

ABSTRACT
Oncolytic viruses (OVs) preferentially target and kill cancer cells without affecting healthy cells through a multi-modal mechanism of action. While historically the direct killing activity of OVs was considered the primary mode of action, initiation or augmentation of a host antitumor immune response is now considered an essential aspect of oncolytic virotherapy. To improve oncolytic virotherapy, many studies focus on increasing virus replication and spread. In this article, we open for discussion the traditional dogma that correlates replication with the efficacy of OVs, pointing out several examples that oppose this principle.
KEYWORDS: Oncolytic virus, oncolysis, antitumor immune response, immunotherapy, productive infection, herpes simplex virus, vaccinia virus, reovirus, vesicular stomatitis virus, newcastle disease virus
Introduction
Widely considered to be a promising modern-age treatment strategy, cancer immunotherapy is playing an increasingly important role in cancer treatment, with more than one thousand clinical trials currently in progress worldwide.1 Among the many immunotherapies being developed, oncolytic viruses (OVs) are gaining traction as potential clinical therapeutic agents, with a Herpes simplex virus type 1 (HSV-1) based OV recently approved by the USA Food and Drug Administration (FDA) for the treatment of melanoma.2 The great potential of OVs to fight cancer is driving different combinatorial approaches to improve oncolytic virotherapy (OVT), most of them predicated on a dual mode of action.3,4 However, how OVs work is complex and still largely unknown. Thus, a deeper understanding of the multi-modal activity of OVs is essential to strategically raise OVT to the next level and to design relevant clinical therapy approaches.
OVs have varying efficacy in treating cancer not only in preclinical models but also in phase I to phase III clinical trials.5 OVs preferentially target and kill cancer cells while having minimal to no detrimental effects on normal cells by exploiting biochemical differences between healthy and transformed cells6 (Figure 1). Cancer cells evolve to resist apoptosis and growth suppression, evade immune system control and proliferate indefinitely, characteristics that favor viral replication.7 Moreover, many cancer cells develop defects in cellular antiviral response pathways, like the type I interferon (IFN) signaling pathway, rendering them more permissive to viral infection.8 This tumor-specific essence of OVs makes them appealing as a cancer therapy since they rarely induce off-target toxicities often seen with conventional therapies. Furthermore, OVs can kill cancer stem cells and replicate in hypoxic environments and in drug-resistant cells.9,10
Figure 1.
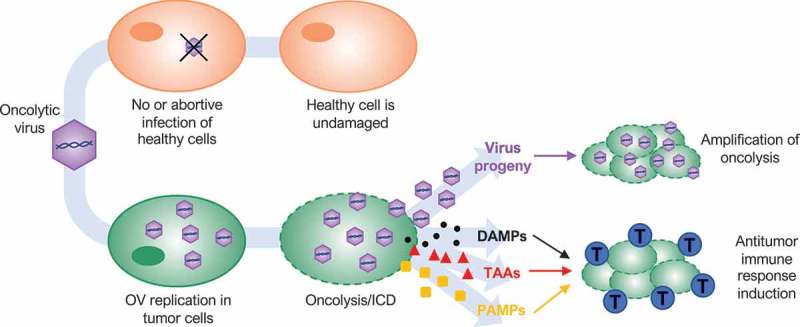
Dual-mode of action of oncolytic viruses (OVs). OVs preferentially target and kill cancer cells while having minimal to no detrimental effects on normal cells. OVs mediate tumor cell destruction by two main mechanisms: direct lysis of infected cells (oncolysis) and indirect augmentation of host antitumor immunity. OVs infect and replicate in cancer cells, inducing tumor cell lysis and release infectious viral progeny that spreads to surrounding tumor cells (amplification of oncolysis). Oncolysis also releases tumor-associated antigens (TAAs), cellular damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) in a highly inflammatory process, termed “immunogenic cell death” (ICD). Cellular detection of viral infection and the products of oncolysis trigger the rapid activation of a host antitumor immune response. The direct recognition and killing of tumor cells are primarily mediated by natural killer cells of the innate immune system and tumor antigen-specific CD8+ cytotoxic T lymphocytes (blue cells) of the adaptive immune system.
In addition to directly killing cancer cells, OVs initiate or augment host innate and adaptive tumor-specific immune responses that exert cytotoxicity to surviving cancer and stromal cells.11,12 Several studies have demonstrated that antitumor immunity plays an important role in the overall efficacy of OVT, inducing long-lasting protection against a variety of cancers, including those with the most dismal outcomes.13 Various approaches have been explored to improve OV antitumoral activity. Many studies focus on increasing virus replication and spread, as traditional dogma correlates replication with efficacy.14,15 However, this dogma has been called into question over the last few years, leaving open the question if productive viral replication, and even oncolysis itself, are essential for OVT. In this article, we will review the historical advances of OVs and the crucial role of host antitumor immunity in OVT. Finally, we will discuss findings that are questioning the importance of productive, lytic infection in the success of OVT.
History of OVs: from a natural viral infection to programmed weapons to target cancer
For more than a century, viruses have been pursued as possible agents of tumor destruction. Even before their full potential was first recognized, viruses demonstrated antitumor activity. Case reports from the early and mid-1900s describe short-lasting cancer remission, typically one or two months, in the context of natural viral infection. Most of the patients were suffering from hematological malignancies such as leukemia or lymphoma, known to be associated with significant suppression of the immune system. Probably one of the most widely cited cases belongs to a woman with myelogenous leukemia that went into remission after a presumed influenza infection, as influenza was identified as a virus more than 30 years later.16 Another case describes the spontaneous regression of lymphatic leukemia in a 4-year-old boy after chickenpox infection.17 Only a few days after developing the classic varicella rash, his white count fell to normal, his platelet count and hemoglobin increased significantly, and examination of his bone marrow confirmed that he was in remission. However, the remission lasted only one month, after which the cancer progressed rapidly until death. More recent clinical reports have described the regression of leukemia, Hodgkin’s disease, and Burkitt’s lymphoma after natural measles infection.18 The picture of an 8-year-old African boy with right orbital swelling due to Burkitt’s lymphoma, whose facial tumor completely regressed after measles infection with a complete remission of more than 4 months, has traveled around the world as a classical case of measles virus natural antitumor activity.19
In the 1950s and 1960s, the understanding of viruses accelerated rapidly due to the development of cell and tissue culture systems that allowed ex vivo virus propagation. Additionally, the advent of xenograft murine cancer models provided the opportunity to test the in vivo antitumor activity of OVs under controlled conditions. While several human and animal viruses were found to cause complete tumor regression in mice, they were less effective in patients and virus virulence was a major concern.20–22 The true potential of OVT was not realized until a couple of decades later when recombinant DNA technology became standard to enhance safety.22 The first description of a virus engineered to replicate selectively in dividing cells was a thymidine kinase-negative mutant of HSV-1 as a potential therapy for gliomas.23 Due to the application of new technology to genetically modify viruses, the field has expanded dramatically. The main question has been which modification should be made to generate the most potent, yet safe, OV. Original dogma correlated virus replication with anticancer efficacy, focusing attention on enhancing the selectivity and potency of direct OV-mediated cancer cell lysis and viral spread through the tumor mass.24 Following this dogma, three types of modifications were investigated: targeting, arming and shielding.25
Targeting modifications introduce or increase cancer cell specificity, improving the safety and efficacy of OVs. For example, deletion of the ICP0 gene of HSV-1 allows selective replication in human cancer cells.26 The ICP0 protein subverts the activity of type I IFN, an anti-proliferative and anti-viral innate immune cytokine. Since mutations within the IFN pathway are a hallmark of cellular immortalization and transformation,27 many cancer cells harbor mutations in IFN-related pathways. Accordingly, ICP0-null oncolytic HSV-1 viruses productively infect cancer cells but are unable to replicate in healthy cells with normal IFN production and responses.26 Another example is oncolytic adenovirus H101, which was the first OV clinically approved by Chinese regulators in 2005 to treat head and neck cancer. H101 has two deletions in the E1B gene and the E3 region that confer the ability to selectively replicate in cancer cells lacking functional p53, which is the most common genetic abnormality identified in human cancer.28,29 Arming OVs refers to the addition of genes that encode molecules that enhance OV oncolytic potency. One example of arming belongs to another oncolytic adenovirus that was engineered to express TRAIL (TNF-related apoptosis-inducing ligand). Expressing TRAIL increases the potency of oncolytic adenovirus in vitro and in animal tumor models by enhancing apoptotic cell death.30,31 While targeting and arming have focused on improving the cytolytic effect of OVs on cancer cells, shielding modifications have been applied to increase OV distribution and spreading by adding coats of polymers surrounding viral particles or changing their envelopes or capsids to avoid neutralizing antibodies detection.25 The potential for improvement with this third modification was not originally appreciated when viruses were predominantly tested in vitro or in immunocompromised xenograft models, but the increasing use of immunocompetent murine tumor models to evaluate OVs highlighted the need to shield some OVs from immune-mediated clearance.32,33
Genetic engineering technologies were pivotal in the history of OVT, particularly to enable the use of human-specific viruses. However, the efficacy of OVT was originally attributed almost exclusively to the ability of the virus to replicate in and spread through the tumor and eliminate all cancer cells through the release of progeny virions. Most of the preclinical studies involved in vitro assays and immunocompromised xenograft models. Indeed, based on this premise, early clinical trials used immunosuppressive drugs to limit the antiviral immune response and allow the OV to replicate to higher levels by increasing OV propagation and survival within infected tumors.34,35 However, this approach failed and drew into question the clinical applicability of OVT. Although it is now appreciated that the tumor microenvironment is immunosuppressive and, therefore, likely to be more permissive for viral replication, the extrapolation from immunocompromised preclinical models to immunocompetent patients was too simplistic. The understanding that the anticancer mode of action of OVs is more complex than simply direct cancer cell lysis is becoming more evident.36
OVT as immunotherapy
While OV replication within tumors was assumed to cause direct lysis of resident cancer cells, leading to clearance of the bulk of the tumor,36 preclinical and clinical data consistently suggest that OVs also utilize other anticancer mechanisms to eradicate tumors. For example, oncolytic vaccinia virus and vesicular stomatitis virus (VSV) have the additional ability of targeting tumor vasculature affecting tumor blood supply and, therefore, tumor progression.37 However, the most relevant mechanism of action is the initiation of a host antitumor immune response, which is now considered an essential aspect of OVT (Figure 1).38–40
The initiation of a host antitumor response can be explained by the ability of many OVs to induce immunogenic cell death (ICD) of cancer cells,41,42 including pyroptosis,43,44 necroptosis,45,46 immunogenic apoptosis,47 and autophagic cell death.43,44 ICD causes the exposure or release of cellular damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) as danger signals, along with tumor-associated antigens (TAAs) (Figure 1). DAMPs attract antigen-presenting cells (APCs) such as dendritic cells (DCs) that engulf TAAs and receive maturation signals. DAMPs also induce DCs to produce proinflammatory cytokines. Mature DCs present TAAs to naïve T cells, initiating antigen-specific immune responses that mediate targeted destruction of residual and recurrent tumor cells.48–52 The direct recognition and killing of tumor cells are primarily mediated by natural killer (NK) cells of the innate system and tumor antigen-specific CD8+ cytotoxic T lymphocytes of the adaptive immune system (Figure 1).13,40
In addition, OVs trigger an antiviral immune activation in tumor cells, even without productive replication,53 that helps to activate antitumor immune stimulation.54 PAMPs activate a cascade of signaling events that stimulate the inflammasome and activate different transcription factors, culminating in the release of pro-inflammatory cytokines and DAMPs. These pro-inflammatory cytokines alter the balance of pro- and anti-inflammatory factors within the tumor microenvironment, which is responsible for its immunosuppressive state.55–58 In addition to counteracting tumor-induced immunosuppression, these compounds mediate the recruitment of cytokine-releasing immune cells with additional effector function, amplifying host antitumor immunity.59,60 Moreover, OV-infected cancer cells process and present virus-specific antigens on their surface, facilitating their identification and destruction by antiviral T cells.12 Thus, antiviral immune stimulation triggered by PAMPs and viral antigens potentiates the antitumor immune response. Indeed, some authors believe that initial antiviral immunological events form the foundation of OV-based cancer immunotherapy rather than restricting OV efficacy by limiting viral replication and spread.54
The fact that OVs induce anticancer immune responses has been known since the late 1990s, when an oncolytic HSV-1 was shown to elicit TAA-specific adaptive immune responses, functioning as an in situ cancer vaccine, in syngeneic murine models of melanoma and colorectal cancer.61 Currently, there is plenty of evidence suggesting that activation of the host immune system is a crucial component of OVT success. The rationale for the design of Talimogene Laherparepvec (T-VEC), the first OV licensed by the FDA as a cancer therapeutic for metastatic melanoma, followed this premise. T-VEC is an oncolytic HSV-1 that has deletions in two genes, ICP34.5 and ICP47, and additionally expresses granulocyte macrophage colony-stimulating factor (GM-CSF). The deletion in ICP34.5 confers selectivity to cancer cells, the deletion in ICP47 removes the inhibition of transporter involved in antigen processing (TAP), improving antigen presentation, and GM-CSF promotes APC recruitment and maturation, stimulating antitumor immunity.62,63 Another OV that also encodes GM-CSF is the oncolytic vaccinia virus pexastimogene devacirepvec (Pexa-Vec) that is now in a phase III trial for the treatment of hepatocellular carcinoma in combination with the chemotherapy Sorafenib.5 The future of immunotherapy will certainly include OVs since their unique mechanism of action shows promise in overcoming obstacles inherent to other immunotherapies.5
Is productive replication essential for OV antitumor efficacy?
Preclinical and clinical studies support the multi-modal mechanism of action of OVs that includes direct oncolysis and indirect induction and augmentation of host antitumor immunity (Figure 1).5 Consequently, the improvement of either or both modes of action by genetic modification of the virus or combining with other therapies should in principle improve OVT. Following this reasoning, Sobol et al. tested in an immune competent murine model of breast cancer OVs with different replication capacities and cytopathic effects in vitro, including HSV-1 OVs harboring a single or double mutation(s) and a VSV M protein mutant.12 Despite significant differences in virus replication, spread and cellular toxicity in vitro (VSV ⋙ HSV single mutant > HSV double mutant), tumor regression and survival were similar with all three viruses in vivo, even when using a 50-fold excess of VSV OV over the HSV-1 OVs, indicating that in vitro cytolytic properties can be poor prognostic indicators of in vivo antitumor activity.12
A similar study found a negative correlation between in vitro replication and in vivo antitumor activity of HSV-1 and HSV-2 harboring one of two different ICP0 mutations.64 In vitro, both HSV-2 OVs showed enhanced replication, cell toxicity and release of the immune stimulatory molecule HMGB1, while both HSV-1 OVs demonstrated almost no replication or toxicity and failed to release HMGB1. Despite showing the lowest “oncolytic” activity in vitro and being cleared most rapidly in vivo, HSV-1 dICP0 treatment was the only treatment to confer a significant survival benefit in a murine breast cancer model. Moreover, treatment with HSV-1 dICP0, but not HSV-2 dICP0, induced HMGB1 release in vivo,64 highlighting the divergence between in vitro and in vivo findings. In a different study, instead of comparing different OVs within the same murine model, three syngeneic murine sarcoma models were evaluated with the same HSV-1 ICP6 mutant OV.65 Interestingly, neither viral permissivity nor cytotoxicity in vitro was predictive of in vivo tumor regression and survival, as the in vivo antitumor effect ranged from no or modest response to complete tumor regression and protection from tumor rechallenge, despite a similar level of tumour cell permissivity in vitro. In both studies, tumor reduction was T-cell mediated with a tumor-specific antigen response.64,65 Thus, at least in the context of oncolytic HSV, the initial stages of virus infection leading to activation of antitumor immunity are more important than virus replication and persistence within the tumor causing direct tumor debulking (Figure 2). In other words, the traditional dogma that correlates productive replication with potency of OVs does not always apply for oncolytic herpesviruses.
Figure 2.
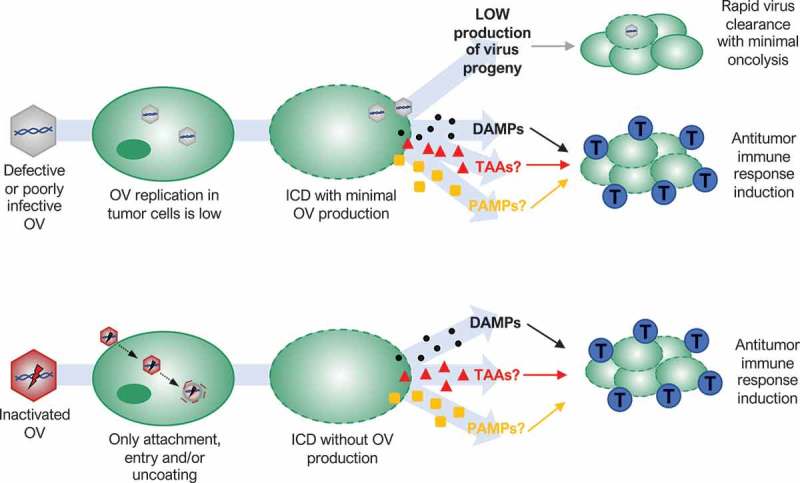
Possible mechanisms of action of oncolytic viruses (OVs) that oppose traditional dogma that correlates replication with efficacy. Defective or poorly infective OVs may produce a small amount of new viral particles after infection of tumor cells, but infection spreading is quickly stopped by a host antiviral response (rapid virus clearance with minimal oncolysis). Nevertheless, several defective, poorly infective or even inactivated OVs are able to kill tumor cells in an immunogenic way. Low virus replication or only virus binding, entry and/or uncoating in tumor cells are enough to induce immunogenic cell death (ICD) releasing damage-associated molecular patterns (DAMPs) that triggers a host antitumor immune response activation or augmentation (blue cells: cytotoxic T lymphocytes). Whether tumor-associated antigens (TAAs) or pathogen-associated molecular patterns (PAMPs) are also involved remains unknown.
Poxviruses are another family of large DNA viruses with a potential antitumor activity that also does not always follow the traditional dogma of OVT. Using heat-inactivation, Dai et al. showed that a non-replicating modified vaccinia virus Ankara (MVA) was more effective than replicating MVA as an OV in vivo.66 Inactivated MVA (iMVA) was able to shrink not only melanoma tumors following direct intratumoral injection, but it was also able to control the growth of non-injected distant tumors, indicating a potent antitumor immunity induction. The effectiveness and adaptive antitumor immune response activation of iMVA were also observed in a murine colon cancer model. In addition, the combination of iMVA and immune checkpoint inhibitors, which relieve T cell inhibitory mechanisms of the suppressive tumor microenvironment, generated synergistic antitumor effects in bilateral tumor implantation models as well as in a unilateral large established tumor model.66 In the case of MVA, only the initial stages of the virus life cycle that precede viral gene expression and genome replication seem to play an important role in MVA therapeutic activity (Figure 2).
Reovirus is a naturally occurring, non-pathogenic double-stranded RNA virus, with selective toxicity toward cells with an activated Ras pathway.67,68 Reovirus is under investigation in phase I and II clinical trials and is considered a potential candidate for phase III trials.69 Although viral life cycles of RNA viruses differ significantly with those of DNA viruses, antitumor activity of reovirus type 3 in vivo was also independent of virus replication in a B16 murine melanoma model.70 In vitro, mouse melanoma cells were resistant to direct oncolysis and failed to support reovirus replication. Limited reovirus replication was also observed in vivo. However, reovirus was able to induce an antitumor immune response and purged lymph node and splenic metastasis in immunocompetent mice, while it failed to reduce tumor burden in immunodeficient mice. Using human cells in vitro, Prestwich et al. also showed that direct reovirus oncolysis is not required to prime antitumor immunity, with UV-inactivated reovirus being similarly immunogenic, suggesting that only the initial stages of reovirus infection play an essential role in antitumor immunity activation (Figure 2).70
Similar, yet different, is the case of another RNA virus, VSV. Single-replication cycle VSV showed to be as effective as fully replicating VSV, while inactivated VSV failed to show therapeutic outcome, indicating that viral gene expression, genome replication or even late stages of the VSV cycle are important for VSV antitumor activity. Following this hypothesis, the authors found a strong correlation between viral gene expression, induction of proinflammatory activity in the tumor and in vivo therapy success, showing that in the case of VSV, viral gene expression is central but a productive, multi-cycle infection is not necessary for antitumor efficacy.71
A third case of an RNA OV that does not match with traditional OV dogma is Newcastle Disease Virus (NDV). This avian virus has interesting anti-neoplastic and pleiotropic immune stimulatory properties and has demonstrated safety in phase II clinical studies.72 According to the properties of the viral fusion protein, NDV can be divided into lytic and non-lytic strains. Lytic strains are able to produce infectious progeny by disrupting the plasma membrane of infected cells, while non-lytic strains mainly stimulate immune responses.73 An interesting observation to highlight is that both strain types have antitumor potential,74 with a non-lytic strain being superior to a lytic strain in some instances.75 Thus, the way tumor cells die, or in other words, the way OVs kill tumor cells, can make a significant difference in terms of OVT success, drawing into question the need for “lytic” outcomes with OVs.
Conclusions
Here, we reviewed the advances of OVs from natural acquired infections to specifically engineered viruses to target cancer in a more selective and potent way.22,25 We also discussed the importance of OV activation of an antitumor immune response to kill, in an indirect manner, not only the primary tumor cells but also metastatic cells that often remain invisible to clinical detection. Moreover, this powerful mechanism of action of OVs is showing promise to overcome obstacles inherent to other immunotherapies.5 OVs have a multiple modes of action that can include direct oncolysis, indirect antitumor immunity induction and other mechanisms such as tumor vasculature disruption.5,37 Alternative mechanisms of OV activity are still under investigation, including OV infection of immune cells such as DCs66 and activation and/or exposure of neoantigens.76 However, oncolysis and antitumor host immune response augmentation are still the main focus of attention. Preclinical and clinical studies have demonstrated that oncolysis alone is not sufficient to completely destroy tumors. Although the tumor microenvironment is locally immunosuppressed, it still has host immune and stromal cells that are competent at controlling viral infections. The clearance of the virus before complete destruction of the tumor, coupled with a complex and dense tumor architecture, is likely the main reason why amplification of viral oncolysis alone is not enough for OVT success.25 Conversely, the induction of host antitumor immunity is crucial for OVT success.36,40 Therefore, the way tumor cells die seems to be the key in OVT, and it is more important than how many tumor cells are killed directly by the virus.40 ICD is by consensus the form of regulated cancer cell death which culminates in the release of DAMPs that induce potent anticancer immunity via specific adaptive immune response against antigens presented on dying cells. However, different pathways regulate the capacity of a particular agent to drive bona fide ICD and the ability of the host to perceive such an instance of cell death as immunogenic.41 Ongoing studies are trying to elucidate which aspects of ICD activate the “right” DAMPs, induce the production of the “right” cytokines and, overall, stimulates the strongest antitumor immune response.77,78
Additionally, we opened for discussion the traditional dogma that correlates replication with efficacy of OVs, pointing out several examples that oppose this principle. While questioning the traditional dogma, we are not intending to suggest that inactivated or defective OVs are always better platforms for OVT. Indeed, our own first HSV-1 OV study found that inactivated virus was significantly less effective than replication competent OV.26 However, it is important to consider that OVs that retain antitumor activity with reduced replication and spread capacity are likely more clinically valuable from a safety perspective. Our intention here is to point out that increasing OV replication may not be always the best approach to improve OVT. Moreover, focusing the attention on improving OV replication or evaluating potential therapeutic combinations in vitro to find the most potent clinical candidate or approach could be problematic since in vitro outcomes can be poor prognostic indicators of in vivo antitumor activity.12,64 Thus, we believe that a better understanding of the complex interactions between OVs, tumor cells, the host immune system and the rest of the tumor microenvironment, currently available only within in vivo models, will be the key to potentiate the next generation of OVT. However, preclinical mouse models of cancer poorly predict patient outcomes given species differences.79 Thus, we believe that the strategic design of phase I clinical trials is of paramount importance. Additionally, closer monitoring of patients’ progression after treatment by assessing tumor volumes, virus replication by imaging,80 immune markers in blood, type of cancer cell death and presence of DAMPs in tumor biopsies, is central to better understand how OVs function within the patient population.
References
- 1.1,496 clinical trials on ClinicalTrials.gov with “cancer AND immunotherapy” as search terms. ClinicalTrials.gov: US National Library of Medicine; 2019. [accessed 2019 January23]. [Google Scholar]
- 2.Ott PA, Hodi FS.. Talimogene laherparepvec for the treatment of advanced melanoma. Clin Cancer Res. 2016;22:3127–3131. [DOI] [PubMed] [Google Scholar]
- 3.Pol J, Buque A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, JM Limacher, et al. Trial watch-oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5:e1117740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pol JG, Levesque S, Workenhe ST, Gujar S, Le Boeuf F, Clements DR, JE Fahrner, Fend L, JC Bell, KL Mossman, et al. Trial watch: oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology. 2018;7:e1503032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH.. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer. 2018;18:419–432. [DOI] [PubMed] [Google Scholar]
- 6.Pikor LA, Bell JC, Diallo JS. Oncolytic viruses: exploiting cancer’s deal with the devil. Trends in Cancer. 2015;1:266–277. [DOI] [PubMed] [Google Scholar]
- 7.Choi AH, O’Leary MP, Fong Y, Chen NG. From benchtop to bedside: a review of oncolytic virotherapy. Biomedicines. 2016;4:pii: E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shmulevitz M, Marcato P, Lee PW. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24:7720–7728. [DOI] [PubMed] [Google Scholar]
- 9.Cuddington BP, Dyer AL, Workenhe ST, Mossman KL. Oncolytic bovine herpesvirus type 1 infects and kills breast tumor cells and breast cancer-initiating cells irrespective of tumor subtype. Cancer Gene Ther. 2013;20:282–289. [DOI] [PubMed] [Google Scholar]
- 10.Zeng W, Hu P, Wu J, Wang J, Li J, Lei L, Liu R. The oncolytic herpes simplex virus vector G47 effectively targets breast cancer stem cells. Oncol Rep. 2013;29:1108–1114. [DOI] [PubMed] [Google Scholar]
- 11.Aurelian L. Oncolytic viruses as immunotherapy: progress and remaining challenges. Onco Targets Ther. 2016;9:2627–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther. 2010;19:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. 2017;7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phan M, Watson MF, Alain T, Diallo JS. Oncolytic viruses on drugs: achieving higher therapeutic efficacy. ACS Infect Dis. 2018;4:1448–1467. [DOI] [PubMed] [Google Scholar]
- 15.Selman M, Ou P, Rousso C, Bergeron A, Krishnan R, Pikor L, Chen A, BA Keller, Ilkow C, JC Bell, et al. Dimethyl fumarate potentiates oncolytic virotherapy through NF-kappaB inhibition. Sci Transl Med. 2018;10:pii: eaao1613. [DOI] [PubMed] [Google Scholar]
- 16.Dock G. The influence of complicating diseases upon leukemia. Am J Med Sci. 1904;127:563–592. [Google Scholar]
- 17.Bierman HR, Crile DM, Dod KS, Kelly KH, Petrakis NL, White LP, MB Shimkin. Remissions in leukemia of childhood following acute infectious disease: staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer. 1953;6:591–605. [DOI] [PubMed] [Google Scholar]
- 18.Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther. 2009;11:43–53. [PMC free article] [PubMed] [Google Scholar]
- 19.Az B, Jl Z. Regression of Burkitt’s lymphoma in association with measles infection. Lancet. 1971;2:105–106. [DOI] [PubMed] [Google Scholar]
- 20.Moore AE. Viruses with oncolytic properties and their adaptation to tumors. Ann N Y Acad Sci. 1952;54:945–952. [DOI] [PubMed] [Google Scholar]
- 21.VIRUSES in treatment of cancer. Br Med J. 1957; 2: 1481–1482. [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. [DOI] [PubMed] [Google Scholar]
- 23.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. [DOI] [PubMed] [Google Scholar]
- 24.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med. 2001;7:781–787. [DOI] [PubMed] [Google Scholar]
- 25.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hummel JL, Safroneeva E, Mossman KL. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther. 2005;12:1101–1110. [DOI] [PubMed] [Google Scholar]
- 27.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu W, Zheng S, Li XF, Huang JJ, Zheng X, Li Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: a pilot phase II clinical trial. World J Gastroenterol. 2004;10:3634–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98:298–300. [DOI] [PubMed] [Google Scholar]
- 30.Sova P, Ren XW, Ni S, Bernt KM, Mi J, Kiviat N, Lieber AA. A tumor-targeted and conditionally replicating oncolytic adenovirus vector expressing TRAIL for treatment of liver metastases. Mol Ther. 2004;9:496–509. [DOI] [PubMed] [Google Scholar]
- 31.Dong F, Wang L, Davis JJ, Hu W, Zhang L, Guo W, Teraishi F, Ji L, Fang B. Eliminating established tumor in nu/nu nude mice by a tumor necrosis factor-alpha-related apoptosis-inducing ligand-armed oncolytic adenovirus. Clin Cancer Res. 2006;12:5224–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN, Henry SM, Peng KW, Russell SJ. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87:3752–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009;20:975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004;11:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, Yasmeen A, TA Bismar, Kirn D, Falls T et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105:14981–14986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, Vile RG. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angarita FA, Acuna SA, Ottolino-Perry K, Zerhouni S, McCart JA. Mounting a strategic offense: fighting tumor vasculature with oncolytic viruses. Trends Mol Med. 2013;19:378–392. [DOI] [PubMed] [Google Scholar]
- 38.Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Workenhe ST, Verschoor ML, Mossman KL. The role of oncolytic virus immunotherapies to subvert cancer immune evasion. Future Oncol. 2015;11:675–689. [DOI] [PubMed] [Google Scholar]
- 40.van Vloten JP, Workenhe ST, Wootton SK, Mossman KL, Bridle BW. Critical interactions between immunogenic cancer cell death, oncolytic viruses, and the immune system define the rational design of combination immunotherapies. J Immunol. 2018;200:450–458. [DOI] [PubMed] [Google Scholar]
- 41.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- 42.Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furukawa Y, Takasu A, Yura Y. Role of autophagy in oncolytic herpes simplex virus type 1-induced cell death in squamous cell carcinoma cells. Cancer Gene Ther. 2017;24:393–400. [DOI] [PubMed] [Google Scholar]
- 44.Colunga AG, Laing JM, Aurelian L. The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther. 2010;17:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y, Wang HX, Mao X, Fang H, Wang H, Li Y, Sun Y, Meng C, Tan L, Song C, et al. RIP1 is a central signaling protein in regulation of TNF-alpha/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection. Oncotarget. 2017;8:43201–43217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saga K, Kaneda Y. Oncolytic Sendai virus-based virotherapy for cancer: recent advances. Oncolytic Virother. 2015;4:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinrich B, Klein J, Delic M, Goepfert K, Engel V, Geberzahn L, Lusky M, Erbs P, Preville X, Moehler M. Immunogenicity of oncolytic vaccinia viruses JX-GFP and TG6002 in a human melanoma in vitro model: studying immunogenic cell death, dendritic cell maturation and interaction with cytotoxic T lymphocytes. Onco Targets Ther. 2017;10:2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M., Mignot G., Panaretakis T, Casares N., et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. [DOI] [PubMed] [Google Scholar]
- 50.Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O., Metivier D, Galluzzi L, Perfettini JL et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med. 2015;21:S6–s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Manuel J, Breckpot K, Brough D, Chaurio R, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hare DN, Collins SE, Mukherjee S, Loo YM, Gale M Jr., Janssen LJ, Mossman KL. Membrane perturbation-associated Ca2+ signaling and incoming genome sensing are required for the host response to low-level enveloped virus particle entry. J Virol. 2015;90:3018–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. 2018;39:209–221. [DOI] [PubMed] [Google Scholar]
- 55.Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, De Bono J, Selby P, Coffey M, Vile R et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180:6018–6026. [DOI] [PubMed] [Google Scholar]
- 56.Gujar S, Dielschneider R, Clements D, Helson E, Shmulevitz M, Marcato P, Pan D, Pan LZ, Ahn DG, Alawadhi A et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol Ther. 2013;21:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gujar SA, Lee PW. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front Oncol. 2014;4:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, Coffey M, Selby P, Vile R, Melcher A. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer. 2011;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol. 2010;10:514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385–393. [DOI] [PubMed] [Google Scholar]
- 62.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman K.A., Spitler L.E., Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. [DOI] [PubMed] [Google Scholar]
- 63.Bl Liu, Robinson M, ZQ Han, Rh Branston, English C, Reay P, McGrath Y, SK Thomas, Thornton M, Bullock P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. [DOI] [PubMed] [Google Scholar]
- 64.Workenhe ST, Simmons G, Pol JG, Lichty BD, Halford W, Mossman KL. Immunogenic HSV mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol Ther. 2014;22:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leddon JL, Chen CY, Currier MA, Wang PY, Jung FA, Denton NL, KM Cripe, KB Haworth, MA Arnold, AC Gross, et al. Oncolytic HSV virotherapy in murine sarcomas differentially triggers an antitumor T-cell response in the absence of virus permissivity. Mol Ther Oncolytics. 2015;1:14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dai P, Wang W, Yang N, Serna-Tamayo C, Ricca JM, Zamarin D, Shuman S, Merghoub T, Wolchok JD, Deng L. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol. 2017;2:pii: eaal1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. Embo J. 1998;17:3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gong J, Mita MM. Activated ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol. 2014;4:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Black AJ, Morris DG. Clinical trials involving the oncolytic virus, reovirus: ready for prime time? Expert Rev Clin Pharmacol. 2012;5:517–520. [DOI] [PubMed] [Google Scholar]
- 70.Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, Thompson J., Galivo F, Harrington KJ, Pandha HS, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15:4374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, Melcher A, Vile R. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schirrmacher V. Fifty years of clinical application of newcastle disease virus: time to celebrate! Biomedicines. 2016;4:pii: E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ginting TE, Suryatenggara J, Christian S, Mathew G. Proinflammatory response induced by newcastle disease virus in tumor and normal cells. Oncolytic Virother. 2017;6:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zamarin D, Palese P. Oncolytic newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol. 2012;7:347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schirrmacher V, Griesbach A, Ahlert T. Antitumor effects of newcastle disease virus in vivo: local versus systemic effects. Int J Oncol. 2001;18:945–952. [DOI] [PubMed] [Google Scholar]
- 76.Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, MP Manns, TC Wirth, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23:1630–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Showalter A, Limaye A, Oyer JL, Igarashi R, Kittipatarin C, Copik AJ, Khaled AR. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. 2017;97:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pound P, Ritskes-Hoitinga M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J Transl Med. 2018;16:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galanis E, Atherton PJ, Maurer MJ, Knutson KL, Dowdy SC, Cliby WA, Haluska P, HJ Long, Oberg A, Aderca I, et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res. 2015;75:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]