

ABSTRACT
Vascular-targeted photodynamic therapy (VTP) induces rapid destruction of targeted tissues and is a promising therapy for prostate cancer. However, the resulting immune response, which may play an important role in either potentiating or blunting the effects of VTP, is still incompletely understood. Myeloid cells such as myeloid-derived suppressor cells (MDSCs) and macrophages are often found in tumors and are widely reported to be associated with cancer angiogenesis, tissue remodeling, and immunosuppression. These cells are also known to play a critical role in wound-healing, which is induced by rapid tissue destruction. In this study, we investigated the effects of VTP on the recruitment of tumor-infiltrating myeloid cells, specifically MDSCs and tumor-associated macrophages (TAMs), in the Myc-Cap and TRAMP C2 murine prostate cancer models. We report that VTP increased the infiltration of myeloid cells into the tumors, as well as their expression of CSF1R, a receptor required for myeloid differentiation, proliferation, and tumor migration. As anti-CSF1R treatment has previously been used to deplete these cells types in other murine models of prostate cancer, we hypothesized that combining anti-CSF1R with VTP therapy would lead to decreased tumor regrowth and improved survival. Importantly, we found that targeting myeloid cells using anti-CSF1R in combination with VTP therapy decreased the number of tumor MDSCs and TAMs, especially M2 macrophages, as well as increased CD8+ T cell infiltration, decreased tumor growth and improved overall survival. These results suggest that targeting myeloid cells via CSF1R targeting is a promising strategy to potentiate the anti-tumor effects of VTP.
KEYWORDS: Prostate cancer, CSF-1R, immunotherapy, MDSCs, myeloid cells, WST11, vascular-targeted photodynamic therapy
Introduction
Vascular-targeted photodynamic (VTP) therapy using padeliporfin (TOOKAD® Soluble (WST11)) is a promising therapeutic option currently being assessed for prostate cancer (PCa) treatment.1,2 VTP ablates targeted tissues by using WST11 as a photosensitizer which reacts with a near-infrared laser in the presence of oxygen. The photosensitizer is infused intravenously while the targeted prostate zone is illuminated by trans-perineal optical fibers.3 The photosensitizer absorbs light and transfers energy to oxygen molecules creating reactive oxygen species and inducing irreversible endothelial damage which is quickly followed by thrombosis, blood stasis, and vessel occlusion, ultimately leading to tumor necrosis.4–6 Results from multiple phase II clinical trials in prostate cancer patients treated by VTP hemiablation show that a high rate of patients have no detectable tumors years after therapy compared to the untreated population.2,7
Prostate tumors and other subsets of solid tumors have significant infiltration of immunosuppressive myeloid cells which promote tumor progression, metastasis, and therapeutic resistance.8–10 These include myeloid-derived suppressor cells (MDSCs),8 as well tumor-associated macrophages (TAMs), which can differentiate into either pro-inflammatory and anti-tumor M1 macrophages, or into immunosuppressive and pro-tumor M2 macrophages.11–13 Importantly, MDSCs and M2 macrophages are also recruited to sites of wound healing.14,15 In addition, they have been shown to generate a favorable microenvironment for tumors, at steady state and in therapeutic settings, by suppressing the adaptive immune response against cancer cells and by promoting tumor growth through angiogenesis or by producing tumorigenic growth factors.12,15–20 A study in a preclinical mouse model of colon cancer suggests that myeloid cells may be recruited to VTP-treated tumors.21 However, the effects of VTP on myeloid cell infiltration and the subsequent effects of these myeloid cells on tumor eradication require further in-depth investigation. The macrophage colony-stimulating factor (M-CSF or CSF1) has a critical role in the differentiation, proliferation, and migration of myeloid cells to tumors via signaling through the CSF1 receptor (CSF1R).22–24 Studies in preclinical mouse models by our group and others show that blocking CSF1R potentiates the anti-tumor immune response, alone and in the context of immune checkpoint blockade.18,25–31
In this study, we sought to study the effect of myeloid cell modulation in the context of VTP-based treatment. We show that VTP increases the infiltration of immunosuppressive MDSCs and M2 macrophages, as well as their expression of CSF1R in the Myc-Cap and TRAMP C2 prostate cancer preclinical mouse models. We then demonstrate that targeting myeloid cells using anti-CSF1R treatment blocks the VTP-mediated influx of myeloid cells. The reduction in MDSCs correlated with an increase in tumor-infiltrating CD8+ T cells, as well as a decrease in tumor progression and improved survival. We therefore conclude that anti-CSF1R therapy potentiates the effects of VTP therapy.
Results
VTP treatment increases total and CSF1R+ myeloid cell infiltration
To determine whether VTP induced myeloid cell infiltration in tumors, Myc-Cap tumor-bearing mice were treated with VTP and assessed ex-vivo for myeloid cell infiltration of tumor by immunofluorescence (IF). We observe that VTP is associated with an increase in CD11b+ myeloid cells when compared to control tumors, including a trend towards more F4/80+ macrophages (Figure 1(a,b)). Furthermore, when we examined macrophage localization by Iba1 immunohistochemistry (IHC) staining, we found that macrophages were enriched in both the peritumoral and intratumoral areas (Figure 1(c,d)). Finally, we found elevated levels of CSF1R+ cell (Figure 1(a,b)), a receptor typically found on TAMs and MDSCs.32,33 These observations confirmed that myeloid cells were being recruited to Myc-Cap tumor in response to VTP.
Figure 1.
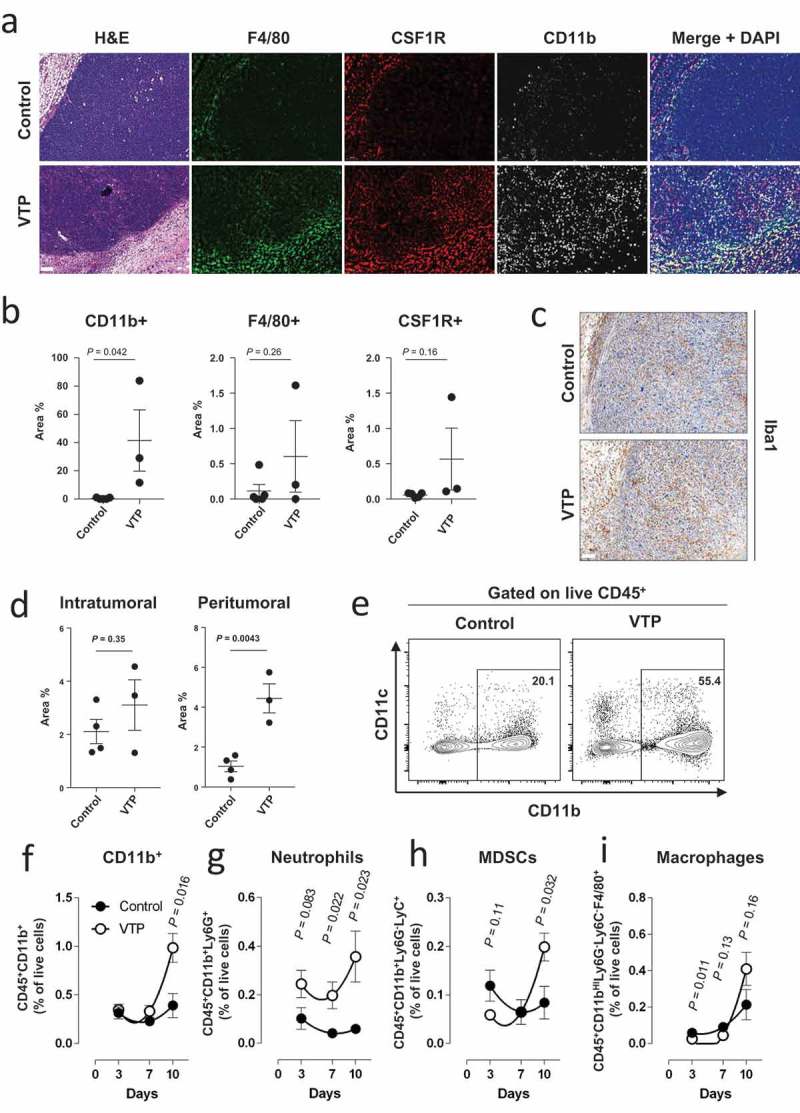
VTP therapy increases myeloid cell infiltration in Myc-Cap tumors.
(a) Representative images of Myc-Cap tumor sections from control or VTP treated tumors 10 days after treatment and stained for CD11b, F4/80 and CSF1R by IF. (b) Quantification of CD11b, F4/80 and CSF1R positive cells as percentage of area covered. (c) Representative tumor section images of macrophage IHC staining for Iba1 positive cells from control or VTP treated Myc-Cap tumors 10 days after treatment, as well as (d) quantification of Iba1 from c). (e) Representative flow cytometry dot plot of the expression of CD11b and CD11c from live CD45+ cells in tumors 10 days after control or VTP therapy. CD11b positive cell gate is indicated. (f) Quantification of CD11b+CD45+ myeloid cells, (g) neutrophils (Ly6C+CD11b+CD45+), (h) MDSCs (Ly6C+Ly6G−CD11b+CD45+) and (i) macrophages (F4/80hiCD11bhiLy6C−Ly6G−CD45+) as a percentage of total live cells over time from Myc-Cap tumors (n = 5 per group) 10 days after control or VTP therapy. All scale bars represent 100 µm. Data shown are a single representative of experiments performed at least 3 times.
Myeloid cell infiltration induced by VTP treatment express markers of immunosuppressive cells
We next wanted to determine whether the influx of CSF1R+ cells in response VTP was associated with immunosuppressive myeloid cells such as TAMs and MDSCs. To do this end, we profiled the myeloid cell populations by flow cytometry (Supplementary Figure 1) at multiple time points (3, 7 and 10 days) after VTP treatment. Consistent with the imaging results, we found a significant increase of CD11b+ myeloid cells in VTP-treated tumors over time as a percentage of total live cells (Figure 1(e,f)). When we examined subsets of CD11b+ cells, we found an increase of neutrophils as early as 3 days after therapy, which was maintained until day 10 (Figure 1(g)). This is likely due to the high level of necrosis induced by VTP, which is known to recruit neutrophils.34 Importantly, we found that CD11b+ myeloid cells expressing Ly6G−Ly6C+, generally considered MDSCs, were found in lower numbers early after treatment, likely as a direct consequence of VTP-mediated cell death, but accumulated subsequently over time compared to control tumors (Figure 1(h)). In addition, the same kinetics were observed for macrophages (Figure 1(i)). Since macrophages can differentiate into either pro-inflammatory M1 macrophages or immunosuppressive M2 macrophages,9 we profiled these macrophages for surface markers of M1 or M2 polarization (Supplementary Figure 1), which we have previously confirmed to be associated with the expression of known immunosuppressive factors.13 We observe that while both the anti-tumor M1 macrophages and the pro-tumor M2 macrophages broadly followed the same kinetics as MDSCs on a per cell basis, there was a consistent skewing toward more M2 macrophages in VTP-treated tumors (Supplementary Figure 2). Finally, we also showed that neutrophils, MDSCs, and macrophages had increased CSF1R expression in VTP-treated tumors compared to control tumor (Supplementary Figure 3). We therefore concluded that VTP therapy leads to an influx of myeloid cells, including neutrophils and immunosuppressive MDSCs and M2 macrophages.
CSF1R blockade enhances VTP treatment efficacy and improves survival in correlation with the number of macrophages and MDSCs in tumors
CSF1R signaling is required for the differentiation and survival of myeloid cells, including MDSCs and macrophages,24 which can be targeted using CSF1R inhibitors.18,25–29 We therefore hypothesized that CSF1R blockade would enhances the anti-tumor effects of VTP by targeting immunosuppressive myeloid cells. To test this, we treated two murine prostate tumor models, Myc-Cap and TRAMP C2, with VTP and anti-CSF1R antibody treatment. As hypothesized, we observed that the combination treatment led to significant tumor reduction and improved survival compared to control or mono-therapies alone in both Myc-Cap (Figure 2(a–c), Supplementary Table 1 and 2) and TRAMP C2 (Figure 2(d–f), Supplementary Table 3 and 4) models. In order to test whether the resulting improved outcome from the combination therapy correlated with a reduction in myeloid cells, these cells were quantified in post-treatment tumors by immunostaining and flow cytometry. We observed that the CSF1R treatment reversed the myeloid cell increase associated with the VTP treatment. In the combination group, there was a notable reduction in CD11b+, F4/80+ and CSF1R+ myeloid cells by IF compared to VTP mono-therapy in Myc-Cap (Figure 3(a,b)) and TRAMP C2 (Supplementary Figure 4) tumors. The reduction of macrophages in the combination therapy group compared to VTP alone was also seen by Iba1 IHC staining in Myc-Cap (Figure 3(c,d)) and TRAMP C2 (Supplementary Figure 5) tumors, as well as by flow cytometry in Myc-Cap tumors (Figure 3(e)). Furthermore, when macrophages were further examined in Myc-Cap tumors for markers of polarization, we observed that anti-CSF1R alone or in combination with VTP led to an equal reduction of M1 and M2 macrophages (Supplementary Figure 6). We also observed a considerable reduction in the number of CD11b+ cells, including neutrophils and MDSCs in the tumor, when mice were given anti-CSF1R antibody alone or in combination with VTP (Figure 3(f–h)). Finally, our group has previously shown that VTP obliterates the tumor vasculature in PC-3 xenografts immediately post-VTP treatment35 and we wanted to determine if combining anti-CSF1R with VTP therapy had any effect on the vasculature at later time points. However, we found that while the vasculature in VTP-treated tumors mostly recovered by day 10 after therapy, combining anti-CSF1R to VTP therapy had modest to little effect, which contrasted with a greater increase in vasculature in the tumors only receiving anti-CSF1R (Supplementary Figure 7).
Figure 2.
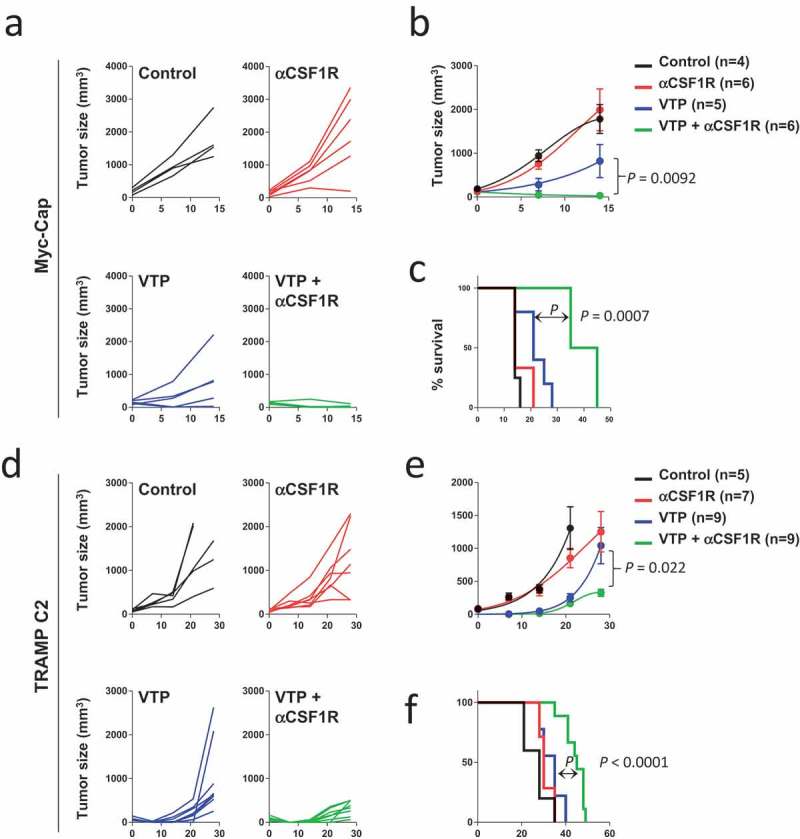
Combination anti-CSF1R and VTP therapy decreases tumor growth and improves survival.
(a) Individual and (b) average Myc-Cap tumor growth in mice treated with control, VTP alone, anti-CSF1R alone or VTP with anti-CSF1R combination therapy. (c) Overall survival of Myc-Cap tumor-bearing mice from all treatment groups. (d) Individual and d) average TRAMP C2 tumor growth in mice treated with control, VTP alone, anti-CSF1R alone or VTP with anti-CSF1R combination therapy. (f) Overall survival of TRAMP C2 tumor-bearing mice from all treatment groups. Statistical differences between tumor growth were measured by performing a t test of the area under the curve. Survival significance was performed by Log–Rank test. Multiple comparison test for A-D between all groups is shown in Supplementary Tables 1–4. Data shown are a single representative of experiments performed at least twice.
Figure 3.
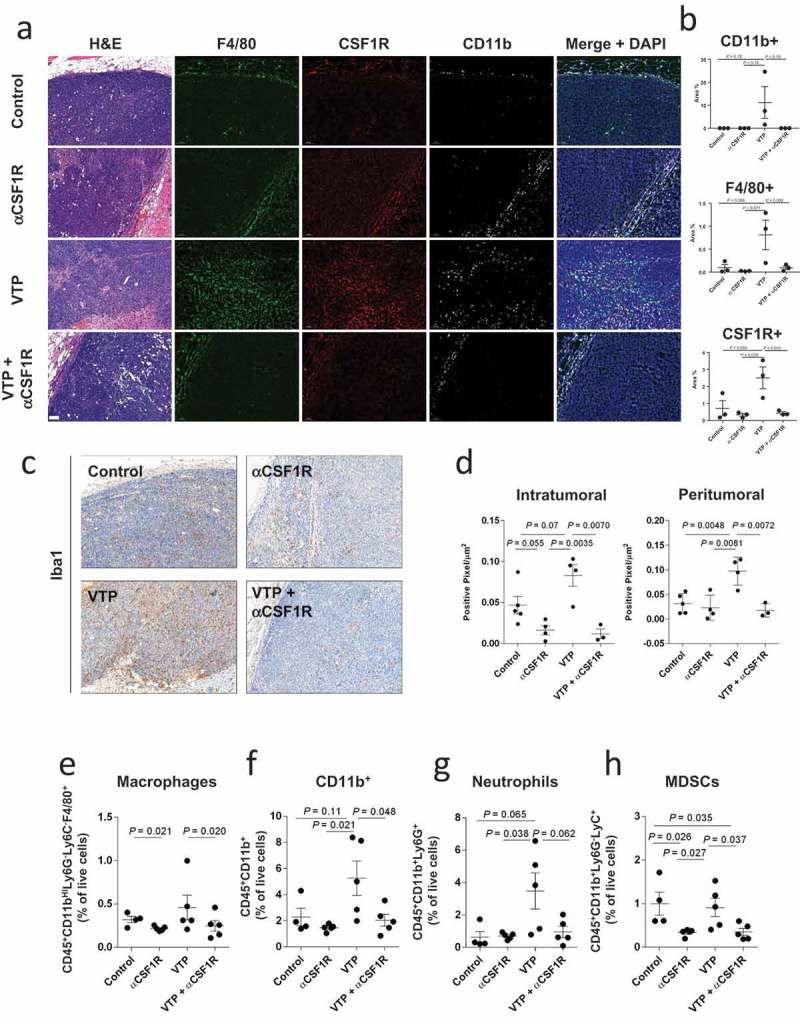
Anti-CSF1R treatment diminishes Myc-Cap tumor myeloid infiltration in control and VTP-treated mice.
Together, our data demonstrate that while the anti-CSF1R therapy alone could deplete macrophages and MDSCs in the Myc-Cap model, a reduction of tumor growth and an increase in overall survival was only seen when anti-CSF1R therapy was combined with VTP treatment.
CSF1R blockade increases CD8+ T cell infiltration in VTP treated tumors
MDSCs and M2 macrophages are known to inhibit T cell tumor infiltration and diminish their effector potential.8,30,31,36 We therefore hypothesized that anti-CSF1R-mediated MDSC and macrophage depletion in VTP-treated tumors would correlate with an increase in infiltrating T cells and effector function. To test this hypothesis, we examined T cell infiltration in Myc-Cap tumors collected 5 and 10 days post-treatment (VTP alone or VTP with anti-CSF1R). We found a significant increase in CD8+ T cells at both time points in tumors treated with the combination therapy compared to tumors treated with VTP alone in the Myc-Cap (Figure 4(a,b) and Supplementary Figure 8a) and TRAMP C2 (Supplementary Figure 8b) models. Increased numbers of CD8+ T cells, but not CD4+ T cells, was further confirmed by flow cytometry (Figure 4(c)). This also correlated with a modest increase in granzyme B expression by CD8+ T cell in tumors from mice treated with the combination of VTP and anti-CSF1R (Figure 4(d,e)). We conclude that when combined with VTP therapy, anti-CSF1R therapy decreases the number of MDSCs and macrophages and is associated with increased CD8+ T cell infiltration and cytotoxic potential, and that this could be a potential mechanism contributing to the diminished tumor growth and improved survival.
Figure 4.
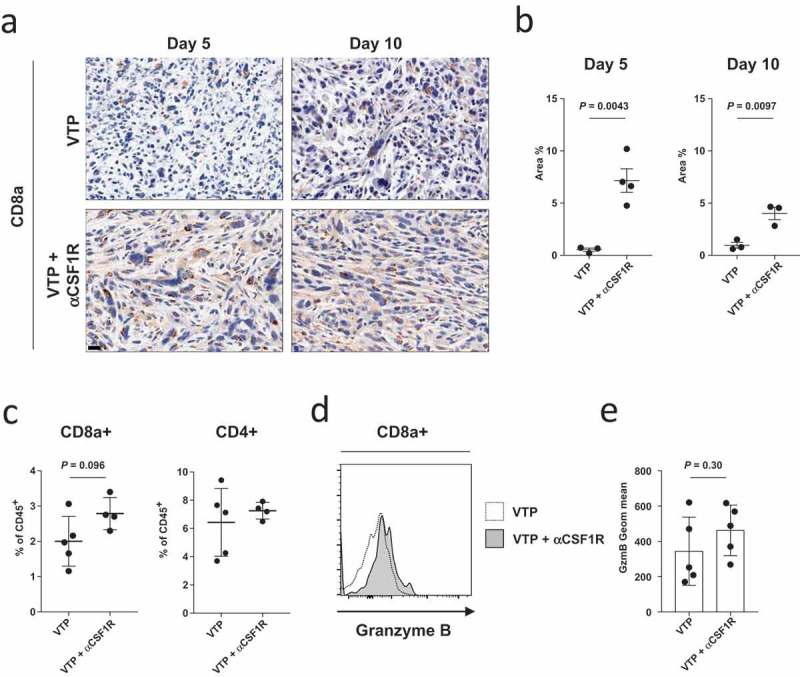
Anti-CSF1R treatment increases CD8+ T cell infiltration in VTP-treated Myc-Cap tumors.
(a) Representative tumor section images of CD8+ T cell staining from tumors 5 and 10 days after VTP therapy alone or VTP therapy with anti-CSF1R. Scale bars represent 20 µm and (b) quantification of intratumoral CD8+ T cell from (a) as a percentage of area covered on days 5 and 10 after treatment. (c) Quantification of CD8+ and CD4+ T cells as a percentage of live CD45+ cell 10 days after treatment as assessed by flow cytometry. (d) Expression of granzyme B in CD8+CD45+ T cells as assessed by flow cytometry on day 10 after treatment and (e) quantification of granzyme B expression in CD8+CD45+ T cells from d). Data shown are a single representative of experiments performed at least twice.
Discussion
Immunosuppressive myeloid cells such as MDSCs and M2 macrophages have been reported to inhibit the adaptive immune response against tumors and to promote tumor growth.12,15–20 Notably, M2 macrophages, as opposed to M1 macrophages, are associated with less favorable outcomes in a wide variety of tumor types.9 In this study, we show that VTP induces the recruitment of myeloid cells in treated tumors, including MDSCs and M2 macrophages, which would generate an immunosuppressive microenvironment. In the case of VTP therapy, we hypothesized that when VTP fails to completely eradicate all tumor cells, the influx of immunosuppressive myeloid cells theoretically inhibits anti-tumor T cells from eliminating the remaining surviving tumors cells. As we observed that VTP therapy increases the expression of CSF1R on infiltrating myeloid cells, we predicted that anti-CSF1R-mediated myeloid cell targeting could be a beneficial complementary treatment to delay the natural healing process after VTP treatment. Importantly, we confirmed that while CSF1R blockade inhibited myeloid tumor infiltration in all conditions, anti-CSF1R influenced tumor growth and survival only in combination with VTP therapy. The use of two murine prostate cancer models in this study attests to the reproducibility of this strategy. Taken together, our observations suggest that targeting pro-tumor myeloid cells such as MDSCs and M2 macrophages through CSF1R blockade is a promising approach for combination treatments for focal therapies such as VTP.
Interestingly, similar results using different therapeutic approaches have been reported. In a study by Xu et al.,29 where the authors showed that irradiation induced elevated levels of tumor-derived CSF1 secretion followed by enhanced TAM infiltration, and that this provided additional growth and survival factors to the tumor. However, when a selective CSF1R inhibitor was combined with the radiotherapy regimen, it resulted in suppressed tumor growth. In another study, androgen blockade therapy (ABT) was found to induce elevated levels of tumor-derived CSF1 and TAM infiltration in both human prostate cancer patients and preclinical mouse models of prostate cancer.37 However, when ABT was combined with anti-CSF1R treatment, the combination treatment led to reduced numbers of TAMs, diminished tumor progression and durable therapeutic benefit.37 Our work and the previous studies highlight the importance of CSF1/CSF1R signaling in the recruitment of tumor-infiltrating myeloid cells that can limit the efficacy of localized therapies for prostate cancers.
While we clearly show that targeting myeloid cells using anti-CSF1R potentiates VTP therapy, it is unclear whether this is due to removing a source of growth factors, releasing the inhibition on T cells, a combination of both or through other unexplored mechanisms. For example, other mechanisms of immunologic suppression were not addressed in this study, such as hypoxia, the presence of newly identified immunosuppressive cancer-associated fibroblasts, as well as other mechanisms.10,38,39 Nevertheless, it has been shown that CSF1R inhibition attenuates the turnover rate of TAMs while increasing the number of CD8+ T cells that infiltrate cervical and breast carcinomas,40 a correlation also observed in this study. Potentiating anti-tumor CD8+ T cells is quickly becoming a central focus when developing new therapies and we believe that this could be one of the main mechanisms driving the additive effect seen when anti-CSF1R is combined with VTP therapy. However, other pathways that were not addressed here could be targeted in future studies to determine if these could have an even greater effect on CSF1R-targeted therapies in combination with VTP. For example, in a recent study examining a castration-resistant prostate cancer murine model, it was found that IL-23 production by MDSCs was driving androgen receptor pathway activity in prostate tumor cells in androgen-deprived conditions and these cells could not be targeted by anti-CSF1R therapy.41 It would therefore be interesting to see if targeting these cells could lead to even greater benefit compared to the VTP plus anti-CSF1R therapy.
In conclusion, combining VTP therapy with anti-CSF1R treatment improves survival and decreased tumor growth in two different murine models of prostate cancer. These effects correlated with the decrease of immunosuppressive myeloid cells and an increase in granzyme B+ CD8+ T cells in the tumor. Our observation constitutes the basis for the rational design of clinical trials combining CSF1R blockade with VTP therapy. In addition, the increase in T cell function in this setting warrants further combinations with immune checkpoint blockade.
Materials and methods
Reagents and antibodies
Lyophilized WST11 was obtained from Steba Biotech (Cedex, France). Myc-Cap cells were cultured in DMEM supplemented with 10% fetal calf serum, 2-ME, pen/strep, L-glut (FCS; Mediatech, Inc.). TRAMP C2 cells were cultured in DMEM supplemented with 5% fetal calf serum (FCS; Mediatech, Inc.), 5% Nu Serum IV (BD Biosciences) HEPES, 2-ME pen/strep, L-glutamine, 5 μg/mL insulin (Sigma), and 10 nmol/L DHT (Sigma). All cells were periodically authenticated by morphologic inspection and tested negative for mycoplasma contamination by PCR. Therapeutic anti-mouse CSF1R (clone AFS98) was obtained from BioXcell (NH, USA). The following antibodies and dilutions were used for flow cytometric analyses: CSF-1R PE (eBioscience; clone AFS98; 1:100), F4/80 PerCP-Cyanine5.5 (eBioscience; clone BM8; 1:100), CD45 Alexa Fluor 700 (eBioscience; clone 30-F11; 1:200), CD8a BV650 (eBioscience; clone 53–6.7; 1:200), CD11b PE-Texas Red (Invitrogen; clone M1/70.15; 1:200), CD11c-FITC (BD Pharmingen; clone HL3; 1:200), Ly6C-PE-Cy7 (BD Pharmingen; clone AL-21; 1:200), Ly6G v450 (BD Horizon; clone 1A8; 1:200), CD206 Alexa Fluor647 (Biolegend; clone C068C2; 1:200) and I-A/I-E APC/Cy7 (Biolegend; clone M5/114.15.2; 1:200). The following antibodies and dilutions were used for IHC: CD8a (eBioscience; clone 4SM15; 1:1000), CD31 (Dianova; clone SZ31; 1:250) and Iba1 (Abcam; polyclonal ab5076; 1:2500). The following antibodies and dilutions were used for IF: CD11b (Abcam; clone EPR1344; 1 µg/mL), F4/80 (Abcam; clone CI:A3-1; 2 µg/mL) and CSF1R (Santa Cruz, clone C-20; 0.5 µg/mL).
Mice
All animal work was performed in accordance with a protocol approved by the IACUC of Memorial Sloan Kettering Cancer Center. Myc-Cap prostate cancer cell tumors were established by injecting 4 × 105 cells into the hindlimb of male 6 to 8-week old FVB/6 mice (Taconic, Hudson, NY). TRAMP C2 prostate cancer cell tumors were established by injecting 7.5 × 105 cells into the hindlimb of male 6 to 8-week old C57BL/6 mice (Taconic, Hudson, NY). Tumor growth was monitored by caliper measurement weekly. When the volume of tumors reached approximately 100 mm3, the animals were randomly assigned to different cohorts for further experiments.
Treatment
For VTP treatment, an anesthetic cocktail of 150 mg/kg ketamine and 10 mg/kg xylazine was administered intraperitoneally prior to treatment and was supplemented with inhaled isoflurane. A single dose of carprofen (5 mg/kg) and 1 mL of subcutaneous warm saline was administered. WST11 was reconstituted in sterile 5% dextran in water at 2 mg/mL under light protected condition and the aliquots were stored at −20°C. On the day of VTP treatment, an aliquot was thawed and filtered through 0.2 μm disc syringe filter (Sartorius Stedin Biotech North America, Bohemia, NY). The mice were intravenously infused with WST11 (9 mg/kg) followed immediately by 10-min laser (Ceramoptec, Bonn, Germany) illumination (755 nm, 150 mW/cm) through a 1 mm frontal fiber (MedLight SA, Ecublens, Switzerland). The light field was arranged to cover the entire tumor area plus 1 mm rim using red-light aiming beam. For anti-CSF1R treatment, mice were injected with 250 µg intraperitoneally on days 1, 4, 7, 10, 13.
Histology
All tumor specimens were fixed in 10% buffered formalin (Fisher Scientific, Pittsburgh, PA), embedded in paraffin, sectioned at five-micron thickness, and stained with hematoxylin-eosin (H&E). IHC staining was performed on FFPE sections at the Laboratory of Comparative Pathology at MSKCC on a Leica Bond RX automated stainer (Leica Biosystems, Buffalo Grove, IL). Following HIER at pH 9.0, the primary antibodies followed by application of an anti-goat IgG secondary antibody and a polymer detection system (DS9800, Novocastra Bond Polymer Refine Detection, Leica Biosystems). The chromogen was 3,3 diaminobenzidine tetrachloride (DAB), and sections were counterstained with hematoxylin. Slides were scanned with Pannoramic Flash (3DHistech, Hungary) using 20x/0.8NA objective, and regions of interest were drawn using CaseViewer (3DHistech, Hungary). Image quantification for Iba1 and CD31 IHC were performed using QuPath 0.1.2 software. Positive area (%) was calculated as follows from the output of Positive pixel count analysis: Positive area (%) = (Positive pixel count (downsampled pixels) x Downsample factor2)/(Area (µm2)/µm2 per pixel). The immunofluorescent staining was performed at Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 min with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 min (Ventana Medical Systems). Multiplex immunofluorescent stainings were performed as previously described (http://dx.doi.org/10.1038/srep09534). Sections were stained according to the following steps: first, slides were incubated with anti-F4/80 (Abcam, cat#ab6640, 2 ug/ml) for 4 h, followed by 60-min incubation with biotinylated goat anti-rat IgG (Vector Labs, cat# PK4004) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa Fluor 488 (Invitrogen, cat# T20922) prepared according to manufacturer instruction with predetermined dilutions. Next, sections were incubated with anti-CSF1R (Santa Cruz, cat#sc-692, 0.5 ug/ml) for 5 h, followed by 60-min incubation with biotinylated goat anti-rabbit IgG (Vector, cat # PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide CF594 (Biotium, cat# 92174) prepared according to manufacturer instruction with predetermined dilutions. Next, sections were incubated with anti-CD8 (Cell Signaling, cat#98941, 2.4 ug/ml) for 5 h, followed by 60-min incubation with biotinylated goat anti-rabbit IgG (Vector, cat # PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide CF543 (Biotium, cat# 92172) prepared according to manufacturer instruction with predetermined dilutions. Finally, sections were incubated with anti-CD11b (Abcam, cat#ab133357, 1 ug/ml) for 5 h, followed by 60-min incubation with biotinylated goat anti-rabbit IgG (Vector, cat # PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa 647 (Invitrogen, cat# T20936) prepared according to manufacturer instruction with predetermined dilutions. After staining, slides were counterstained with DAPI (Sigma Aldrich, cat# D9542, 5 ug/ml) for 10 min and coverslipped with Mowiol. Slides were scanned with Pannoramic Flash (3DHistech, Hungary) using 20x/0.8NA objective, and regions of interest were drawn using CaseViewer (3DHistech, Hungary). The images were then analyzed using ImageJ/FIJI (NIH) to count colocalized cells with F4/80, CSF1R, CD8, and CD11b. The DAPI channel was used to obtain the cell nuclear mask by thresholding and cell segmentation. The other channels were thresholded and checked to see co-localization and cell counts for the combination of channels were measured.
Flow cytometry
Single cell suspensions were collected from tumors by physically disrupting tumors through a 70 µm nylon mesh in PBS. Cell suspensions were incubated in Fc block (2.4G2; MSKCC antibody and bioresource core) for 10 min on ice in 0.5% bovine serum albumin and 2 mM EDTA in PBS, followed by antibodies at previously noted dilutions plus Fixable viability dye eFluor 506 (eBioscience; cat# 65–0866-14) for 30 min on ice. For CD8+ T cell staining, single cell suspensions were first stained, without any additional restimulation, with αCD8 and αCD45 as described above, followed by fixation and permeabilization using the eBioscienceTM Foxp3/Transcription Factor Staining Bugger Set (Invitrogen) according to manufacturer’s instructions and then stained αGZMB for 30 min on ice in 1X permeabilization buffer provided by the commercial kit. Data were acquired using the LSRII Flow cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar). Gating strategy is reported in Supplementary Figure 1.
Statistical analysis
Data are represented as means ± SEM and differences between groups were calculated using the unpaired two-tailed Student t-test. Differences in tumor growth were calculated by measuring the area under the curve and followed by an unpaired two-tailed Student t-test. Log Rank tests were used to compare differences in overall survival. Statistical analysis was performed with the Prism 7.0.2 software (GraphPad Software, Inc.).
Funding Statement
This study was supported by NIH grants R01CA056821, and MSKCC Cancer Center Core Grant P30CA008748. This study was also supported in part by funding from Ludwig Institute for Cancer Research, Swim Across America and Parker Institute for Cancer Immunotherapy and the Thompson Family Foundation.
Acknowledgments
We would like to thank Swim Across America, the Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, the NIH/NCI Cancer Center Support Grant (P30 CA008748) for their support of some of the coauthors (MG, TM, JDW) and the Thompson Family Foundation. Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Weill Cornell Medicine, New York, NY 10065, USA.
Disclosure of Potential Conflicts of Interest
AR. Azzouzi is proctor for Steba Biotech.
K. Kim, A. Scherz, and J.A. Coleman are listed as co-inventors on a provisional patent application on VTP and combination therapy that is owned by Memorial Sloan Kettering Cancer Center and the Weizmann Institute of Science.
A. Scherz is the inventor of WST11 that has been licensed by “Yeda”, the commercial brunch of Weizmann Institute to Steba Biotech with royalties agreement.
T. Merghoub is a consultant for: Immunos Therapeutics and Pfizer; is co-founder and has equity in: IMVAQ therapeutics; has research support from: Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Inc., Peregrine Pharmaceuticals, Inc., Adaptive Biotechnologies, Leap Therapeutics, Inc., and Aprea; is an inventor on patent applications related to work on Oncolytic Viral therapy, Alpha Virus Based Vaccine, Neo Antigen Modeling, CD40, GITR, OX40, PD-1 and CTLA-4.
Wolchok is consultant for: Adaptive Biotech; Advaxis; Amgen; Apricity; Array BioPharma; Ascentage Pharma;Astellas; Bayer; Beigene; Bristol Myers Squibb; Celgene; Chugai; Elucida; Eli Lilly; F Star; Genentech; Imvaq; Janssen; Kleo Pharma; Linneaus; MedImmune; Merck; Neon Therapuetics; Ono; Polaris Pharma; Polynoma; Psioxus; Puretech; Recepta; Trieza; Sellas Life Sciences; Serametrix; Surface Oncology; Syndax. Research support: Bristol Myers Squibb; Medimmune; Merck Pharmaceuticals; Genentech. Equity in: Potenza Therapeutics; Tizona Pharmaceuticals; Adaptive Biotechnologies; Elucida; Imvaq; Beigene; Trieza; Linneaus. Honoraium: Esanex. Patents: Xenogeneic DNA Vaccines; ALPHAVIRUS REPLICON PARTICLES EXPRESSING TRP2; Myeloid-derived suppressor cell (MDSC) assay; NEWCASTLE DISEASE VIRUSES FOR CANCER THERAPY; Genomic Signature to Identify Responders to Ipilimumab in Melanoma; Engineered Vaccinia Viruses for Cancer Immunotherapy; Anti-CD40 agonist mAb fused to Monophosphoryl Lipid A (MPL) for cancer therapy; CAR+ T cells targeting differentiation antigens as means to treat cancer; Anti-PD1 Antibody; Anti-CTLA4 antibodies; Anti-GITR antibodies and methods of use there of.
No potential conflicts of interest were disclosed for other authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Azzouzi AR, Vincendeau S, Barret E, Cicco A, Kleinclauss F, van der Poel HG, Stief CG, Rassweiler J, Salomon G, Solsona S, et al. Padeliporfin vascular-targeted photodynamic therapy versus active surveillance in men with low-risk prostate cancer (CLIN1001 PCM301): an open-label, phase 3, randomised controlled trial. Lancet Oncol. 2017;18:181–191. doi: 10.1016/S1470-2045(16)30661-1. [DOI] [PubMed] [Google Scholar]
- 2.Lebdai S, Bigot P, Leroux PA, Berthelot LP, Maulaz P, Azzouzi AR. Vascular targeted photodynamic therapy with padeliporfin for low risk prostate cancer treatment: midterm oncologic outcomes. J Urol. 2017;198:335–344. doi: 10.1016/j.juro.2017.03.119. [DOI] [PubMed] [Google Scholar]
- 3.Azzouzi AR, Lebdai S, Benzaghou F, Stief C.. Vascular-targeted photodynamic therapy with TOOKAD(R) soluble in localized prostate cancer: standardization of the procedure. World J Urol. 2015;33:937–944. doi: 10.1007/s00345-015-1535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borle F, Radu A, Fontolliet C, van Den Bergh H, Monnier P, Wagnières G. Selectivity of the photosensitiser TOOKAD for photodynamic therapy evaluated in the Syrian golden hamster cheek pouch tumour model. Br J Cancer. 2003;89:52320–52326. doi: 10.1038/sj.bjc.6601428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brandis A, Mazor O, Neumark E, Rosenbach-Belkin V, Salomon Y, Scherz A. Novel water-soluble bacteriochlorophyll derivatives for vascular-targeted photodynamic therapy: synthesis, solubility, phototoxicity and the effect of serum proteins. Photochem Photobiol. 2005;81:983–993. doi: 10.1562/2004-12-01-RA-389. [DOI] [PubMed] [Google Scholar]
- 6.Ashur I, Goldschmidt R, Pinkas I, Salomon Y, Szewczyk G, Sarna T, Scherz A. Photocatalytic generation of oxygen radicals by the water-soluble bacteriochlorophyll derivative WST11, noncovalently bound to serum albumin. J Phys Chem A. 2009;113:8027–8037. doi: 10.1021/jp900580e. [DOI] [PubMed] [Google Scholar]
- 7.Azzouzi AR, Barret E, Bennet J, Moorre C, Taneja S, Muir G, Villers A, Coleman J, Allen C, Scherz A, et al. TOOKAD(R) Soluble focal therapy: pooled analysis of three phase II studies assessing the minimally invasive ablation of localized prostate cancer. World J Urol. 2015;33:945–953. doi: 10.1007/s00345-015-1505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 10.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, Rice PL, Shroyer KR, Malkinson AM. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am J Pathol. 2010;176:2972–2985. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. 2017;85:117–125. doi: 10.1016/j.jaut.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 13.De Henau O, Raush M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu H, Ghosh A, Pink M, Tchaicha J, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12:170–180. doi: 10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- 15.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22:275–281. doi: 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Priceman S. J., Sung J. L., Shaposhnik Z., Burton J. B., Torres-Collado A. X., Moughon D. L., Johnson M., Lusis A. J., Cohen D. A., Iruela-Arispe M. L., et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 21.Preise D, Oren R, Glinert I, Kalchenko V, Jung S, Scherz A, Salomon Y. Systemic antitumor protection by vascular-targeted photodynamic therapy involves cellular and humoral immunity. Cancer Immunol Immunother. 2009;58:71–84. doi: 10.1007/s00262-008-0527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 24.Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J ImmunoTher Cancer. 2017;5:53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aharinejad S, Paulus P, Sioud M, Hofmann M, Zins K, Schäfer R, Stanley ER, Abraham D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004;64:5378–5384. doi: 10.1158/0008-5472.CAN-04-0961. [DOI] [PubMed] [Google Scholar]
- 26.Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–527. doi: 10.2119/molmed.2011.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SD, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber T, West BL, Bollag G, Ribas A Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74:153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J., Escamilla J., Mok S., David. J., Priceman S., West B., Bollag G., McBride W., Wu L.. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50–58. doi: 10.1016/j.ebiom.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, Murphy M, Schaer D, Wolchok JD, Merghoub T. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5:e1151595. doi: 10.1080/2162402X.2016.1151595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caescu CI, Guo X, Tesfa L, Bhagat TD, Verma A, Zheng D, Stanley ER Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood. 2015;125:e1–13. doi: 10.1182/blood-2014-10-608000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–120. [DOI] [PubMed] [Google Scholar]
- 34.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim K, Zhang H, La Rosa S, Jebiwott S, Desai P, Kimm S, Scherz A, O’Donoghue JA, Weber WA, Coleman JA. Bombesin antagonist-based radiotherapy of prostate cancer combined with WST-11 vascular targeted photodynamic therapy. Clin Cancer Res. 2017;23:3343–3351. doi: 10.1158/1078-0432.CCR-16-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 37.Escamilla J, Schokrpur S, Liu C, Priceman SJ, Moughan D, Jiang G, Pouliot F, Magyar C, Sung JL, Xu J, et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015;75:950–962. doi: 10.1158/0008-5472.CAN-14-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 39.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., Sirven P., Magagna I., Fuhrmann L., Bernard C., et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463–479 e410. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, De Bernadis G, Losa M, Mirenda M, Pasquini E, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559:363–369. doi: 10.1038/s41586-018-0266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.