

ABSTRACT
In combination cancer immunotherapies, consideration should be given to designing treatment schedules that harmonize with the immune system’s natural timing. An efficacious temporally programmed combination therapy of extended half-life interleukin 2 (eIL2), tumor targeting antibody, and interferon (IFN) α was recently reported; however, tumor-ablative efficacy was associated with significant toxicity. In the current work, altering the order and timing of the three agents is shown to decouple toxicity from efficacy. Delaying the administration of eIL2 to be concurrent with or after IFNα eliminates toxicity without affecting efficacy in multiple syngeneic tumor models and mouse strains. The toxicity resulting from eIL2 administration before IFNα is dependent on multiple systemic inflammatory cytokines including IL6, IL10, IFNγ, and tumor necrosis factor α. Natural killer (NK) cells are the main cellular contributor to toxicity, but are not essential for tumor control in this system. When pre-conditioned with eIL2, splenic NK cells became hyper-activated and upregulate IFNα signaling proteins that cause an excessive, toxic response to subsequent IFNα exposure. This work illustrates an example where accounting for the temporal dynamics of the immune system in combination therapy treatment schedule can favorably decouple efficacy and toxicity.
KEYWORDS: Dose scheduling, timing, toxicity, cytokines
Introduction
Improvement in efficacy of cancer immunotherapy is generally expected to require combination therapies. For example, recent combination of Nivolumab and Ipilimumab resulted in a 58% rate of objective response in updated results of the phase 3 Checkmate 067 trial in untreated advanced melanoma.1 These two agents act on adaptive immunity via T cells2 ; so perhaps non-T-cell-inflamed cancers could be made responsive by enlisting components of the innate immune system.3 However, maintaining acceptable therapeutic index while increasing the number of administered agents is a challenge. For example, the combination arms of the Checkmate 067 trial resulted in grades 3 or 4 adverse events in 59% of patients and 39% of combination-treated patients had a treatment-related adverse event leading to discontinuation of therapy.
The inflammatory cytokines interleukin (IL) 2 and interferon (IFN) α are promising options for combination therapies because they are FDA approved agents capable of enhancing both innate and adaptive immunity.4,5 As a monotherapy, high dose IL2 engenders 10–20% response rates against renal cancer and melanoma6 and is capable of enhancing innate mechanisms like antibody-dependent cell-mediated cytotoxicity (ADCC)7,8 as well as stimulating T cell activation and proliferation.9(p2) Unfortunately, high dose IL2 therapy must be administered under intensive care conditions due to significant toxicities including fevers and severe hypotension from vascular leak syndrome,10 causing 50% of patients to cease treatment after one of four courses according to a study of metastatic renal cell carcinoma treatment.11 Similar to IL2, IFNα also promotes a multi-faceted anti-cancer response12,13 including direct anti-proliferative effects on cell growth14 and enhanced communication between innate and adaptive immunity.15–17 Beyond any direct tumoricidal effects,18 IFNα is a key cytokine in the stimulator of interferon genes (STING) pathway response and augments maturation of antigen presenting cells (APCs) like dendritic cells (DCs).19,20 Given in high dose in an adjuvant setting in surgically resected melanoma, IFNα improved relapse-free survival (RFS) and had variable effects on overall survival.21–23 Despite improvements in RFS of adjuvant IFNα, toxicity resulted in between 10% and 37% rates of treatment cessation.24,25 Although IL2 and IFNα continue to improve survival in some adjuvant and primary cancer indications, motivation for development of either IL2 or IFNα as new monotherapies has slowed due to these unfavorable therapeutic indices.
Combinations of IL2 and IFNα with each other or with different types of clinically approved immunotherapies have been explored but generally do not have the therapeutic window to offer adequate tumor control. A small clinical trial concurrently dosing these two agents 3 times per week against various cancer types showed very limited efficacy.4 In a later clinical trial for metastatic melanoma that assessed whether the combination of IFNα and IL2 improved chemotherapy, the cytokines were administered continuously over the course of 4 days and added no survival advantage5 Additionally, occurrence of grades 3 and 4 toxicities tripled including nausea, diarrhea, and drops in white blood cell counts below 3000 per mm3 in the combination cytokine group.5 Since efficacy for single agents is often linked to higher dose level and number of doses received, combination therapy treatment schedule follows that precedent. The elements of innate and adaptive immunity believed to be most important for the mechanisms of action of IL2 and IFNα (e.g. CD8 T cell clonal expansion and DC maturation, respectively) can occur at differing times during the steps of the anti-cancer immune response.26 This indicates that the therapeutic window might be improved by dosing agents at different times in treatment schedules rationally designed to match the current immune system response. In fact, emerging evidence suggests that a shorter exposure burst to type I or type II IFNs could prevent tumors from acquiring resistance, while chronic treatment might paradoxically foster resistance to checkpoint blockade.27 Critical thinking is needed to re-direct schedule design away from the paradigm of sustained maximum-tolerated dose (MTD) treatment and towards dosing at the proper timescale and duration for therapeutic efficacy of each individual agent.
Recent preclinical studies using an extended half-life version of IL2 (eIL2)28 and IFNα in combination with tumor targeting antibodies have successfully used temporally designed treatment schedules to significantly improve therapeutic efficacy.29 Staggering the IFNα to be 2 days after a tumor-targeting agent and eIL2 instead of concurrently raised the cure rate from 0% to 60–100% for large, established syngeneic mouse tumor models. Despite the high cure rates from delayed IFNα administration, toxicity remained a significant drawback, with mice losing 10–20% of their body weight. In this work, altering the order of cytokine administration was shown to eliminate toxicity without affecting therapeutic efficacy by tempering the strength of innate inflammation. Although eIL2 was dosed in prior work29 at the same time as the tumor targeting antibody to potentiate ADCC and maintain pharmacological presence during subsequent T cell proliferation days later, the current work was motivated to investigate whether eIL2’s most important role is expansion of activated lymphocytes in a therapy engaging adaptive immunity. When IFNα administration was staggered after a tumor-targeting antibody, treating with eIL2 at the same time or after IFNα eliminated weight loss toxicity without compromising therapeutic efficacy in two tumor models. The reduction in toxicity conferred by dosing eIL2 concurrently instead of before IFNα was mediated by lower systemic levels of inflammatory cytokines and reduced natural killer (NK) cell activation state, which were not essential to therapeutic efficacy. This study underscores the potential importance of order and timing in treatment schedule design to decouple efficacy and toxicity in combination immunotherapy.
Results
The order of eIL2 with respect to IFNα decouples efficacy and toxicity
To investigate ways to lower toxicity of the efficacious combination of tumor targeting antibodies, eIL2, and staggered IFNα, the timing of eIL2 was varied while keeping 1-day separation between antibody and IFNα (Figure 1(a)). Mouse weights were monitored every day or every 2 days and maximal weight loss was observed 2 and 3 days after IFNα (Figure S1a). Subsequent weight change is reported as percent decrease between first day of treatment (0%) and 2 or 3 days after IFNα treatment unless otherwise noted. In C57BL/6 mice with B16F10 tumors treated with the anti-TRP1 antibody TA99, administering eIL2 concurrent with or after IFNα eliminated the 10–20% weight loss seen when eIL2 was given before IFNα (Figure 1(b)). This stark difference in toxicity did not lower the therapeutic efficacy from 20% to 40% survival, even when eIL2 was given as late as 3 days after treatment initiation (Figure 1(c)). We confirmed that IFNα synergized with the eIL2 and tumor targeting antibody to increase efficacy against B16F10 tumors by eliminating IFNα treatment but delivering the eIL2 concurrently with TA99 or 1, 2 or 3 days after TA99 (Figure S1b-d). Our previous work confirmed the importance of both antibody and eIL2 to therapeutic efficacy29 and that monotherapies of eIL2 or TA99 do not lead to tumor cures,28 so we did not repeat these conditions in this work. In this work, the memory response of pooled, surviving mice from B16F10 tumors originally treated with all three agents was examined. Consistent with prior studies,29 rechallenged mice had extended survival compared with age-matched, naïve mice, with around 25–50% cure rate (Figure S1e). Importantly, there was not a statistically significant difference in survival in this rechallenge study between mice originally treated with the staggered (toxic) and concurrent (non-toxic) eIL2 and IFNα regimens. Weight loss curves without IFNα show that weight loss did not occur without this agent (Figure S1a). Since weight loss was eliminated by leaving out IFNα or by giving the eIL2 at or after IFNα, we did not quantify weight loss of antibody alone or under conditions with eIL2. Individual tumor area curves for every treatment condition confirm the survival results (Figure S1f-m).
Figure 1.
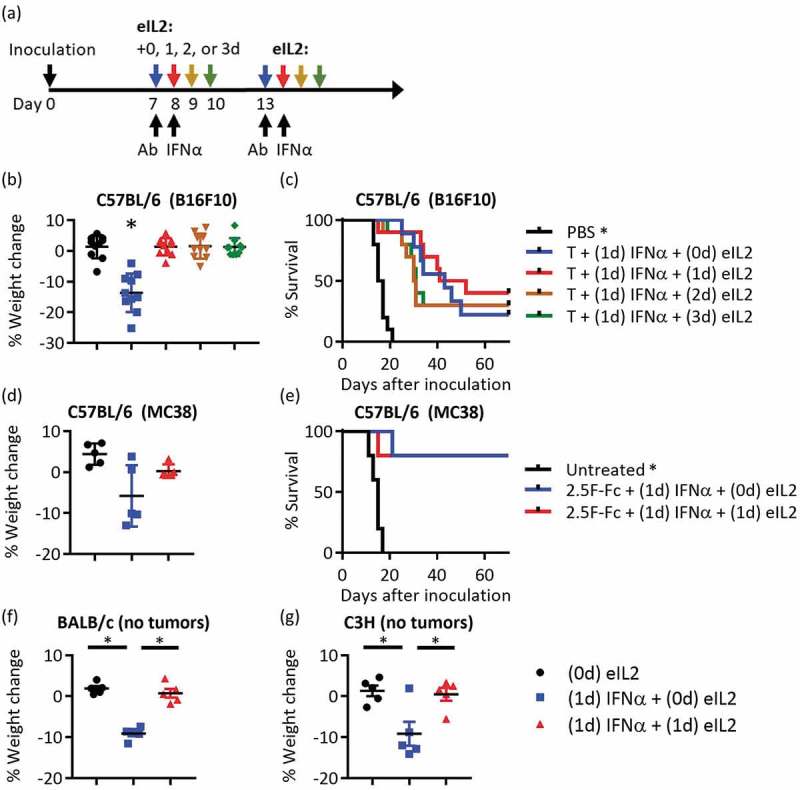
Delaying eIL2 eliminated the weight loss toxicity but did not affect therapeutic efficacy (a) Schematic of experimental design is shown. Two courses of therapy were given beginning on days 7 and 13, keeping the timing of tumor targeting antibody (Ab) and IFNα 1 day apart. In the four groups of treated mice, eIL2 was administered at the same time as Ab (0d), 1d, 2d, or 3d after Ab. (b,c) Percent weight change and survival curves are shown for four different times of eIL2 administration using the TA99 Ab (T) against B16F10 tumors. Reported weight loss is 4 days after TA99. Data are from two independent experiments totaling 9–10 mice per group. (d,e) eIL2 is administered with or 1 day after Ab (concurrent with IFNα) using the 2.5F-Fc antibody-like construct against MC38 tumors. Reported weight loss is 3 days after 2.5F-Fc. p = 0.11 comparing (0d) eIL2 and (1d) eIL2 weight change. Data are from one independent experiment totaling five mice per group. (f,g) In non-tumor bearing BALB/c or C3H mice, percent weight change plots are shown for eIL2 given 1 day before or concurrent with IFNα. No Ab was used for this study. Data are from one independent experiment totaling five mice per group. *Indicates p < 0.05 and error bars are ± SEM.
The same toxicity trends occurred in a different tumor model and in two different mouse strains. In the MC38 colon carcinoma model, toxicity was reduced and efficacy was maintained when eIL2 was given concurrently with IFNα, following treatment with an antibody-like construct (2.5F-Fc) that targets integrins overexpressed on many mouse and human tumors,30 (Figure 1(d,e). Weight loss curves over time (Figure S2a) as well as individual tumor area curves (Figure S2b-f) support the summarized weight loss and survival results. This trend of weight loss from staggered eIL2 and IFNα held independent of the presence of tumors and in different mouse strains. Giving eIL2 at the same time as IFNα in non-tumor-bearing BALB/c and C3H mice ameliorated weight loss observed when eIL2 was given 1 day before IFNα (Figure 1(f,g) and Figure S2g-h), confirming that the observed toxicity is independent of both tumor burden or mouse strain.
Inflammatory serum cytokines are partially responsible for toxicity of staggered eIL2 and IFNα
The sources of differential toxicity between giving eIL2 prior to versus simultaneous with IFNα were investigated through looking for organ-level toxicity and then systemic levels of inflammatory cytokines or chemokines.
Blood clinical chemistry, serum protein and enzyme levels, and organ pathology were assessed at multiple time points after treating mice with the toxic staggered eIL2 and IFNα or giving these agents at the same time. In order to look at time points weeks after treatment began and to avoid conflating tumor-related toxicity with treatment-related toxicity, these studies were completed in non-tumor-bearing C57BL/6 mice. Complete blood count (CBC) analysis revealed an increased proportion of monocytes as a percent of white blood cells in the staggered eIL2 and IFNα-treated group 3 days after the first round of treatment began (Figure S3a), which could reflect systemic over-activation of innate immunity. Although not outside normal ranges, mice from the toxic, staggered treatment had low red blood cell counts and increased mean corpuscular hemoglobin, red cell distribution width, and mean platelet volume (Figure S3c-e). Coupled with a trend toward decreased (not statistically significant) mean cellular hemoglobin concentration (Figure S3f) in both treated groups after the first treatment, the staggered eIL2 and IFNα-treated mice could be suffering from mild anemia, which has been seen in clinical immune-related toxicities.31 The decreased activity of the liver enzyme alkaline phosphatase (Figure S3g) could be indicative of malnourishment, but it was found in both the more and less toxic treatment groups. Decreased blood protein levels and albumin-to-globulin ratio (Figure S3h-j) may be indicative of mild liver or kidney damage, which could be associated mainly with eIL2 treatment. Organ pathologies in the lung, kidney, liver, and spleen seem associated with proximity to eIL2 treatment (Figure S3k-n). Overall, these results do not explain the weight loss differences between the staggered and concurrent eIL2 and IFNα.
Since the toxicity associated with staggered treatment-related weight loss was not manifested in bloodwork or organs examined, inflammatory cytokines and chemokines were assessed using a Luminex multiplex bead-based assay. Mouse serum was collected from B16F10 tumor-bearing mice at multiple time points before and after treatment comparing administering eIL2 1 day before and concurrently with IFNα. Using each time point (excluding the 0+ hours point present for only one treatment condition), the area under the curve (AUC) was calculated for each individual factor for a given treatment condition. The ratios of the AUCs for the toxic versus non-toxic treatment conditions were then plotted for the various factors (Figure 2(a)). This AUC analysis revealed increases in multiple agents including leukemia inhibitory factor (LIF), IL6, IL15, and KC in the toxic (staggered eIL2) treatment group (Figure 2(a)). It was not unexpected that LIF and IL6 both had high AUC ratios, since LIF signals through the same, shared gp130 signaling receptor subunit as IL6 and IL6 plays a well-documented role in autoimmune related toxicities.32 KC, also known as chemokine ligand 1, is a neutrophil chemoattractant and activator that shares signaling with many other chemokines.33 Although not statistically significant, other cytokines or chemokines such as IFNγ, IL5, IL10, tumor necrosis factor (TNF) α, macrophage colony-stimulating factor (M-CSF), and IL12(p40) also were increased in the toxic group in this AUC ratio analysis (Figure 2(a)). The partial least squares regression (PLSR) analysis scores plots for the cytokines/chemokines (variables) (Figure S4a) and weight changes (response) (Figure S4b) indicated the separation of the toxic treatment group (group 2) along both components compared to non-toxic, concurrent IFNα and eIL2 (group 3), toxic and non-toxic NK depletions (groups 7 and 8), and leaving out each of the individual cytokines (groups 4–6). The loadings plot confirmed the importance of KC, IL6, and IL10 with the weight loss toxicity because these AUCs were anti-correlated with weight change (Figure S4c). Thus, increases in these cytokines accompany decreases in weight. LIF, IFNγ, TNFα (upper left cluster of points), IL5, and IL12(p40) were all also anti-correlated with the weight change on the loading plot at least along the first component. IL6, IL10, IFNγ, and TNFα are upregulated in clinical cytokine release syndromes,32 so their contributions to weight loss in the toxic treatment group are not surprising. The average levels over time of every cytokine and chemokine used for the AUC and PLSR analysis were also plotted for every treatment condition, supporting the conclusions of the prior analysis (Figure S5). Note that IL2 was excluded from the AUC and PLSR analysis because eIL2 was given exogenously in most of the treatment courses, and thus the measured IL2 levels did not reflect toxic response of the mice to treatment. Spikes corresponding to eIL2 administration can be seen in the IL2 time course plot in Figure S5 and are absent in the treatment condition without any eIL2. Based on the AUC analysis, the loadings and scores plots from the PLSR analysis, the individual time course plots, and the feasibility of neutralizing these soluble factors, IL5, IL6, IL10, IFNγ, and TNFα were chosen for follow-up studies.
Figure 2.
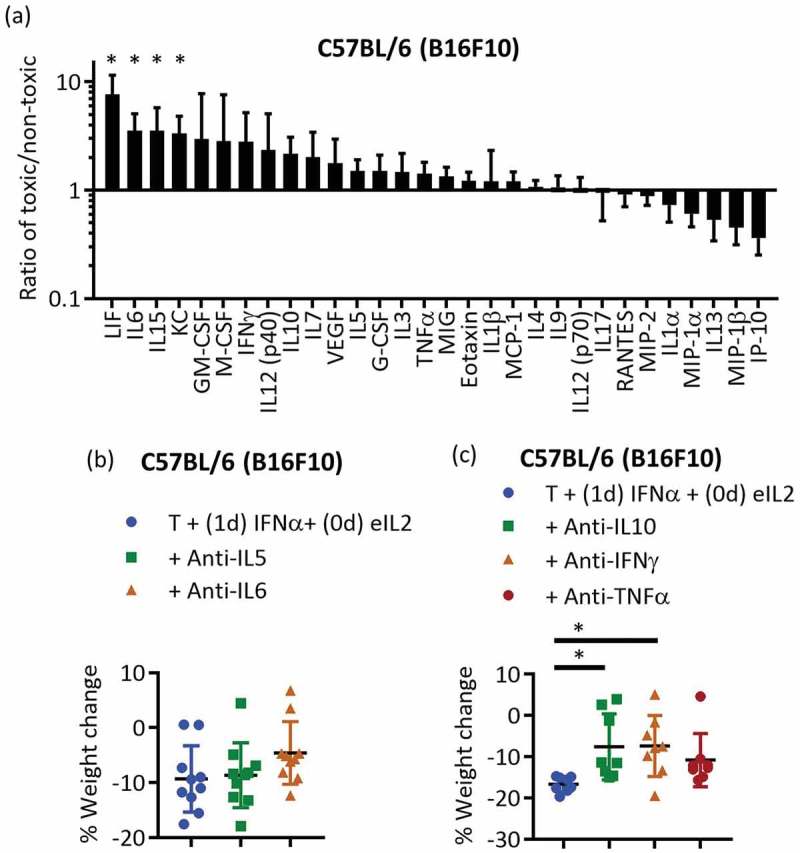
A variety of inflammatory agents were responsible for toxicity of staggered eIL2 before IFNα (a) The ratios of the AUCs over time for individual cytokine levels in the staggered eIL2 and IFNα (toxic) and concurrent eIL2 and IFNα (non-toxic) treatment conditions were plotted. Data are from one independent experiment in B16F10 tumor-bearing mice with three mice per treatment group. (b,c) Cytokine neutralization studies in B16F10 tumor-bearing mice started treatment day 6 (10 mice per group) (b) or day 7 (7–8 mice per group) (c). Percent weight change is shown 3 days after IFNα treatment. Each plot contains data from one independent experiment. *Indicates p < 0.05 and error bars are ± SEM.
Antibody neutralization of IL10 and IFNγ in B16F10 tumor-bearing mice mildly but statistically significantly reduced weight loss toxicity (Figure 2(c)), while TNFα, IL5, and IL6 neutralization did not significantly affect weight loss (Figure 2(b,c), although the full-time course of the weight losses for these agents suggests that all five cytokines may contribute to toxicity (Figure S6a-c). IL5 and IL6 neutralizations were continued during two treatment courses in B16F10 tumor-bearing mice. Survival analysis and individual tumor area curves (Figure S6d-k) do not indicate that IL5 or IL6 contributed to efficacy. Consistent with the Luminex multiplex analysis, no individual cytokine seemed predominantly responsible for the weight loss, characteristic of a broader cytokine storm in response to eIL2 administered before IFNα.
NK cells contribute to toxicity but not efficacy
Once several inflammatory cytokines were identified that contributed to autoimmune-related toxicity, cellular depletions were performed to assess contribution to weight loss toxicity. From an extensive panel of cellular depletions and neutralizations explored to examine weight loss, only depletion using anti-NK1.1 resulted in a statistically significant reduction in weight loss (Figure S7a). Despite the strong correlation of the neutrophil-attracting chemokine KC with weight loss (Figure S4c), depletion of neutrophils through anti-Ly6G did not significantly reduce weight loss.
Depleting NK cells in the toxic treatment schedule of staggered eIL2 and IFNα significantly reduced the weight loss in B16F10-bearing C57BL/6 mice 2 days after IFNα treatment (Figure 3(a)). At all other time points other than the time point 2 days after IFNα, the mice given staggered eIL2 and IFNα with depletion of NK cells have comparable weight change to untreated mice (Figure S7b). Despite the documented role of NK cells in tumor control in other model systems,34,35 NK cells remarkably did not contribute to the 20–40% survival of tumor-bearing mice either in the toxic, staggered eIL2 and IFNα treatment schedule or the non-toxic, concurrent eIL2 and IFNα schedule (Figure 3(b) and Figure S7e-i). Although depletion using the anti-NK1.1 antibody would deplete both NK cells and NK T cells, depleting with the anti-Asialo-GM1 antibody specific to NK cells (and not NK T cells)36 showed that NK cells themselves were responsible for the weight loss (Figure 3(c), Figure S7d). Although anti-Asialo-GM1 also depletes basophils and heterogeneous Asialo-GM1 expression is found on T cell subsets,37 using this antibody in a separate group from anti-NK1.1 showed that NK T cells were not responsible for the decreased weight loss of the NK1.1 depletion. Verification of NK depletion showed that at least 95% of splenic NK cells were depleted (Figure S7c). Previous studies emphasizing a role for NK cells in tumor control were often performed in mice lacking T cells, raising questions of their relevance to immunotherapy with an intact immune system. This particular antibody-plus-cytokine treatment protocol has been shown previously to strongly require CD8+ T cells, macrophages, and CD8+ DCs for efficacy.29
Figure 3.
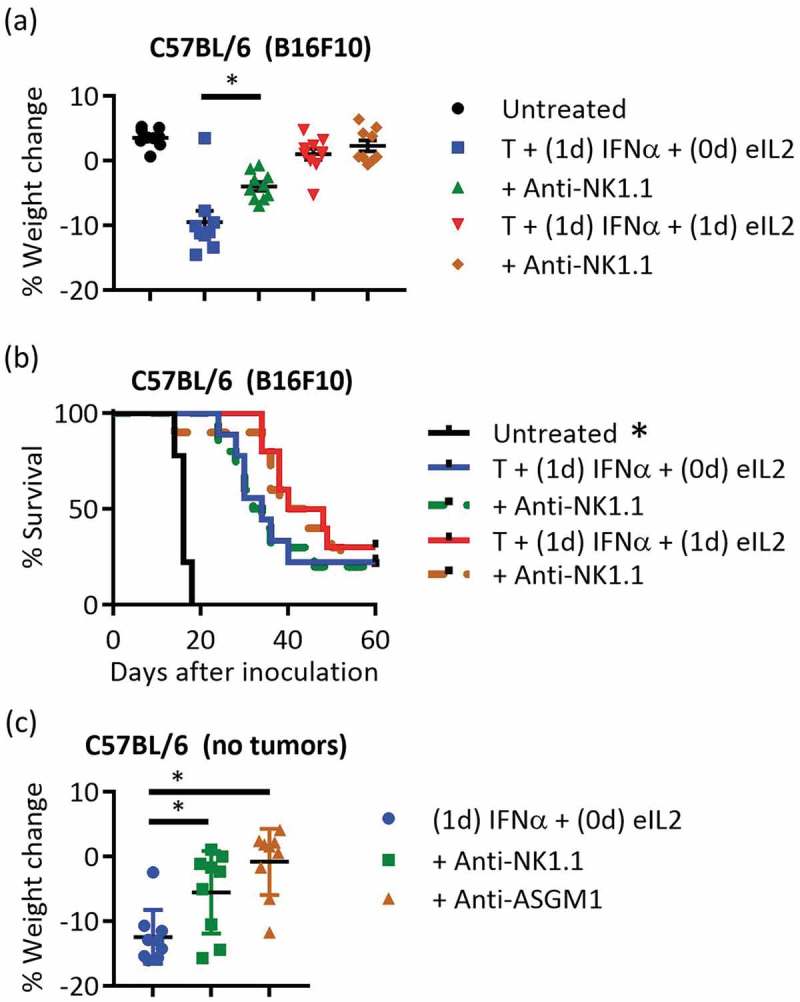
NK cells were important for toxicity but not for therapeutic efficacy (a,b) Percent weight change and survival plots were compared for the toxic and non-toxic treatment conditions with and without NK cell depletion in the B16F10 tumor model. Data are from two independent experiments totaling nine or ten mice per group. Treatment in this study began day 6 instead of day 7, so TA99 (T) was dosed days 6 and 12, IFNα was dosed days 7 and 13, and eIL2 was dosed days 6 and 12 or 7 and 13. (c) Non-tumor bearing mice were given anti-NK1.1 or anti-ASGM1 depleting antibodies before treatment with 1-day staggered eIL2 and IFNα. Percent weight changes were quantified 2 days after IFNα. Data are from one independent experiment totaling nine mice per group. *Indicates p < 0.05 and error bars are ± SEM.
Staggering eIL2 before IFNα increases NK cell activation and sensitivity to IFNα
To determine how NK cells contributed to toxicity when treated with eIL2 prior to IFNα, splenic NK cells were characterized using flow cytometry 1 day after treatment with eIL2, which was when IFNα was typically administered. Representative flow cytometry plots for each treatment condition are shown in Figures S8-9. NK cell activation markers described previously such as CD11c, CD43, CD44, CD69, and B22038 were significantly increased in spleens of B16F10 tumor-bearing mice 1 day after antibody and eIL2 treatment compared with treatment with antibody alone (Figure 4(a–e)). At a higher activation state, NK cells would be poised to produce more inflammatory cytokines and react more strongly to IFNα. In addition to increasing activation marker expression, eIL2 caused an increase in expression of signaling proteins important to the IFNα response. One day after eIL2 administration, splenic NK cells had increased expression of the IFNα receptor IFNΑR1 (Figure 4(f)). Since IFNα signaling is propagated through JAK/STAT pathways,39 levels of STAT1 and STAT3 proteins were examined and also shown to be increased on NK cells 1 day after eIL2 administration (Figure 4(g,h).
Figure 4.
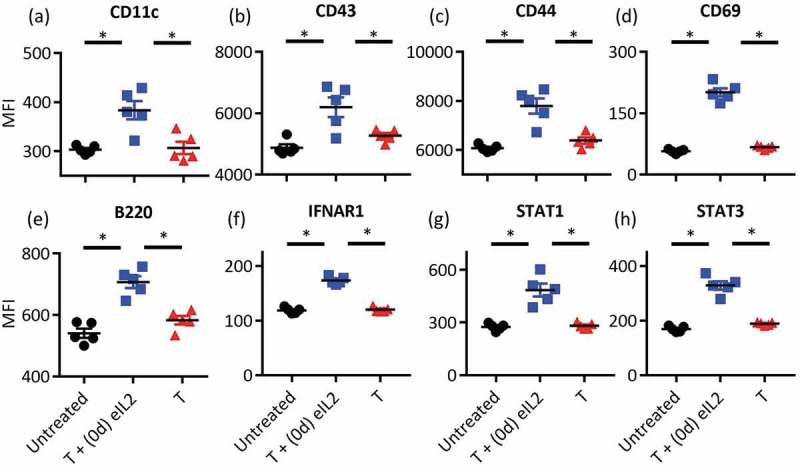
Staggering eIL2 before IFNα increased the NK cell activation prior to exposure to IFNα Splenic NK cells were harvested from mice bearing established B16F10 tumors 1 day after the indicated treatment and NK cell activation markers (a–e) and IFNα-responsive signaling proteins (f–h) were quantified by flow cytometry. Median fluorescence intensity (MFI) of all single, live, CD3 negative, NK1.1 positive cells is plotted on the y-axis. Data are from one independent experiment totaling five mice per group, with separate staining panels for NK activation markers (a–e) and IFN signaling proteins (f–h). *Indicates p < 0.05 and error bars are ± SEM.
Discussion
Combination cancer immunotherapies hold promise for improving clinical response rates, but strategies to mitigate the associated auto-immune toxicities are needed. Our work used the cytokines IFNα and IL2 in syngeneic mouse tumor models to demonstrate that therapeutic efficacy and toxicity may sometimes be decoupled by alterations in the treatment schedule. Although translation of the exact murine timescales into the clinic would be challenging, several lessons can be learned from this work.
The common clinical practice in combination therapy of using the treatment schedule of each individual agent until toxicity ends treatment should be considered carefully in the context of temporally designed treatment schedules. For example, a short burst of IFNα after a tumor-targeting antibody is significantly more efficacious than starting to dose IFNα earlier because it allows DCs to take up antigen before maturation by IFNα.29 Clinically, IFNα is infused daily or even PEGylated to increase exposure time,40 but in vitro studies looking at DCs’ ability to activate CD4 T cells indicate that shorter bursts of DC activation induce a type 1 T cell helper phenotype much more effectively than longer chronic exposure.41 In a clinical trial of advanced metastatic melanoma, testing chemotherapy and IFNα with or without the addition of IL2 resulted in no clinical improvement from IL2.42 Although the results presented in the current work suggest that the order of IFNα before IL2 in the clinical trial would minimize toxicity of these two agents in combination, the lengthy, high-dose of IFNα and failure to stagger IFNα after the chemotherapy could be responsible for the lack of therapeutic synergy of these cytokines. Emerging evidence suggesting that the STING pathway is critical to the success of therapies like checkpoint blockade43 and radiation44 underscores the importance of lessons about IFNα timing. Although local administration of STING agonists can circumvent toxicity,45 carefully choosing the timing of combination therapy partners to avoid pre-activation of NK cells is critical, given the toxic strength of their IFNα response when pre-activated by IL2. Agents intended to support T cell expansion and function, such as IL2, may be safest and most efficacious after IFNα administration, whereas delivery of antigen-generating agents like tumor targeting antibodies, chemotherapy, or radiation will be important before IFNα and DC maturation.
Generating a milder burst of initial innate activation using tumor targeting antibodies without initial IL2 may avoid early systemic toxicity, while being sufficient to release tumor debris to be presented to the adaptive immune system. This manuscript demonstrates that cytokines like IL10 and IFNγ contribute to the early systemic toxicity of staggered eIL2 and IFNα therapy. Additional cytokine depletions of IL5 and IL6, while not statistically significant compared to their control group in Figure 2(b), may still have some contribution toward weight loss. Although cytokines like IL246 and IFNs47 activate NK cells and these cells can exhibit potent anti-tumor activity,48 other innate cell types are capable of initiating more mild inflammation in response to tumor targeting antibodies. Macrophages are capable of antibody-mediated attack of tumor cells and subsequent antibody-enhanced DC uptake of tumor antigens can initiate a productive adaptive response.49 Antibodies can also engage with eosinophils to generate respiratory burst in response to tumor cells as well as utilize neutrophils or the complement system to attack cancer.28 Indeed, the same tumor targeting antibody used in this work, TA99, induced respiratory burst in B16F10 tumors within the first few days after treatment as quantified by a luminol assay in the previous work.28 With initial antibody-driven innate inflammation, IL2 could be given as late as 3 days after tumor targeting antibody in this work and maintain the same efficacy. The toxicities of IL2 are well documented,50 so delaying IL2 to avoid excessive, toxic antagonism of innate inflammation from a different combination therapy agent like tumor targeting antibodies, radiation, or chemotherapy would be ideal. In addition to contributing to toxicity, prematurely dosing IL2 prior to T cell priming could preferentially activate Tregs because of constitutive CD25 expression51,52 and may also induce anergy in helper T cells.53 Although IL2 has traditionally been characterized as an important NK cell stimulator,46 IL2’s most important role in cancer immunotherapy may be in driving adaptive instead of innate inflammation. Clinical analysis of IL2 immunotherapy responses to melanoma and renal cancer have shown durable responses indicative of adaptive immunity54 and IL2 is commonly used to enhance T cell proliferation and survival in adoptive cell therapies.55,56 NK cells and IL2 may be superfluous to the initial innate melee caused by a tumor-targeting antibody as long as adaptive immunity can be expanded at a later time by IL2 treatment.
This work illustrates that an MTD-focused chronic treatment paradigm should be reconsidered to balance toxicity and efficacy. Although IL2 has traditionally been characterized as an important NK cell stimulator,46 IL2’s most important role in cancer immunotherapy might instead be to drive adaptive immunity later in the treatment cycle after mild innate inflammation is initiated by other agents. Re-design of treatment schedules with careful attention to order and timing may decouple efficacy and toxicity, opening the therapeutic window of previously discarded cytokine-based therapeutic combinations like IL2 and IFNα.
Methods
Mice
Female C57BL/6 (Taconic), C3H (Taconic), and Balb/c mice (Jackson) were used at 6–10 weeks of age. Mice were housed in pathogen free environments. All procedures were approved by the MIT Division of Comparative Medicine (DCM) and consistent with federal, state, and local regulations using animal protocol number 0515-043-18 under MIT’s Institutional Animal Care and Use Committee.
Cell lines and media
B16F10 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS and Penicillin-Streptomycin and MC38 cells were maintained in DMEM with 10% FBS.
Tumor treatment experiments
For experiments with B16F10 and MC38 tumors, mice were inoculated subcutaneously in the right flank with 106 cells. The following agents were administered through intraperitoneal (IP) injection at the following days after tumor inoculation unless otherwise noted: 100 µg TA99 (B16F10 tumors, days 7 and 13), 500 µg 2.5F-Fc30 (MC38 tumors, days 7 and 13), 30 µg eIL2 (days 7 and 13 or 8 and 14), and 10 µg IFNα (days 8 and 14). Tumor areas were calculated as the product of the two perpendicular tumor diameters and mice were euthanized when tumor area exceeded 100 mm2. The few mice that died from reasons other than tumor area (low body condition due to ulceration or weight loss exceeding 20%) are noted in figure legends and are not included in the survival study analysis or plots.
For tumor rechallenge studies, age-matched naïve mice and cured mice pooled from B16F10 tumor studies treated with TA99, eIL2, and IFNα were rechallenged with 105 B16F10 cells on the left flank at least 100 days after cessation of the original treatment. Rechallenged mice were not included in this study if they had any type of depletion during original treatment.
As opposed to more invasive assessments of toxicity, weight loss is our primary metric for toxicity in tumor studies. It can be regularly quantified over time without weakening a mouse to take serum or sacrificing the mouse to look in the organs. Weight loss is generally acceptable as a metric of mouse body condition and has been used to monitor immunotherapy toxicity in mice57 and cynomolgus macaques.58
Depletion and neutralization studies
All depleting or neutralizing antibody injections were purchased from Bio X Cell and injected IP unless otherwise noted. Anti-IL5 (TRFK5) was given at 1 mg per dose 1 day before each round of treatment began. Anti-IL6 (MP5-20F3) was given at 400 µg per dose every 2 days starting 1 day before treatment began. Anti-IL10 (JES5-2A5) was given at 500 µg per dose every 3 days starting 1 day before treatment began. Antibodies to neutralize IFNγ (XMG1.2) or TNFα (XT3.11) were given at 600 µg per dose every 2 days starting 1 day before treatment began. Anti-NK1.1 (PK136) was given at 400 µg per dose every 4 days starting 2 days before treatment began for a total of four doses during survival studies. Anti-Asialo-GM1 (Poly21460, Biolegend) was given at 50 µg per dose every 4 days starting 2 days before treatment began. Schedules and dosing for depletions or neutralization were chosen to be at least as frequent and as high of doses as previous work.29,59–61 Although we recognize that verifying NK cell depletion is problematic when done with the same clone, other strong antibody clones used to stain for NK1.1 do not exist and so prior work has demonstrated depletion by staining with the same clone.61
For the supplemental depletion and neutralization study, depletions not already described were done by giving anti-NK1.1, anti-Ly6G (1A8), anti-CD8α (2.43), anti-CD4 (GK1.5), or anti-CD19 (1D3) at 600 µg per dose every 3 days starting 2 days before treatment began. Anti-CSF1R (AFS98) antibody was given at 600 µg per dose every 2 days starting 2 days before treatment began.
Protein production and purification
All therapeutic proteins were made in house. TA99 (mouse IgG2c heavy chain and kappa light chain) was purified from a stably transfected HEK293 F cell line grown in FreeStyle 293 media (Life Technologies). 2.5F-Fc (mouse IgG2c heavy chain) and eIL2 (a fusion of mouse serum albumin and IL2 (MSA-IL2))28 were purified 6–8 days after transfecting gWiz plasmids into HEK293 cells using polyethylenimine in FreeStyle supplemented with OptiPro (Life Technologies). For TA99 and 2.5F-Fc, supernatant from centrifuged HEK293 cells was purified through rProtein A Sepharose Fast Flow resin according to manufacturer’s instructions (GE Healthcare). MSA-IL2 was purified from HEK293 cell supernatant using TALON® Metal Affinity Resin according to manufacturer’s instructions (Clontech) and subsequent gel chromatography on a HILOAD 16/600 Superdex 200 PG column on an AKTApurifier 10 fast protein liquid chromatography machine (GE Healthcare). IFNα was produced in Rosetta-gami 2 (DE3) competent E. coli (EMD Millipore) as previously described.29 All proteins were tested to be below 0.1 EU/dose by the QCL-1000 chromogenic LAL assay (Lonza) and run on reducing and non-reducing SDS PAGE 4–12% Bis-Tris gels (Life Technologies) to confirm size. Fc-IL2 fusion protein used as eIL2 for the supplemental depletion and neutralization study was made by transfecting HEK293 cells and purified in two steps using TALON resin and anti-FLAG M2 Affinity Gel (Sigma-Aldrich) as previously described.28
Flow cytometry
For analysis of NK cell activation, mouse spleens were collected with no treatment or 1 day after treatment with TA99 and eIL2 or TA99 alone. After dissociation through a 70um filter and ACK lysis, single cell suspensions were stained and run on a LSRFortessa flow cytometer (BD Biosciences) at the Koch Institute Swanson Biotechnology Center Flow Cytometry Core Facility. eBioscience™ Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific) and TruStain fcX (Biolegend) were used for viability and Fc block prior to antibody staining. Antibodies against CD3 (145-2C11), NK1.1 (PK136), B220 (RA3-6B2), CD11c (N418), CD43 (1B11), CD44 (IM7), CD69 (H1.2F3), and IFNAR-1 (MAR1-5A3) were purchased from Biolegend. Antibodies against STAT1 (Clone 1/Stat1) and STAT3 (232209) were purchased from BD Biosciences and Thermo Fisher Scientific, respectively. For intracellular staining of STAT proteins, the cells were first fixed and permeabilized using the BD Biosciences Cytofix/Cytoperm kit according to manufacturer instructions. For the gating strategy, NK cells were gated first for single cells and viability, then on CD3 low and NK1.1 high, before plotting the resulting NK cell population median fluorescence intensity.
Systemic cytokine and chemokine levels
Blood was collected from three B16F10 tumor-bearing mice per treatment group into BD Microtainer Clot Activator/SST Gel tubes at time points 3, 24, 27, 51, and 75 h after injection of TA99. The second and third time points are before and 3 h after injection of IFNα. eIL2 was injected either concurrently with TA99 or concurrently with IFNα. Serum was collected after blood centrifugation, diluted 1:2 with PBS, and flash frozen. Luminex multiplex assays were completed by Eve Technologies using the Mouse Cytokine Array/Chemokine Array 31-Plex panel. IL2 was excluded from the analysis because IL2 was injected systemically so it would not be indicative of a toxic response.
All data were analyzed using Microsoft Excel or Matlab (Mathworks). To analyze the data from the multiplex assay, AUCs of concentration versus time were calculated for each cytokine/chemokine across the different treatment conditions for all time points. The ratios of these AUCs for each cytokine/chemokine were plotted for TA99 and 1-day staggered IFNα comparing eIL2 given with TA99 versus given with IFNα.
PLSR analysis with 3 components and sevenfold cross-validation was also done to assess which chemokine and cytokine AUCs best explained the weight loss for each mouse. The AUCs using only the time points 3 h after IFNα up to 2 days after IFNα were used for best explanation of the weight loss, quantified as change from treatment start to 2 days after IFNα. AUCs and weight losses were z-scored prior to the PLSR. The first two components explained 52% and 19% of the variation in weight change, so plots shown were composed of loadings and scores for just the first two components.
Organ and blood toxicity
Mice were euthanized at indicated time points after first treatment with TA99 and whole blood samples or serum from non-tumor-bearing C57BL/6 mice were submitted at to the diagnostics facility at the MIT DCM. This facility performed automated CBC analysis and sent frozen serum samples to be screened on a Chem 11 panel by IDEXX Laboratories. Organs were also taken at the same time points for histology by the Histology Core Facility at the Koch Institute’s Swanson Biotechnology Center. Kidneys, spleen, and liver were fixed in 10% neutral buffered formalin before being transferred to 70% ethanol for preservation prior to sectioning and H&E staining. During necropsy, lungs were first inflated with 10% neutral buffered formalin through intratracheal injection prior to excision, fixation, sectioning, and staining. Each organ was assessed for level of inflammatory infiltrates by pathologist Dr. Roderick Bronson at the Histology Core.
Statistical analysis
All statistical analysis was done using Graphpad Prism statistical software. Kaplan Meyer tumor survival curves were compared using a Log-rank (Mantel-Cox) test. Comparison between the toxic and each other treatment group in weight loss plots was done using one-way ANOVA with Dunnett’s multiple comparisons test. Flow cytometry plots compared all groups with each other through one-way ANOVA with Tukey’s multiple comparisons test. AUC ratios were assessed for significance using a two-way ANOVA with Bonferroni’s multiple comparisons test. Organ toxicity study results were examined using one t-test per time point for each metric, then correcting for multiple comparisons using the Holm-Sidak method with an alpha of 0.05. All *indicates p < 0.05 for the statistical test and all error bars are ± SEM.
Funding Statement
This work was supported by the National Cancer Institute [CA174795]
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-A-AS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD.. Combination immunotherapy: a road map. J ImmunoTher Cancer. 2017;5:16. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budd GT, Osgood B, Barna B, Boyett JM, Finke J, Medendorp SV, Murthy S, Novak C, Sergi J, Tubbs R.. Phase I clinical trial of interleukin 2 and alpha-interferon: toxicity and immunologic effects. Cancer Res. 1989;49(22):6432–6436. [PubMed] [Google Scholar]
- 5.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Seipp CA, Einhorn JH, White DE, Steinberg SM. Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. J Clin Oncol. 1999;17(3):968–975. doi: 10.1200/JCO.1999.17.3.968. [DOI] [PubMed] [Google Scholar]
- 6.Skrombolas D, Frelinger JG. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev Clin Immunol. 2014;10(2):207–217. doi: 10.1586/1744666X.2014.875856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flieger D, Spengler U, Beier I, Kleinschmidt R, Hoff A, Varvenne M, Sauerbruch T, Schmidt-Wolf I. Enhancement of antibody dependent cellular cytotoxicity (ADCC) by combination of cytokines. Hybridoma. 1999;18(1):63–68. doi: 10.1089/hyb.1999.18.63. [DOI] [PubMed] [Google Scholar]
- 8.Hank JA, Robinson RR, Surfus J, Mueller BM, Reisfeld RA, Cheung NK, Sondel PM. Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant interleukin 2. Cancer Res. 1990;50(17):5234–5239. [PubMed] [Google Scholar]
- 9.Cousens LP, Orange JS, Biron CA. Endogenous IL-2 contributes to T cell expansion and IFN-gamma production during lymphocytic choriomeningitis virus infection. J Immunol. 1995;155(12):5690–5699. [PubMed] [Google Scholar]
- 10.Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, Atkins MB. High dose interleukin-2 (Aldesleukin) – expert consensus on best management practices-2014. J ImmunoTher Cancer. 2014;2:26. doi: 10.1186/s40425-014-0026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allard CB, Gelpi-Hammerschmidt F, Harshman LC, Choueiri TK, Faiena I, Modi P, Chung BI, Tinay I, Singer EA, Chang SL. Contemporary trends in high-dose interleukin-2 use for metastatic renal cell carcinoma in the United States. Urol Oncol. 2015;33(11):496.e11–496.e16. doi: 10.1016/j.urolonc.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gresser I, Bourali C, Lévy JP, Fontaine-Brouty-Boyé D, Thomas MT. Increased survival in mice inoculated with tumor cells and treated with interferon preparations. Proc Natl Acad Sci USA. 1969;63(1):51–57. doi: 10.1073/pnas.63.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilcek J. Fifty years of interferon research: aiming at a moving target. Immunity. 2006;25(3):343–348. doi: 10.1016/j.immuni.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Paucker K, Cantell K, Henle W. Quantitative studies on viral interference in suspended L cells. Virology. 1962;17(2):324–334. doi: 10.1016/0042-6822(62)90123-X. [DOI] [PubMed] [Google Scholar]
- 15.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu Y-X. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell. 2014;25(1):37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidler IJ, Heicappell R, Saiki I, Grutter MG, Horisberger MA, Nuesch J. Direct antiproliferative effects of recombinant human interferon-alpha B/D hybrids on human tumor cell lines. Cancer Res. 1987;47(8):2020–2027. [PubMed] [Google Scholar]
- 19.Luft T, Pang KC, Thomas E, Hertzog P, Hart DN, Trapani J, Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161(4):1947–1953. [PubMed] [Google Scholar]
- 20.Santodonato L, D’Agostino G, Nisini R, Mariotti S, Monque DM, Spada M, Lattanzi L, Perrone MP, Andreotti M, Belardelli F, et al. Monocyte-derived dendritic cells generated after a short-term culture with IFN-alpha and granulocyte-macrophage colony-stimulating factor stimulate a potent Epstein-Barr virus-specific CD8+ T cell response. J Immunol. 2003;170(10):5195–5202. doi: 10.4049/jimmunol.170.10.5195. [DOI] [PubMed] [Google Scholar]
- 21.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern cooperative oncology group trial EST 1684. J Clin Oncol. 1996;14(1):7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 22.Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE, Ernstoff MS, Smith TJ, Rao U, Steele M, Blum RH. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000;18(12):2444–2458. doi: 10.1200/JCO.2000.18.12.2444. [DOI] [PubMed] [Google Scholar]
- 23.Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS, Ernstoff MS, Rao U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol. 2001;19(9):2370–2380. doi: 10.1200/JCO.2001.19.9.2370. [DOI] [PubMed] [Google Scholar]
- 24.Eggermont AMM, Suciu S, Santinami M, Testori A, Kruit WHJ, Marsden J, Punt CJA, Salès F, Gore M, Mackie R, et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet. 2008;372(9633):117–126. doi: 10.1016/S0140-6736(08)61033-8. [DOI] [PubMed] [Google Scholar]
- 25.Tarhini AA, Gogas H, Kirkwood JM. IFN-α in the treatment of melanoma. J Immunol. 2012;189(8):3789–3793. doi: 10.4049/jimmunol.1290060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Benci JL, Xu B, Qiu Y, Tj W, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, Kauke MJ, Moynihan KD, Angelini A, Williams RT, et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell. 2015;27(4):489–501. doi: 10.1016/j.ccell.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzeng A, Kauke MJ, Zhu EF, Moynihan KD, Opel CF, Yang NJ, Mehta N, Kelly RL, Szeto GL, Overwijk WW, et al. Temporally programmed CD8α(+) DC activation enhances combination cancer immunotherapy. Cell Rep. 2016;17(10):2503–2511. doi: 10.1016/j.celrep.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwan BH, Zhu EF, Tzeng A, Sugito HR, Eltahir AA, Ma B, Delaney MK, Murphy PA, Kauke MJ, Angelini A, et al. Integrin-targeted cancer immunotherapy elicits protective adaptive immune responses. J Exp Med. 2017;214(6):1679–1690. doi: 10.1084/jem.20160831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan U, Ali F, Khurram MS, Zaka A, Hadid T. Immunotherapy-associated autoimmune hemolytic anemia. J ImmunoTher Cancer. 2017;5. doi: 10.1186/s40425-017-0214-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121(24):4930–4937. doi: 10.1182/blood-2013-02-486217. [DOI] [PubMed] [Google Scholar]
- 34.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18(9):1004–1015. doi: 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 35.López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–154. doi: 10.1016/j.ccell.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13(4):459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- 37.Nishikado H, Mukai K, Kawano Y, Minegishi Y, Karasuyama H. NK cell-depleting anti-asialo GM1 antibody exhibits a lethal off-target effect on basophils in vivo. J Immunol. 2011;186(10):5766–5771. doi: 10.4049/jimmunol.1100370. [DOI] [PubMed] [Google Scholar]
- 38.Elpek KG, Rubinstein MP, Bellemare-Pelletier A, Goldrath AW, Turley SJ. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Ralpha complexes. Proc Natl Acad Sci USA. 2010;107(50):21647–21652. doi: 10.1073/pnas.1012128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 40.Patel JN, Walko CM. Sylatron: a pegylated interferon for use in melanoma. Ann Pharmacother. 2012;46(6):830–838. doi: 10.1345/aph.1Q791. [DOI] [PubMed] [Google Scholar]
- 41.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1(4):311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 42.Keilholz U, Punt CJA, Gore M, Kruit W, Patel P, Lienard D, Thomas J, Proebstle TM, Schmittel A, Schadendorf D, et al. Dacarbazine, cisplatin, and interferon-alfa-2b with or without interleukin-2 in metastatic melanoma: a randomized phase III trial (18951) of the European organisation for research and treatment of cancer melanoma group. J Clin Oncol. 2005;23(27):6747–6755. doi: 10.1200/JCO.2005.03.202. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114(7):1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-R, Lemmens E, Banda T, Leong JJ, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henney CS, Kuribayashi K, Kern DE, Gillis S. Interleukin-2 augments natural killer cell activity. Nature. 1981;291(5813):335–338. doi: 10.1038/291335a0. [DOI] [PubMed] [Google Scholar]
- 47.Swann JB, Hayakawa Y, Zerafa N, Sheehan KCF, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178(12):7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- 48.de Andrade LF, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, Tey S-K, Takeda K, Zitvogel L, Martinet L, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74(24):7298–7308. doi: 10.1158/0008-5472.CAN-14-1339. [DOI] [PubMed] [Google Scholar]
- 49.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 51.Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, Höfer T. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci USA. 2010;107(7):3058–3063. doi: 10.1073/pnas.0812851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, Abbas AK. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. 2009;183(1):332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sckisel GD, Bouchlaka MN, Monjazeb AM, Crittenden M, Curti BD, Wilkins DEC, Alderson KA, Sungur CM, Ames E, Mirsoian A, et al. Out-of-sequence signal 3 paralyzes primary CD4+ T cell dependent immunity. Immunity. 2015;43(2):240–250. doi: 10.1016/j.immuni.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang JC, Sherry RM, Steinberg SM, Topalian SL, Schwartzentruber DJ, Hwu P, Seipp CA, Rogers-Freezer L, Morton KE, White DE, et al. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J Clin Oncol. 2003;21(16):3127–3132. doi: 10.1200/JCO.2003.02.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nat Rev Clin Oncol. 2011;8(10):577–585. doi: 10.1038/nrclinonc.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo Y, Luan L, Rabacal W, Bohannon JK, Fensterheim BA, Hernandez A, Sherwood ER. IL-15 Superagonist–mediated immunotoxicity: role of NK Cells and IFN-γ. J Immunol Author Choice. 2015;195(5):2353–2364. doi: 10.4049/jimmunol.1500300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selby MJ, Engelhardt JJ, Johnston RJ, Lu L-S, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, In vitro functional studies, and cynomolgus macaque toxicology. PLoS One. 2016;11(9):e0161779. doi: 10.1371/journal.pone.0161779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530(7591):434–440. doi: 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- 60.Liang Y, Yang K, Guo J, Wroblewska J, Fu Y-X, Peng H. Innate lymphotoxin receptor mediated signaling promotes HSV-1 associated neuroinflammation and viral replication. Sci Rep. 2015;5:10406. doi: 10.1038/srep10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22(12):1402–1410. doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.