

ABSTRACT
Single-agent immunotherapy, including with immune checkpoint inhibition with anti-PD-1 antibody, has not extended survival in patients with malignant glioma. However, PD-1 inhibition may still play a role in combination immunotherapy with multiple agents. In this study, we evaluated anti-PD-1 antibody treatment in combination with multiple approaches, including vaccination and agonist anti-OX40 immunotherapy, as well as triple combination immunotherapy with each of the above agents in a murine glioma model. Treatments were delivered on days 3,6, and 9 after intracranial implantation of glioma cells in the right frontal lobes of the mice. Vaccination consisted of subcutaneous implantation of irradiated GL261 cells engineered to express GM-CSF. We harvested splenocytes and brain tissue 18 days after glioma implantation and analyzed them by ELISPOT and flow cytometry, respectively. Treated mice surviving for 120 days were challenged with implantation of large numbers of GL261 cells and either followed for survival or sacrificed for study of the memory response. Survival was assessed by the Kaplan-Meier method and the log-rank test. Means were compared by the 2-tailed student’s t-test. We report that combining anti-PD-1 immunotherapy with either vaccination or agonist anti-OX40 immunotherapy improves survival in GL261-bearing mice compared with any of the above as monotherapy. Triple combination immunotherapy with vaccination, anti-PD-1 antibody, and agonist anti-OX40 antibody results in long-term survival in all mice. Triple combination immunotherapy resulted in an elevated CD4+/CD8 + T lymphocyte ratio amongst tumor-infiltrating lymphocytes as well as a diminished fraction of regulatory T lymphocytes, likely reflective of a more vigorous Th1 antitumor response.
KEYWORDS: Glioma, glioblastoma, immunotherapy, vaccination, PD-1, OX40, immune checkpoint, GL261
Introduction
Malignant glioma remains an incurable disease. Advances in surgical technique, chemotherapy penetration into brain tumors, and understanding of the genetic underpinnings of glial tumors have led to relatively little improvement in overall outcome over the past generation. Since the Food and Drug Administration approval of temozolomide for glioblastoma in 2005, only the use of tumor-treating fields applied externally has been associated with improved survival, both for patients with newly diagnosed and recurrent disease.1,2 Median overall survival for patients with glioblastoma is currently estimated to be between 14 and 18 months.3,4
As has recently been the case with cancer, in general, there has been much interest in the use of immunotherapy against glioblastoma. Preclinical demonstration of the efficacy of vaccination has led to several randomized phase 2 and phase 3 clinical studies, including with peptides,5 dendritic cells,6 and heat shock proteins derived from patient tumor specimens.7,8 While much important work continues, initial results from several high-profile trials have been negative.
Likewise, clinical studies of immune checkpoint inhibition have not, to date, demonstrated improved overall survival for patients with glioblastoma. Most notable of these is the Checkmate −143 study (NCT02017717), which, after an initial dosing exploratory phase,9 randomized patients with recurrent glioblastoma to receive either anti-PD1 immunotherapy with nivolumab or anti-VEGF treatment with bevacizumab. Though toxicity was acceptable, there were no differences in overall survival, 12-month overall survival, or progression-free survival – leading to much disappointment in the neuro-oncology community.10,11
We propose that effective immunotherapy against glioblastoma is unlikely to be achieved by monotherapy, particularly by single-agent checkpoint inhibition. Glioblastoma-associated immunosuppression operates on many fronts, and overcoming it will require targeted reversal of diverse systemic and intratumoral immunodeficiencies in these patients, including lack of antigen recognition, systemic immunosuppression of T lymphocytes, and a hostile tumor microenvironment that drives local T cell dysfunction. We have previously demonstrated in murine glioma models that combining whole tumor cell vaccination with immune checkpoint-active molecules improves survival via complementary mechanisms.12 Likewise, in melanoma patients, combination immunotherapy with anti-CTLA-4 immunoglobulin and anti-PD-1 immunoglobulin is highly efficacious – more so than single-agent treatment.13 To build upon these concepts for the use of anti-PD1 immunotherapy in glioma, we sought to examine the effects of combining anti-PD1 antibody with whole tumor cell vaccination (GVAX) or with agonist anti-OX40 monoclonal antibody.
OX40 is a member of the Tumor Necrosis Factor Receptor (TNFR) superfamily that is upregulated upon antigen-specific stimulation of T cells, particularly on CD4+ cells.14 Upon ligation, there is increased clonal proliferation, expression of stimulatory cytokines, and expansion of memory populations. Furthermore, OX40 ligation on effector T cells can render them insensitive to suppression by regulatory T cells and, likewise, ligation on OX40-expressing regulatory T cells themselves directly inhibits their suppressive capacity.15
We have shown that vaccination of patients with recurrent malignant glioma with irradiated autologous tumor cell mixed with irradiated GM-K562 cells (analogous to GVAX) leads to lymphocyte activation, specifically marked by significant upregulation of OX40 on CD4+ lymphocytes.16 We, therefore, hypothesized that antigen-specific stimulation by whole tumor vaccination might synergize with OX40 ligation, which we confirmed in recently published preclinical work.12 In the current study, we examine triple combination immunotherapy in the GL261 model, simultaneously treating with vaccination, anti-PD-1 antibody, and agonist anti-OX40 antibody.
Results
PD-1 inhibition in combination with GVAX improves survival in mice bearing established intracranial glioma
As suggested in prior work,17 also using the GL261 glioma model, PD-1 inhibition alone improved survival in comparison to both control and vaccinated glioma-bearing mice and was typically associated with about 20% long-term survival and cure. Combination immunotherapy with GVAX and anti-PD-1 antibody improved long term survival to 50% of the animals, statistically significantly better than either treatment alone (Figure 1a).
Figure 1.
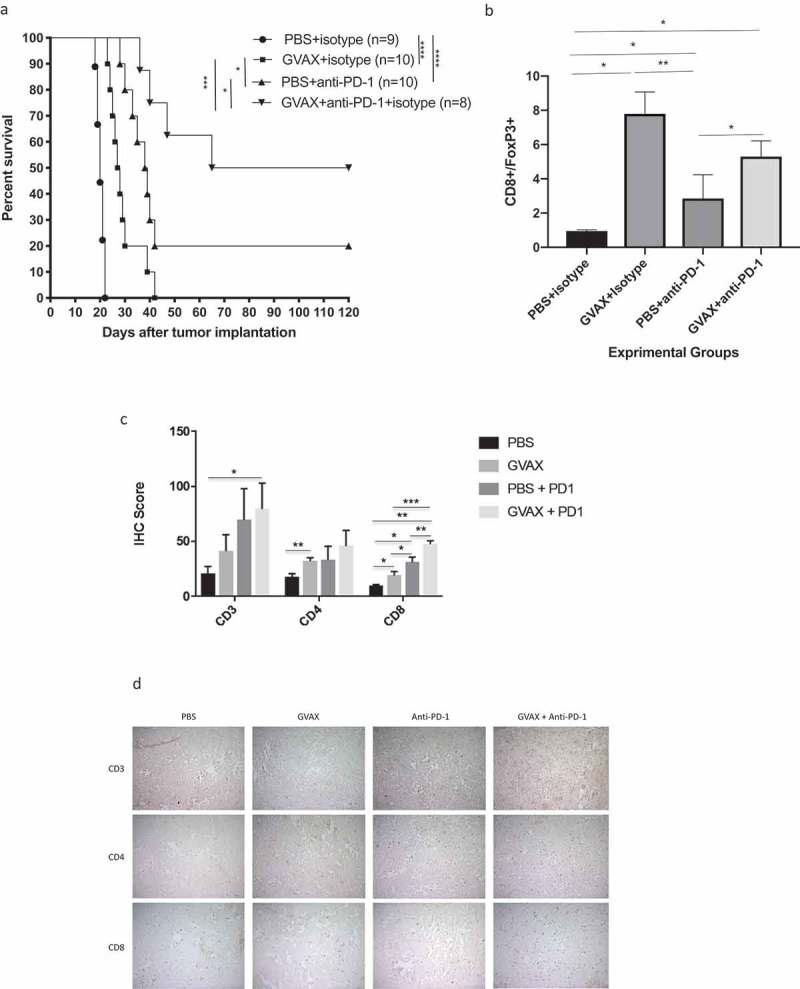
Combining GVAX with anti-PD-1 immunotherapy enhances antitumor immunity and improves survival in syngeneic mice, when compared to either as monotherapy. (a) Up to 50% of mice treated GVAX + anti-PD1 achieved long-term survival. Mice treated with either GVAX or PD-1 blockade as monotherapy lived significantly longer than control animals, but long-term survival was relatively rare. (b) Flow cytometry shows that the CD8 + T lymphocyte/FoxP3 + T lymphocyte ratio is very low (<1.0) in the brains of control-treated mice and markedly elevated (ABOUT 8.0) after vaccination. (c) Quantitative analysis by immunohistochemistry shows that combination GVAX + anti-PD-1 immunotherapy significantly augments lymphocyte infiltration into the brains, most significantly in the CD8+ subset. (d) Representative IHC figures in all treatment groups, staining for CD3, CD4, or CD8– magnification is 20x. (* = p < .05, ** = p < .005, *** = p < .0005, IHC = immunohistochemistry).
Consistent with our findings in previous studies12, flow cytometry of the brain tissue showed that GVAX alone very significantly improves the CD8+/FoxP3 + T lymphocyte ratio – from a ratio of 0.95 in control-treated animals to 7.79 (Figure 1b). PD-1 inhibition alone does so as well, but to a lesser degree (ratio = 2.85). The CD8+/Foxp3 + T lymphocyte ratio in the brains of combination-treated animals was likewise high (5.3), not statistically separable from that of vaccination alone. Immunohistochemistry (Figure 1d) of the brain tumor tissue for CD3, CD4, and C8 strengthened the association between GVAX + anti-PD-1 combination immunotherapy and increased CD8+ lymphocyte infiltration, as the change for this subset was greater than and more significant that it was for CD4+ cells (4.64x vs. 2.58x, Figure 1c).
PD-1 inhibition in combination with agonist anti-OX40 antibody improves survival in mice bearing intracranial GL261 tumors
We have recently demonstrated that OX40 ligation by an agonizing anti-OX40 antibody improves survival in GL261-bearing syngeneic mice, via effects that are largely mediated by promoting Th1 antitumor immunity, both systemically and within the brain tumor18. Furthermore, we demonstrated very high rates of expression of PD-1 on brain infiltrating lymphocytes in these animals. Up to 50% of both CD4+ and CD8 + T lymphocytes within GL261 tumors expressed PD-1 at 10 days after tumor implantation. PD-1 inhibition is thought to exert effects primarily by extending activation of CD8+ cells14. Therefore, we reasoned that OX40 ligation would be complementary with anti-PD-1 immunotherapy in murine glioma models, which has been observed in other tumor models.
Combining anti-PD1 monoclonal antibody with agonist anti-OX40 monoclonal antibody consistently improved survival when compared to either therapy alone, though not always significantly so (p = 0.2, Figure 2a). Anti-PD-1 + agonist anti-OX40 immunotherapy was consistently associated with a 30–40% cure rate in these mice. The intratumoral CD8+/FoxP3 + T lymphocyte ratio (8:1) was greatest in the combination-treated animals, which was the only group to reach significance when compared to the same ratio in the brain tumors of untreated animals (Figure 2b).
Figure 2.
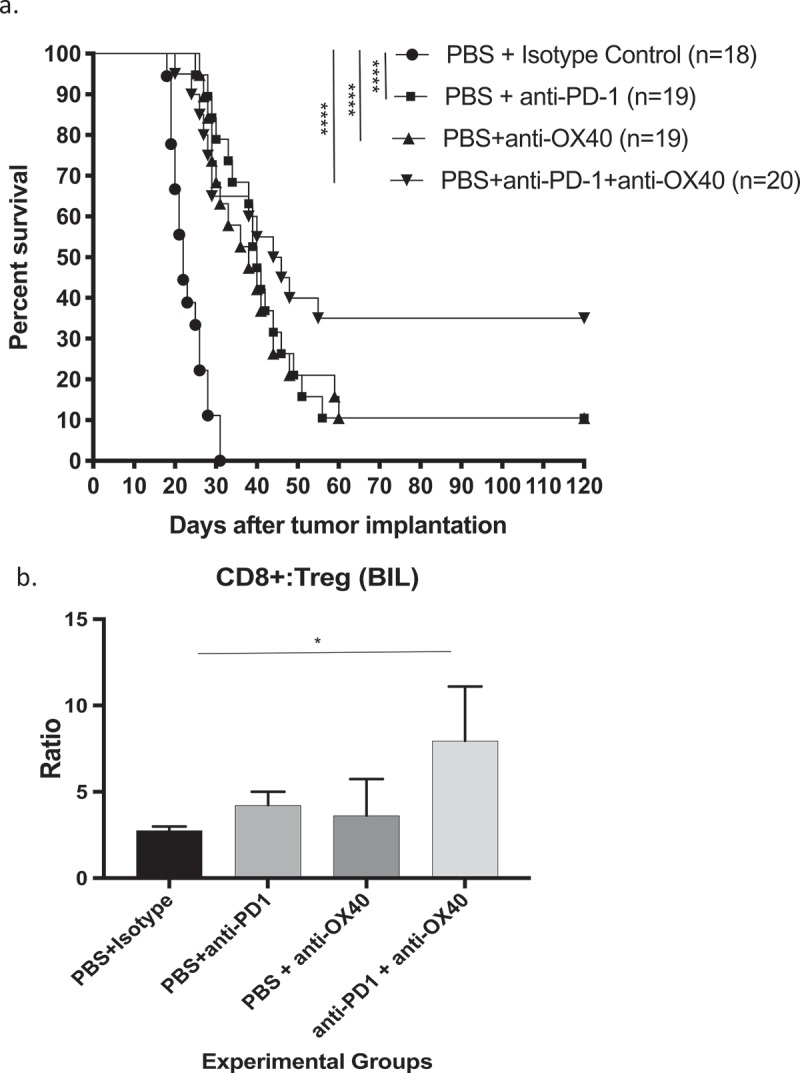
Immunotherapy with anti-PD-1 antibody combined with agonizing anti-OX40 antibody significantly improves survival (a) and (b) increases the CD8+/FoxP3+ lymphocyte ratio in the brains of syngeneic mice bearing GL261 gliomas, as measured by flow cytometry. (* = p < .05, ** = p < .005, *** = p < .0005, **** = p < .0001).
Triple combination immunotherapy with GVAX + anti-PD-1 antibody + agonizing anti-OX40 antibody leads to long-term survival in syngeneic mice bearing intracranial GL261 tumors and is associated with vigorous type 1 helper T cell response
Our accumulation of survival data and mechanistic support reflected that vaccination with irradiated tumor cells expressing GM-CSF is complementary with either anti-PD-1 immunotherapy or with agonist anti-OX40 immunotherapy. We also observed that the combination of anti-PD-1 antibody and agonist anti-OX40 resulted in more effective therapy than either treatment alone. We, therefore, considered combining GVAX with anti-PD-1 antibody and agonist anti-OX40 antibody simultaneously. In these experiments, monotherapy was associated with modest survival advantage against control animals. Each double combination was more effective than its components as monotherapy, as we had previously seen. Triple combination therapy was highly effective, as concurrent delivery of subcutaneous vaccination, intraperitoneal anti-PD-1 monoclonal antibody and intraperitoneal agonist anti-OX40 antibody on days 3,6, and 9 after intracranial implantation of tumor consistently lead to cure in all treated animals (Figure 3 and Supplementary Table 1).
Figure 3.
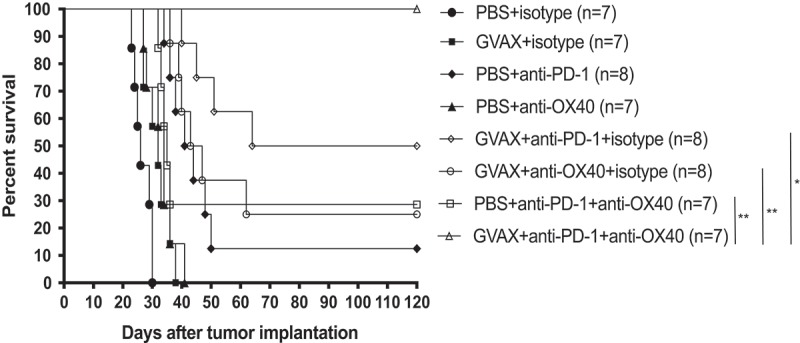
Triple Combination Immunotherapy with GVAX, anti-PD-1, and agonizing anti-OX40 antibody is curative in GL261 tumor-bearing C57/BL6 mice. Depicted statistical comparisons are limited to triple immunotherapy vs. double combinations (* = p < .05, ** = p < .005, *** = p < .0005). Supplementary Table 1 contains statistical associations between each of the treatment groups.
We next performed ELISPOT analyses of splenocytes harvested from all treatment groups on day 18 (Figure 4a). Upon in vitro stimulation by irradiated GL261 cells, splenocytes harvested from triple combination-treated animals expressed significantly higher amounts of IFN-gamma and IL-2 (Supplementary Tables 2 and 3), consistent with stronger development of a systemic Th1 immune response. With regard to splenocyte expression of IL-2, treatment with agonist anti-OX40 appears to be particularly impactful: among the monotherapies, splenocytes from agonist anti-OX40 antibody-treated mice were most likely to express IL-2 (mean 35 cells ± 5), cells from the GVAX + agonist anti-OX40 double combination expressed more IL-2 than cells from the GVAX + anti-PD-1 combination group (61 cells ± 5.5 vs. 12 cells ± 1.7), and splenocytes from the anti-PD-1 + agonist anti-OX40L group expressed far more than those from the anti-PD-1 monotherapy group (24.5 cells ± 5.8 vs 5.8 cells ± 3.3).
Figure 4.
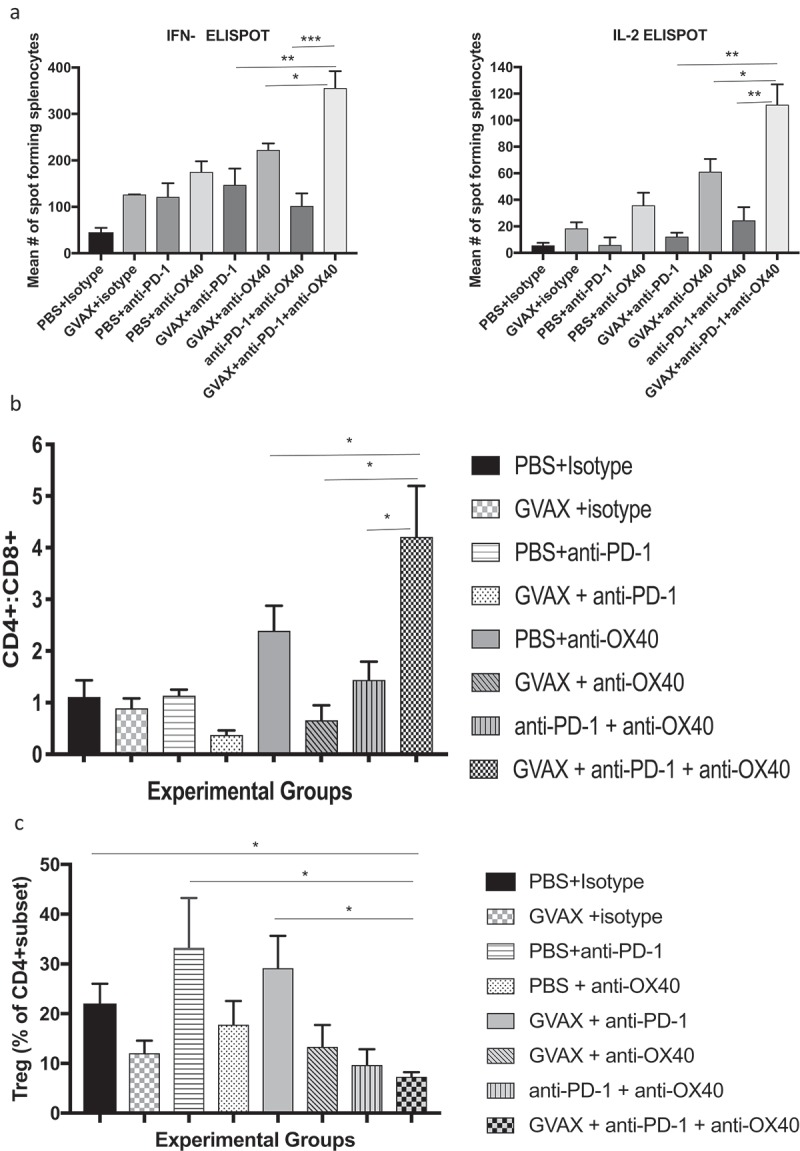
(a) Triple Combination Immunotherapy with GVAX, anti-PD-1, and agonizing anti-OX40 antibody increases expression of Th1 cytokines by splenocytes restimulated by exposure to glioma cells. ELISPOT analyses demonstrated that expression of interferon-gamma and IL-2 were highest by splenocytes harvested from animals treated with combination immunotherapy. Depicted statistical comparisons are limited to triple immunotherapy vs. double combinations. Remaining statistical comparisons are shown in Supplementary Tables 2 and 3. (b) Flow cytometry of brain infiltrating lymphocytes revealed that triple combination immunotherapy drove the highest CD4+/CD8+ lymphocyte ratios within the brain (Depicted statistical comparisons are limited to triple immunotherapy vs. double combinations – see Supplementary Table 4 for the remainder), while (c) also being associated with lowest fraction of regulatory T lymphocytes within the CD4+ subset (* = p < .05, ** = p < .005, *** = p < .0005).
Positive change in the tissue-resident CD4+/CD8 + T lymphocyte ratio is a biomarker for the vigor of a Th1 immune response19 and the overcoming of immune senescence.20 By flow cytometry of mouse brains, we found that triple combination immunotherapy most dramatically increased this ratio – by a factor of 3.7 compared to baseline and nearly twice as high as any other treatment group suggesting intratumoral shifting toward Th1 immunity (Figure 4b and Supplementary Table 4). Notably, this shift in T lymphocyte dynamics was not associated with an increased proportion or infiltrate of immunosuppressive regulatory T lymphocytes. In fact, flow cytometry of brain infiltrating lymphocytes revealed that the percentage of CD4 + T lymphocytes that were also FoxP3+ (the regulatory group) was lowest in the triple combination – treated group, at 7%, and this was only treatment group for which this percentage was significantly lower than in control-treated animals. Anti-PD-1 immunotherapy, in the absence of anti-OX40, trended heavily towards increasing the percentage of regulatory T lymphocytes within the CD4+ compartment (Figure 4c).
Triple combination immunotherapy with GVAX, anti-PD-1 antibody, and agonizing anti-OX40 immunotherapy led to long-term survival in all mice bearing intracranial GL261 tumors, augmented systemic Th1 immune responses to tumor, and shifted the CD4+:CD8 + T lymphocyte ration within the tumor microenvironment. The percentage of regulatory T-lymphocytes was driven significantly lower by triple combination immunotherapy.
Triple combination immunotherapy for animals with intracranial glioma tumors is associated with strong CD4 + T lymphocyte memory responses
Surviving animals were subject to intracranial injection of 400,000 GL261 cells in the left cerebral hemisphere, contralateral to the original injection site, 120 days after initial implantation of tumor. At the same time, tumor-naïve and age-matched controls also underwent implantation of tumor. By 25 days, all the naïve mice had succumbed to brain tumor progression. We observed continued long-term survival and apparent rejection of GL261 re-challenge in animals that had been initially cured with triple combination immunotherapy (Figure 5a) or with double combination anti-PD-1 + agonist anti-OX40 immunotherapy. Hematoxylin and eosin immunohistochemistry of brains 11 days after tumor rechallenge revealed widespread and aggressive growth of tumors in the naïve animals and relatively little tumor in the treated groups (Figure 5b). Subjectively, the volume of tumor was visibly greater in members of anti-PD-1 + agonist anti-OX40 group when compared to the scant tumor tissue present in the mice treated with triple combination immunotherapy. These small tumor remnants were also heavily infiltrated by lymphocytes.
Figure 5.
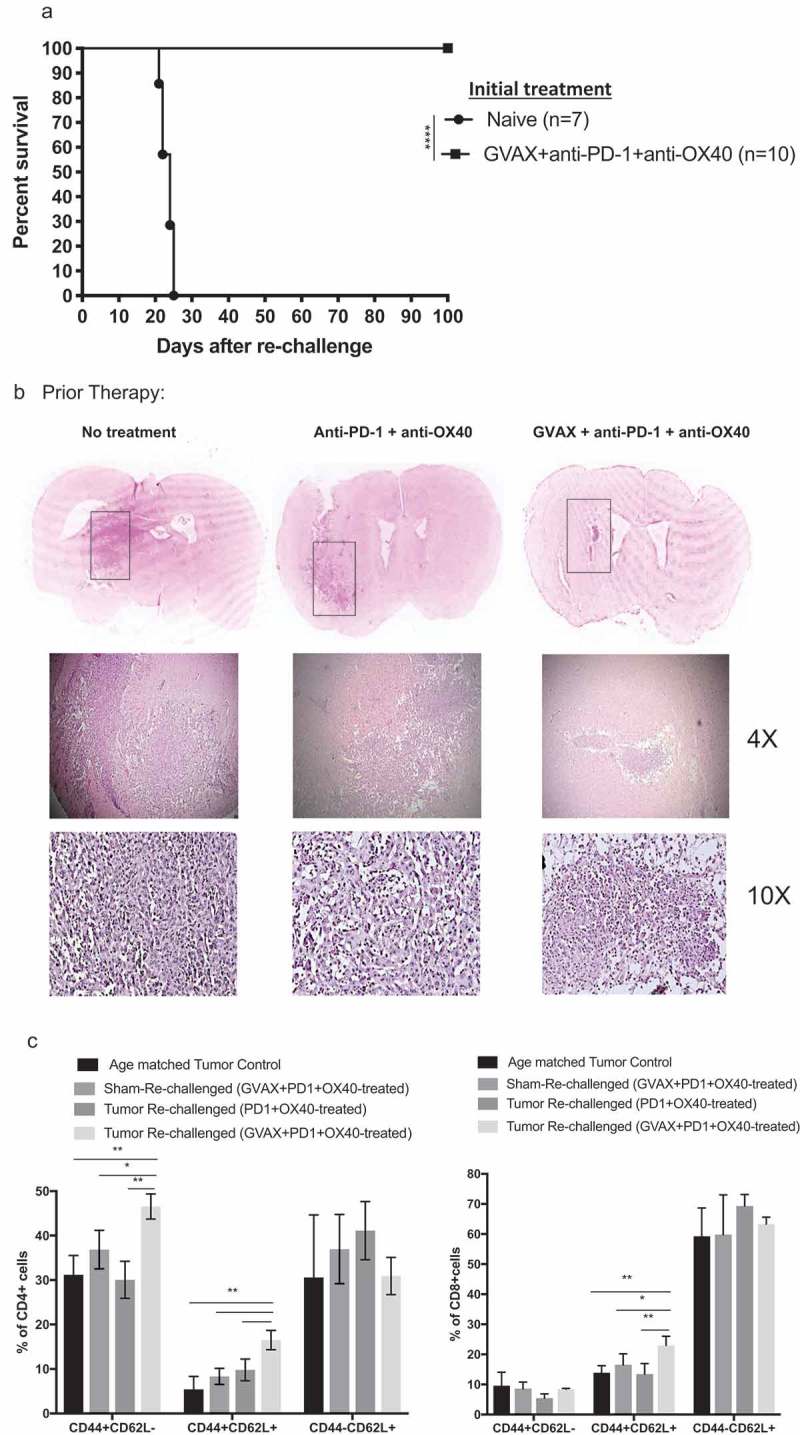
Mice cured by immunotherapy reject glioma rechallenge. (a) All mice that had been cured of GL261 tumor by triple combination immunotherapy survived GL261 rechallenge by day 120 injection of 400,000 tumor cells, reflecting memory. Age-matched animals that had not been treated with immunotherapy all succumbed to tumor progression within 24 days. (b) Hematoxylin and eosin immunohistochemistry of brain from each challenged group harvested 11 days after GL261 re-implantation, showing progression of tumor grossly, at 4x magnification, and at 10x magnification. Brains from the treatment-naïve group show a large mass of hypercellular tumor, while brain from the triple immunotherapy-treated animals show scant tumor cells, heavily infiltrated with lymphocytes. (c) Bar graphs derived from flow cytometry analyses of brain infiltrating lymphocytes show that the percentage of CD4 + T lymphocytes (left panel) and CD8 + T lymphocytes (right panel) that were comprised by CD44+ CD62L- (effector memory), CD44+ CD62L+ (central memory), and CD44-CD62L+ (naïve) subsets (* = p < .05, ** = p < .005, **** = p < .0001).
At the same 11-day timepoint, we performed flow-cytometry of the brains, focused on CD4+ lymphocyte and CD8+ lymphocyte memory T cell subsets (naïve, effector memory, central memory). We also examined brains from a group of animals whose tumors had been eradicated by triple combination immunotherapy and subsequently underwent “sham” rechallenge with intracranial injection of saline, rather than GL261 cells (Figure 5c). We found that 11 days following tumor rechallenge, triple combination immunotherapy – treated animals had a greater intracranial infiltrate of CD4+ CD44hiCD62Llo effector memory cells than did anti-PD-1 antibody + agonist anti-OX40 double immunotherapy-treated animals. Also, within the CD4+ memory compartment, triple combination immunotherapy led to a significantly increased fraction of CD44hiCD62Lhi central memory cells as well after tumor rechallenge. Regarding CD8 + T lymphocyte memory subsets, triple combination immunotherapy leads to increased numbers of CD44hiCD62Lhi central memory cells, but there were no differences in CD8+ effector memory or naïve T cell subsets.
Discussion
The promise of immunotherapy has yet to be realized for patients with malignant glioma. Primary brain tumors present therapeutic challenges for immunotherapy, as they have a relatively low antigenic load, and they have evolved, through immunoediting, to evade immune detection by actively suppressing both systemic and local antitumor immunity. While global systemic immunity is severely impaired – including by T cell lymphopenia,21 disproportionately high numbers of regulatory T cells, and impaired T effector responses to antigen22 – the tumor milieu is also heavily suppressive to T cell activation and cytotoxic antitumor efficacy.23 Glioma-associated negative immune regulation, directed specifically against intratumoral lymphocytes, includes expression of PD-L1,24 high numbers of regulatory T lymphocytes, significant numbers of suppressive myeloid cells, and the induction of T-lymphocyte dysfunction, chiefly through exhaustion.23 We have shown in preclinical models, including the GL261 line used in this study, that, as gliomas progress, the intratumoral CD4+ T lymphocytes are heavily skewed towards a Th2 orientation and exist in a state of exhaustion, defined by high co-expression of PD-1 and either TIM-3 or LAG-3.12 Given the above defects, which are a partial representation of the entire array of glioma immunosuppressive effects, it should be unsurprising that single-agent checkpoint inhibition has been clinically ineffective and that phase 3 immunotherapy studies have been negative.
A combination immunotherapy approach may yield improved outcomes. This has been exemplified in patients with advanced melanoma, for whom significantly longer survival, with impressive durability, can be achieved by combing anti-CTLA-4 monoclonal antibody treatment with anti-PD-1.13 While the actions of either agent may have similar impact on T-cell activation, they can also be complementary and even synergistic.
OX40 ligation may be a useful adjunct in anti-glioma immunotherapy. Several clinical studies have demonstrated safety for this approach in patients with cancer,25 and combination studies with anti-PD-1 or anti-PD-L1 agents are underway as well. In preclinical models, we have demonstrated anti-glioma activity with systemic delivery of an agonistic anti-OX40 antibody and further enhanced survival in combination with GVAX. In these studies, agonist anti-OX40 antibody drove Th1 antitumor responses systemically and intratumorally and prevented development of intra-glioma T lymphocyte exhaustion. While the combination significantly improved survival, most mice did eventually succumb to disease at the study dose and schedule.12
In this report, we build upon our prior analysis of agonist anti-OX40 immunotherapy by combining it with anti-PD-1 antibody, the first demonstration of these agents together in a glioma model. We also examined, for the first time, the combination of anti-PD-1 immunotherapy with GVAX, analogous to our prior reports with anti-CTLA-4 and agonist anti-OX40 antibodies. Combining anti-PD-1 immunotherapy with either agonist anti-OX40 or GVAX led to better survival than any of the agents alone, and we achieved long-term survival for 30% to 50% of glioma-bearing mice. We have consistently seen that GVAX improves the intratumoral CD8+/FoxP3 + T lymphocyte ratio in a way that we do not typically see with checkpoint active molecules alone – thereby, improving the infiltrate of the tumor microenvironment and setting the stage for enhanced survival through brisker lymphocyte activation, for example, by PD-1 inhibition acting on CD8+ cells. Agonist anti-OX40 treatments are probably most relevant in overcoming CD4-targeted immunosuppression.26 Given the both overlapping and distinct mechanisms of antitumor immune activation by GVAX, anti-PD1 antibody, and agonist anti-OX40 antibody, we studied “triple combination immunotherapy” with simultaneous delivery of each agent on days 3,6, and 9 after intracranial implantation of GL261 cells.
In the GL261 model, the combination of GVAX, anti-PD-1 monoclonal antibody, and agonizing anti-OX40 monoclonal antibody cured tumor-bearing C57/Bl6 mice. While GL261 seems to be responsive to an array of experimental immunotherapeutic approaches, complete eradication is rarely seen. The high level of efficacy here underscores the likelihood that impactful glioma immunotherapy should be multipronged. As a further demonstration of this tendency, Kim, et al. likewise employ a three-armed approach to cure intracerebral GL261 tumors with a combination of anti-PD-1, anti-TIM3, and focal stereotactic radiation.27 A likely oversimplified, but reasonable, summary of our triple combination approach posits that whole tumor cell vaccination (GVAX) expands the number and diversity of activated tumor-specific T cells and increases the number of intratumor CD8 + T cells, while PD-1 inhibition further invigorates the effects of those mobilized cells, as OX40 ligation continues to skew systemic and tumor microenvironments towards Th1 immunity, suppresses regulatory T lymphocytes, and prevents T lymphocyte exhaustion. Our data, best shown in Figure 4, suggest that addition of OX40 ligation to the GVAX + anti-PD-1 combination results in greater systemic elaboration of Th1 cytokines, a much higher CD4+/CD8+ ratio within the tumor, and a significantly reduced percentage of regulatory T cells within the intratumoral CD4+ population.
In many ways, GL261 is an excellent syngeneic murine model for human glioma.28 The tumors are microscopically and macroscopically invasive, and we have shown a skew towards systemic and intratumoral Th2 immune responses.12 However, they may be more responsive to immunotherapy than are human tumors, as the GL261 antigenic load significantly exceeds that of human glioblastoma29
Triple therapy, in this case, addresses both systemic and intratumoral deficiencies in cellular antitumor immunity. There are likely overlapping impacts of each of these therapies – cellular responses to stimuli are complex and dynamic, and continued study is required to better understand the full impacts of these therapies in different cancer environments. Furthermore, our report, and our body of work in general, focuses on the impacts on immunotherapy on T lymphocyte-driven responses, and other arms of the immune response may be at play. Clinical immunotherapy failures in glioma patients have highlighted the role that tumor-associated macrophages and myeloid-derived suppressor cells may play in tumor microenvironment negative immune regulation.30 Innate responses are impaired as well.31 Direct engagement of more than the T-cell mediated arm of antitumor immunity may be required for optimized responses.
However, tumor-specific activated CD8 + T lymphocytes, whose activity is promoted by activated CD4+ Th1 cells, are the ultimate tumoricidal arm of the immune system, and the mechanistic goal of immunotherapy is to bolster their infiltration and their activity in the face of immunosuppressive obstacles. Triple combination immunotherapy with GVAX, PD-1 inhibition, and OX40 ligation is a model for how to rationally modulate antiglioma immunity via a multipronged and effective attack.
Materials and methods
Parental and engineered cell lines
GL261, a syngeneic murine tumor cell line, was obtained from the National Cancer Institute. GL261-GMCSF cells, which were used as a vaccine base, were engineered via retroviral transduction and infection as previously described. The expression and secretion of GMCSF by GL261-GMCSF cells was confirmed by ELISA using manufacturer’s instructions (RayBiotech) and was measured as 442 (95% confidence interval 370–524) nanograms/1 × 106 cells/48 hours. All GL261 cell lines were cultured in DMEM medium (Mediatech Inc.) supplemented with 10% Fetal Bovine Serum (HyClone) at 5% CO2 and 37°C.
Animals
Six to seven-week-old female wild-type C57BL/6 mice were purchased from National Cancer Institute. All mice were maintained and used in accordance with the animal protocol approved by the MGH Institutional Animal Care and Use Committee (IACUC).
Intracranial tumor cell implantation
GL261 parental cells (75,000 cells per mouse) suspended in 3-µl DMEM were stereotactically implanted into the brains (right striatum, 2.5 mm lateral from bregma and 2.5 mm deep) of C57BL/6 mice (7 to 8 weeks of age) using a Hamilton 1701N gastight syringe (Hamilton Co.) and a stereotactic mouse frame (Kopf Instruments)
Vaccination and anti-PD1/agonist anti-OX40 treatment
On days 3, 6 and 9 post-tumor implantation, appropriate groups of mice were subcutaneously injected with 1 × 106 irradiated (35 Gy) GL261-GMCSF cells, referred to as GVAX here, suspended in100 microliters of PBS. Dependent on their groups, mice were also administered 200 µg of anti-PD-1 (clone: RMP1-14, BioXCell) or 250 µg of anti-OX40 (OX86 clone, BioXCell) or isotype control (Rat IgG2a) or isotype control (Rat IgG1) prepared in 100 µl of PBS on days 3, 6 and 9 post-tumor implantation. Mice were followed for survival and sacrificed when neurological symptoms became apparent for survival study. Mice used for immune response studies were euthanized at a particular day post-tumor implantation to collect the brains or spleens.
IFN-γ and IL-2 ELISPOT assays
Mice receiving different treatments were sacrificed, and their spleens were harvested on 18 days post-tumor implantation. Spleens were immediately processed to isolate splenocytes. 1 × 106 splenocytes were stimulated for 48 h in vitro with 1 × 105 irradiated (35 Gy) GL261 cells or in RPMI 1640 medium, supplemented with 10% IFCS, 50 μM 2-ME, 2 mM glutamine, 20 mM HEPES, penicillin-streptomycin in six well tissue culture plates (BD Falcon). 1 × 105 splenocytes from mice in each treatment and control group were loaded in duplicates onto 96-well PVDF-backed 96-well plates coated with anti-mouse IFN-γ and anti-mouse IL-2 monoclonal antibodies (R & D Systems). These plates were then incubated at 5% CO2 and 37°C for 24 h. After incubation, the plates were washed four times with the wash buffer provided by the manufacturer and biotinylated antibody specific for each coated antibody was added to each well and incubated overnight at 4°C. Next morning the plates were washed four times again, and Streptavidin-AP was added into each well and the plates were incubated at room temperature for 2 h. Plates were washed again, and BCIP/NBT chromogen was added to each well and incubated for an hour at room temperature. The contents of the plates were then discarded, and they were then washed with distilled water, dried and read using an ImmunoSpot version 5.1.36 ELISPOT reader (Cellular Technology Limited).
Tissue processing and immunohistochemistry
For immunohistochemistry studies, day 18 mice from each treatment group were sacrificed and their brains harvested and fixed in 10% formalin followed by 70% ethanol. Specimens were subsequently paraffin embedded and cut into 5-μm sections. Sections were deparaffinized in xylene and dehydrated in ethanol followed by microwave treatment in 10 mM sodium citrate buffer (pH 6.0) for 15 min for antigen retrieval. The sections were next treated with 3% H2O2 to block endogenous peroxidase. Bovine serum albumin and antibody specific protein blocking buffer were then used to block non-specific binding for 20 and 30 min respectively. The sections were incubated overnight with anti-CD3 (ab5690, abcam), anti-CD4 (4SM95, eBioscience), and anti-CD8a (4SM15, affymetrix). Blocking buffer was used instead of the primary antibody for negative controls. The sections were then incubated for 30 min at room temperature with the appropriate peroxidase-labeled secondary antibody as instructed by the manufacturer in the kit (Vector Lab ImmPRESS polymer detection kit, Vector labs). Diaminobenzidine (DAB) was used for color development and the sections were counterstained with hematoxylin. Finally, the sections were mounted with Cytoseal-XYL (8312–4, Thermo scientific) and photographed under a light microscope (Nikon optiphot 2) and using spot software.
For quantitative analysis of the immune infiltrate (“histoscore”), three pictures were taken with 20X magnification per cut section of the brain tumors. Stained cells were counted from each picture, and an average from the three was calculated. This average represented the IHC score for that section. Three mice per treatment group were studied in this fashion.
Flow cytometry analysis
Mice receiving different treatments were sacrificed, and their brains were harvested on 18 days post-tumor implantations. Tumor tissues were processed from brains, and single cell suspension of the tumor cells was made for the analysis of tumor-infiltrating lymphocytes. Briefly, mouse brain containing the tumor was transferred to petri dish containing PBS in sterile fashion. Using a sterile scalpel and forceps the specimen was minced into equal sized pieces. The minced sample was transferred from petri dish to a new 50 ml falcon tube. The tube was then centrifuged at 1500 rpm for 5 min and the supernatant was removed. For dissociating the cells 1–2 ml of accutase (Sigma) containing 10 µl/ml DNase I (Roche) was added and incubated for 10 min at 37°C with gently rocking the tube. 15 mL FACS buffer was added and the contents of the tube were passed through a 40 µm cell strainer fixed on a 50 mL falcon tube. The strainer was washed with PBS to get the maximum number of cells. The tube was then centrifuged at 1500 rpm for 5 min and the supernatant was removed. The cells were resuspended in 12 ml PBS and counted. Fluorescently labeled antibodies used for the analysis were purchased from Biolegend (BV 605-CD3, PerCP/Cy5.5-CD4, BV 510- CD8a, Alexa Fluor 647-FOXP3). Dead cells were excluded using the Zombie UV™ Fixable Viability kit (Biolegend). Flow cytometry data were acquired using LSRII and analyzed using Flowjo software and graphs were generated using Prism 6 software (GraphPad Software).
For calculation of the CD8+/FoxP3 ratio, the percentage of the entire cellular population that was CD8+ was divided by the percentage of the entire cellular population that was CD4+ FoxP3+.
Tumor challenge in long-term surviving mice and analysis of brain by immunohistochemistry and flow cytometry
Selected mice surviving 120 days after treatment were subjected to injection of 400,000 GL261 cells in the left cerebral hemisphere, contralateral to the original injection site, and followed for survival. On day 11 after tumor re-injection, some of these mice were sacrificed, and their brains were collected and fixed with 10% formalin. Five-micrometer sections were prepared from paraffin-embedded brains. The sections were then deparaffinized in xylene for 10 min, followed by series of 100%, 95% and 70% EtOH dehydration for 5 min each. The sections were stained with 100% hematoxylin for 1 min and rinsed with distilled water followed by wash in running tap water for 3 min. The sections were counterstained with Eosin for 45 s, followed by dehydration in 95% and 100% EtOH for 5 min each. The sections were then dipped in Xylene for 10 min followed by mounting with Cytoseal XYL (8312–4, Thermo scientific). Other mouse brains were prepared for flow cytometry at the same timepoint. Mouse brains containing the tumor were harvested, and single cell suspensions were created. Fluorescently labeled antibodies used for flow cytometry analyses were purchased from Biolegend (BV 605-CD3, PerCP-CD4, BV 510- CD8a, APC-CD62L, and PE-CD44). Dead cells were excluded using the Zombie UV™ Fixable Viability kit (Biolegend). Flow cytometry data were acquired using LSRII and analyzed using Flowjo software and graphs were generated using Prism 6 software (GraphPad Software).
Statistical analysis
Data were analyzed and graphed using Prism 6 (GraphPad Software). Data were expressed as mean ± SD and differences were considered significant at P < 0.05. Individual data sets were compared using Student’s T-test when comparing two groups. Kaplan-Meier analysis was used for mouse survival studies, and the groups were compared using the log-rank test. Flow cytometry data were analyzed using Flowjo software.
Presented data are representative of experiments, each of which was performed at least 3 times. For survival studies, 8–10 animals per group were followed. For studies of immune function, we typically harvested splenocytes and TILs from three animals per group.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Stupp R, Wong ET, Kanner AA, Steinberg D, Engelhard H, Heidecke V, Kirson ED, Taillibert S, Liebermann F, Dbalý V, et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer. 2012;48(14):2192–2202. doi: 10.1016/j.ejca.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318(23):2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–1385. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 6.Srinivasan VM, Ferguson SD, Lee S, Weathers SP, Kerrigan BCP, Heimberger AB.. Tumor vaccines for malignant gliomas. Neurotherapeutics. 2017;14(2):345–357. doi: 10.1007/s13311-017-0522-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JJ, Curry WT, Cahill DP, Dietrich J. Perspectives on investigational drugs and immunotherapies for glioblastoma. Expert Opin Investig Drugs. 2016;25(9):1007–1009. doi: 10.1080/13543784.2016.1213242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloch O, Shi Q, Anderson SK, Knopp M, Raizer J, Clarke J, Waziri A, Colman H, Bruce J, Olson JJ, et al. ATIM-14. Alliance A071101: A phase II randomized trial comparing the efficacy of heat shock protein peptide complex-96 (HSPPC-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology. 2017;19(suppl_6):vi29–vi29. doi: 10.1093/neuonc/nox168.110. [DOI] [Google Scholar]
- 9.Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of checkmate 143. Neuro Oncol. 2018;20(5):674–686. doi: 10.1093/neuonc/nox208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurz SC, Wen PY. Quo vadis-do immunotherapies have a role in glioblastoma? Curr Treat Options Neurol. 2018;20(5):14. doi: 10.1007/s11940-018-0499-0. [DOI] [PubMed] [Google Scholar]
- 11.Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, Phuphanich S, de Souza P, Ahluwalia MS, Lim M, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: checkMate 143. Neuro-Oncology. 2017;19(suppl_3):iii21–iii21. doi: 10.1093/neuonc/nox036.071. [DOI] [Google Scholar]
- 12.Jahan N, Talat H, Curry WT. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro Oncol. 2018;20(1):44–54. doi: 10.1093/neuonc/nox125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 15.Voo KS, Bover L, Harline ML, Vien LT, Facchinetti V, Arima K, Kwak LW, Liu YJ. Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. J Immunol. 2013;191(7):3641–3650. doi: 10.4049/jimmunol.1202752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curry WT, Gorrepati R, Piesche M, Sasada T, Agarwalla P, Jones PS, Gerstner ER, Golby AJ, Batchelor TT, Wen PY, et al. Vaccination with irradiated autologous tumor cells mixed with irradiated GM-K562 cells stimulates antitumor immunity and T lymphocyte activation in patients with recurrent malignant glioma. Clin Cancer Res. 2016;22(12):2885–2896. doi: 10.1158/1078-0432.CCR-15-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- 18.Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother. 2012;35(5):385–389. doi: 10.1097/CJI.0b013e3182562d59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackute J, Zemaitis M, Pranys D, Sitkauskiene B, Miliauskas S, Bajoriunas V, Lavinskiene S, Sakalauskas R. The prognostic influence of tumor infiltrating Foxp3(+)CD4(+), CD4(+) and CD8(+) T cells in resected non-small cell lung cancer. J Inflamm (Lond). 2015;12:63. doi: 10.1186/s12950-015-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saavedra D, García B, Lorenzo-Luaces P, González A, Popa X, Fuentes KP, Mazorra Z, Crombet T, Neninger E, Lage A. Biomarkers related to immunosenescence: relationships with therapy and survival in lung cancer patients. Cancer Immunol Immunother. 2016;65(1):37–45. doi: 10.1007/s00262-015-1773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–1468. doi: 10.1038/s41591-018-0135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, Bigner DD, Dranoff G, Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 23.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res. 2018;24(16):3792–3802. doi: 10.1158/1078-0432.CCR-18-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205. doi: 10.1093/neuonc/nov172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73(24):7189–7198. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gedeon PC, Riccione KA, Fecci PE, Sampson JH. Antibody-based immunotherapy for malignant glioma. Semin Oncol. 2014;41(4):496–510. doi: 10.1053/j.seminoncol.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–136. doi: 10.1158/1078-0432.CCR-15-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN Neuro. 2011;3(3):e00063. doi: 10.1042/AN20110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, Fu Y, Alexandrov A, Mardis ER, Artyomov MN, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res. 2016;4(12):1007–1015. doi: 10.1158/2326-6066.CIR-16-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, Holtman IR, Wang X, Eggen BJ, Boddeke HW, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644. doi: 10.1371/journal.pone.0116644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129(Pt 9):2416–2425. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.