteinase K at 65°C overnight. For normalization, 50 μL input sample was mixed with 350 μL elution buffer. DNA was extracted from both input sample and ChIP elution sample with Phenol:Chloroform:Amyl alcohol (Life Technology) followed by ethanol precipitation. PCR primers were designed based on known promoter sequences and/or Sin3A binding sites (https://epd.vital-it.ch/human/human_database.php and http://www.ag-rehli.de/NGSdata.htm). qPCR reactions on the precipitated DNA was performed with Sybr green dye and the following primers: IFNB1 forward primer (TCCCAGGAACTCAATGAAGG), IFNB1 reverse primer (GTGTCGCAATGGAGTGTTGT), IRF7 forward (TTTTTTGAGACTGAGTCTTGCTCT), IRF7 reverse (CCGCCTGTTCTTATTATTGGAT), IRF8 forward primer (ATTTCTCGGAAAGCAGAGCA), IRF8 reverse primer (GCCCACTGTGCCTACCTG), TNF forward primer (ACACACAAATCAGTCAGTGG), TNF reverse primrer (CTTCTGTCTCGGTTTCTTCTC), CCL3 forward primer (TTATAGCAGCTGAGGAAGCAGAA), CCL3 reverse primer (TCTCTAACTCTCAGCTCTCAACTCAT).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Prism software version 7.0 for Mac OS X (GraphPad Software). For PCR, infection and ELISA experiments, relative values were compared either by a paired t test or by two-way ANOVA followed by Sidak’s multiple comparison test. All the statistical details of experiments, including the statistical tests used, are listed within each figure legend. Data presented are representative of three or more independent experiments, unless otherwise stated in the figure legend, and are expressed as mean ± SD *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is Gene Expression Omnibus (GEO): GSE103829.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-IFIT1 | Santa Cruz | Cat# 82946; RRID: AB_2121945 |
| anti-phospho-p38 | Cell Signaling | Cat# 4511S; RRID: AB_2139682 |
| anti-phospho-p65 | Cell Signaling | Cat# 3033S; RRID: AB_331284 |
| anti-p105 | Santa Cruz | Cat# sc293141 |
| anti-GAPDH | Cell Signaling | Cat# 97166 |
| anti-hnRNPL | Santa Cruz | Cat# sc-32317; RRID: AB_627736 |
| anti-phospho-IRF3 | Abcam | Cat# ab76493; RRID: AB_1523836 |
| anti-phospho-JNK | Cell Signaling | Cat# 9255; RRID: AB_2307321 |
| anti-phospho-ERK | Cell Signaling | Cat# 4370; RRID: AB_2315112 |
| anti-phospho-STAT1 | Cell Signaling | Cat# 9167S; RRID: AB_561284 |
| anti-IFIT1 antibody | Novus | Cat# NBP2-33751 |
| anti-FLAG antibody | Sigma | Cat# F7425; RRID: AB_439687 |
| anti-HA antibody | Abcam | Cat# ab130275; RRID: AB_11156884 |
| Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor®488 Conjugate) | Cell Signaling | Cat# 4412; RRID: AB_1904025 |
| Anti-Mouse IgG (H+L), F(ab’)2 Fragment (Alexa Fluor®488 Conjugate) | Cell Signaling | Cat# 4408; RRID: AB_10694704 |
| anti-Sin3A | Santa Cruz | sc-136318X |
| anti-IRF3 | Santa Cruz | sc-33641X |
| Bacterial and Virus Strains | ||
| Sendai Virus (Cantell) | Gift from Dr. Kathryn Zoon, NIAID, (Bedsaul et al., 2016) | N/A |
| Influenza A (Texas/36/91) | Gift from Dr. Ronald Germain, NIAID, (Brandes et al., 2013) | N/A |
| Burkholderia cenocepacia (J2315) | Gift from Dr. David Greenberg (Al-Khodor et al., 2014) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PMA | Sigma | Cat# P8139-5MG |
| GM-CSF | R & D Systems | Cat# 215-GM-010 |
| M-CSF | R & D Systems | Cat# 416-ML-010 |
| LPS from Salmonella minnesota R595 (Re) (TLRgrade) | ENZO Life Sciences | Cat# ALX-581-008-L002 |
| Poly IC | ENZO Life Sciences | Cat# ALX-746-021-M005 |
| Poly(dA:dT) | InvivoGen | Cat# tlrl-patn |
| Leptomycin B | Abcam | Cat# ab120501 |
| Hoechst 33342 | Sigma | Cat# 14533 |
| Recombinant IFN-β | R & D systems | Cat# 11415-1 |
| Deposited Data | ||
| Microarray Data (Gene Expresion Omnibus under) | This Paper | GSE103829 |
| Experimental Models: Cell Lines | ||
| U937 cells | ATCC | Cat# CRL-1593.2 |
| THP1 cells | ATCC | Cat# TIB-202 |
| HEK293T cells | ATCC | Cat# CRL-3216 |
| THP1 B5 (TNF reporter) cells | (Li etal., 2015) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wild type and Ifit1 —/— C57BL/6 mice | Michael Diamond, Washington University, (Szretter et al., 2012) | N/A |
| Oligonucleotides | ||
| IFNB1 F/R primers TCCCAGGAACTCAATGAAGG/ GTGTCGCA ATGGAGTGTTGT |
IDT DNA | N/A |
|
IRF7 F/R primers TTTTTTGAGACTGAGTCTTGCTCT/ CCGCCT GTTCTTATTATTGGAT |
IDT DNA | N/A |
| IRF8 F/R primers ATTTCTCGGAAAGCAGAGCA/ GCCCACTGTGC CTACCTG |
IDT DNA | N/A |
| TNF F/R primers ACACACAAATCAGTCAGTGG/ CTTCTGTCTCGG TTTCTTCTC |
IDT DNA | N/A |
| CCL3 F/R primers TTATAGCAGCTGAGGAAGCAGAA/ TCTCTAA CTCTCAGCTCTCAACTCAT |
IDT DNA | N/A |
| IFIT1 shRNA#1 CCAGACAATGGATATATTAAG | IDT DNA | N/A |
| IFIT1 shRNA#4 GCCCTGAAGCTTCAGGATGAA | IDT DNA | N/A |
| HA IFIT1 F primer GTGTGGTGGAATTCTGCAGATACCATGGGAT CCACCGGTTACCCATACGATGTTCCAGATACGCTCTCGAGATG CCTGATTTAGAAAACAGAGTCTTGG |
IDT DNA | N/A |
| HA IFIT1 reverse primer CCAGAGGTTGATTGTCGAGCGGCCGCC ACTGTGCTGGATCTAAGGACCTTGTCTCACAGAGTTCTCAAAG |
IDT DNA | N/A |
| NLS-HA-F Primer ACCGACTCTAGTCCAGTGTGGTGGAATTCTGC AGATACCATGGGATCCCCTGCAGCAAAACGTGTGAAACTCGAC ACCGGTTACCCATACGATGTTCCAG |
IDT DNA | N/A |
| IFIT1-R primer CCAGAGGTTGATTGTCGAGCGGCCGCCACTGTGC TGGATCTAAGGACCTTGTCTCACAGAGTTCTCAAAG |
IDT DNA | N/A |
| Software and Algorithms | ||
| Statistics: Prism | Graphpad | N/A |
| Other | ||
| HDAC2 siRNA | Dharmacon | D-003495-05 |
| Ifit3 siRNA | Dharmacon | D-040963-03 |
| IFIT1 siRNA1 | Ambion | s7150 |
| IFIT1 siRNA2 | Ambion | s7151 |
| IFIT1 siRNA3 | Ambion | s7152 |
| Non-targeting (NT) siRNA | Dharmacon | D-001210-05-05 |
| TLR4 siRNA | Dharmacon | M-008088-01-0003 |
Highlights.
An siRNA screen identifies IFIT1 as a negative regulator of pro-inflammatory genes
IFIT1 also positively regulates the induction of interferon-stimulated genes (ISGs)
Nuclear IFIT1 facilitates both Sin3A removal and IRF3 recruitment to ISG loci
A defective IFIT1-dependent interferon response increases susceptibility to bacteria
ACKNOWLEDGMENTS
We thank Timothy Myers and the team at the Genomic Technologies Section of the Research Technology branch of NIAID for running and analyzing the microarray experiments and Dr. Mike Diamond of Washington University for providing the Ifit1−/− mice. Sendai virus (Cantell strain) was a generous gift from Dr. Kathryn Zoon. We thank the NIH Department of Transfusion Medicine for providing human blood-derived monocytes and colleagues in the Laboratory of Systems Biology for helpful discussions and critical reading of the manuscript. This work was generously supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases. B.C. was supported through the NIH Community College Program and Special Projects of the Office of Intramural Training and Education at the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three data files and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.002.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akira S, Uematsu S, and Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 724, 783–801. [DOI] [PubMed] [Google Scholar]
- Al-Khodor S, Marshall-Batty K, Nair V, Ding L, Greenberg DE, and Fraser ID (2014). Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell. Microbiol 16, 378–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. (2009). Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aregger M, Hart T, and Moffat J (2015). Extensive mapping of an innate immune network with CRISPR. Mol. Syst. Biol 11, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assani K, Tazi MF, Amer AO, and Kopp BT (2014). IFN-γ stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS ONE 9, e96681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, and Hellman J (2007). MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol 178, 1164–1171. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay SK, Leonard GT Jr., Bandyopadhyay T, Stark GR, and Sen GC (1995). Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem 270, 19624–19629. [DOI] [PubMed] [Google Scholar]
- Bansal N, Kadamb R, Mittal S, Vig L, Sharma R, Dwarakanath BS, and Saluja D (2011).Tumor suppressor protein p53 recruits human Sin3B/HDAC1 complex for down-regulation of its target promoters in response to genotoxic stress. PLoS ONE 6, e26156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedsaul JR, Zaritsky LA, and Zoon KC (2016). Type I interferon-mediated induction of antiviral genes and proteins fails to protect cells from the cytopathic effects of Sendai virus infection. J. Interferon Cytokine Res 36, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes M, Klauschen F, Kuchen S, and Germain RN (2013). A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttar NS, DeMars CJ, Lomberk G, Rizvi S, Bonilla-Velez J, Achra S, Rashtak S, Wang KK, Fernandez-Zapico ME, and Urrutia R (2010). Distinct role of Kruppel-like factor 11 in the regulation of prostaglandin E2 biosynthesis. J. Biol. Chem 285, 11433–11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantaert T, Baeten D, Tak PP, and van Baarsen LG (2010). Type I IFN and TNFα cross-regulation in immune-mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res. Ther 12, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, and Natoli G (2012). Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 109, E2865–E2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, et al. (2011). Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 147, 853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia J, Goh G, Racine V, Ng S, Kumar P, and Bard F (2012). RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Mol. Syst. Biol 8, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchacovich R, Espinoza CG, Virk Z, and Espinoza LR (2008). Biologic therapy (TNF-alpha antagonists)-induced psoriasis: a cytokine imbalance between TNF-alpha and IFN-alpha? J. Clin. Rheumatol 14, 353–356. [DOI] [PubMed] [Google Scholar]
- Das TK, Sangodkar J, Negre N, Narla G, and Cagan RL (2013). Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 32, 3184–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Schaller AM, Geballe AP, and Malik HS (2016). Evolution-guided functional analyses reveal diverse antiviral specificities encoded by IFIT1 genes in mammals. eLife 5, e14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, and Farzan M (2013). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol 13, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V, and Sen GC (2011). The ISG56/IFIT1 gene family. J. Interferon Cytokine Res 31, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V, and Sen GC (2015). Interferon-induced Ifit proteins: their role in viral pathogenesis. J. Virol 89, 2462–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Ganesan S, and Sajjan US (2012). Host evasion by Burkholderia cenocepacia. Front. Cell. Infect. Microbiol 1, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay NJ, and Gangloff M (2007). Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem 76, 141–165. [DOI] [PubMed] [Google Scholar]
- Gotea V, and Ovcharenko I (2008). DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res 36, W133–W139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes JA, Nielsen SJ, Battaglioli E, Miska EA, Speh JC, Berry DL, Atouf F, Holdener BC, Mandel G, and Kouzarides T (2000). The corepressor mSin3A is a functional component of the REST-CoREST repressor complex. J. Biol. Chem 275, 9461–9467. [DOI] [PubMed] [Google Scholar]
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, and Tschopp J (2011). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. [DOI] [PubMed] [Google Scholar]
- Guo J, Hui DJ, Merrick WC, and Sen GC (2000). A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J 19, 6891–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, and Taniguchi T (2004). Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 101, 15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Meng X, Huang C, Shen C, and Li J (2017). Frontline Science: ATF3 is responsible for the inhibition of TNF-α release and the impaired migration of acute ethanol-exposed monocytes and macrophages. J. Leukoc. Biol. J. Leukoc. Biol 101, 633–642. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Negishi H, and Taniguchi T (2013). The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol 78, 105–116. [DOI] [PubMed] [Google Scholar]
- Kadamb R, Mittal S, Bansal N, Batra H, and Saluja D (2013). Sin3: insight into its transcription regulatory functions. Eur. J. Cell Biol 92, 237–246. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, et al. (2004). Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol 5, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, and Yoshida M (1998). Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res 242, 540–547. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, et al. (2013). IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, and Shu HB (2009). ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 106, 7945–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Wong JJ, Sum C, Sin WX, Ng KQ, Koh MB, and Chin KC (2011). IRF8 and IRF3 cooperatively regulate rapid interferon-β induction in human blood monocytes. Blood 117, 2847–2854. [DOI] [PubMed] [Google Scholar]
- Li N, Sun J, Benet ZL, Wang Z, Al-Khodor S, John SP, Lin B, Sung MH, and Fraser ID (2015). Development of a cell system for siRNA screening of pathogen responses in human and mouse macrophages. Sci. Rep 5, 9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhu Y, Yong WK, Sze NS, Tan NS, and Ding JL (2015). Cutting Edge: Synchronization of IRF1, JunB, and C/EBPβ activities during TLR3-TLR7 cross-talk orchestrates timely cytokine synergy in the proinflammatory response. J. Immunol 195, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman RJ, Iyer A, Fairlie TJ, Cotterell A, Gupta P, Reid RC, Vesey DA, Sweet MJ, and Fairlie DP (2016). Differential Anti-inflammatory activity of HDAC inhibitors in human macrophages and rat arthritis. J. Pharmacol. Exp. Ther 356, 387–396. [DOI] [PubMed] [Google Scholar]
- Miller AH, Vayttaden SJ, Al-Khodor S, and Fraser ID (2015). Assay development for image-based quantification of intracellular bacterial replication and analysis of the innate immune response to infection. Assay Drug Dev. Technol 13, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz IM, MacArtney T, Sanchez-Pulido L, Ponting CP, Rocha S, and Rouse J (2012). Family with sequence similarity 60A (FAM60A) protein is a cell cycle-fluctuating regulator of the SIN3-HDAC1 histone deacetylase complex. J. Biol. Chem 287, 32346–32353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, and Lanzavecchia A (2005). Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol 6, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau GJ, Schlesinger A, Richmond JF, and Young RA (2003). Cumulative Toll-like receptor activation in human macrophages treated with whole bacteria. J. Immunol 170, 5203–5209. [DOI] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015). A genomewide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabò A, Amati B, Ostuni R, and Natoli G (2017). Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat. Immunol 18, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Lassnig C, Eberle CA, Gòrna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, et al. (2011). IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol 12, 624–630. [DOI] [PubMed] [Google Scholar]
- Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, Bossuyt X, Boutten A, Bienvenu J, Duquesne A, et al. (2011). A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis 70, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud JM, Kim DY, Atasheva S, Rasalouskaya A, White JP, Diamond MS, Weaver SC, Frolova EI, and Frolov I (2015). IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLoS Pathog 11, e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser H, Bekisz J, and Zoon KC (2014). New function of type I IFN: induction of autophagy. J. Interferon Cytokine Res 34, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Seki E, and Brenner DA (2006). Toll-like receptor signaling in the liver. Gastroenterology 130, 1886–1900. [DOI] [PubMed] [Google Scholar]
- Seki E, and Brenner DA (2008). Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. ; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 110, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiio Y, Rose DW, Aur R, Donohoe S, Aebersold R, and Eisenman RN (2006). Identification and characterization of SAP25, a novel component of the mSin3 corepressor complex. Mol. Cell. Biol 26, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, Rebres R, Roach T, Seaman W, Simon MI, and Fraser ID (2006).A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. USA 103, 13759–13764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, and Liu B (2003). Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol 3, 900–911. [DOI] [PubMed] [Google Scholar]
- Sin WX, Li P, Yeong JP, and Chin KC (2012). Activation and regulation of interferon-β in immune responses. Immunol. Res 53, 25–40. [DOI] [PubMed] [Google Scholar]
- Suet Ting Tan R, Lin B, Liu Q, Tucker-Kellogg L, Ho B, Leung BP, and Ling Ding J (2013). The synergy in cytokine production through MyD88-TRIF pathways is co-ordinated with ERK phosphorylation in macrophages. Immunol. Cell Biol 91, 377–387. [DOI] [PubMed] [Google Scholar]
- Sun J, Li N, Oh KS, Dutta B, Vayttaden SJ, Lin B, Ebert TS, De Nardo D, Davis J, Bagirzadeh R, et al. (2016). Comprehensive RNAi-based screening of human and mouse TLR pathways identifies species-specific preferences in signaling protein use. Sci. Signal 9, ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Katz S, Dutta B, Wang Z, and Fraser ID (2017). Genome-wide siRNA screen of genes regulating the LPS-induced TNF-α response in human macrophages. Sci. Data 4, 170007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M Jr., Virgin HW, Klein RS, Sen GC, and Diamond MS (2012). 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog 8, e1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, Gabriele L, and Ozato K (2007). The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, and Akira S (2003). Toll-like receptors. Annu. Rev. Immunol 21, 335–376. [DOI] [PubMed] [Google Scholar]
- Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, and Smale ST (2016). A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell 165, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oevelen C, Bowman C, Pellegrino J, Asp P, Cheng J, Parisi F, Micsinai M, Kluger Y, Chu A, Blais A, et al. (2010). The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol. Cell. Biol 30, 5686–5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergunst AC, Meijer AH, Renshaw SA, and O’Callaghan D (2010). Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect. Immun 78, 1495–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, and Zhao K (2009). Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang E, and Lu M (2015). Toll-like receptor (TLR)-mediated innate immune responses in the control of hepatitis B virus (HBV) infection. Med. Microbiol. Immunol. (Berl.) 204, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gao X, Cao Y, Jiang H, Wang T, Song X, Guo M, and Zhang N (2015). Selenium deficiency facilitates inflammation through the regulation of TLR4 and TLR4-related signaling pathways in the mice uterus. Inflammation 38, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Zhu X, Santat LA, Chang MS, Liu J, Zavzavadjian JR, Wall EA, Kivork C, Simon MI, and Fraser ID (2007).A versatile approach to multiple gene RNA interference using microRNA-based short hairpin RNAs. BMC Mol. Biol 8, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the microarray data reported in this paper is Gene Expression Omnibus (GEO): GSE103829.