

ABSTRACT
Although immune checkpoint inhibitors have shown improvement in survival in comparison to chemotherapy in urothelial bladder cancer, many patients still fail to respond to these treatments and actual efforts are made to identify predictive factors of response to immunotherapy. Understanding the tumor-intrinsic molecular basis, like oncogenic pathways conditioning the presence or absence of tumor-infiltrating T cells (TILs), should provide a new rationale for improved anti-tumor immune therapies.
In this study, we found that urothelial bladder cancer from human samples bearing PIK3CA gene mutations was significantly associated with lower expression of a defined immune gene signature, compared to unmutated ones. We identified a reduced 10-gene immune gene signature that discriminates muscle-invasive bladder cancer (MIBC) samples according to immune infiltration and PIK3CA mutation. Using a humanized mouse model, we observed that BKM120, a pan-PI3K inhibitor, significantly inhibited the growth of a human bladder cancer cell line bearing a PIK3CA mutation, associated to increased immune cell infiltration (hCD45+). Using qRT-PCR, we also found an increase in the expression of chemokines and immune genes in PIK3CA-mutated tumors from mice treated with BKM120, reflecting an active immune infiltrate in comparison to untreated ones. Moreover, the addition of BKM120 rendered PIK3CA-mutated tumors sensitive to PD-1 blockade. Our results provide a relevant rationale for combination strategies of PI3K inhibitors with immune checkpoint inhibitors to overcome resistance to immune checkpoint inhibitors.
Keywords: Bladder cancer, PIK3CA mutation, targeted therapy, immunotherapy
Introduction
Bladder cancer is considered as a major source of mortality worldwide, with an estimate of 429,800 new cases and 165,100 deaths worldwide in 2012.1 About two-thirds of newly diagnosed urothelial bladder cancers are non-muscle-invasive bladder cancer (NMIBC) that can further evolve to muscle-invasive tumors in about 10% of the cases; and one-third of the newly diagnosed cases are muscle-invasive bladder cancer (MIBC).2 Despite chemotherapy treatments, the prognosis of metastatic urothelial carcinoma remains poor after the first line platinum-based regimen and there is an urgent need for new effective strategies in this setting.3,4 Immunotherapy, such as immune checkpoint inhibitors, offers as a new therapeutic option in advanced urothelial carcinoma resulting in survival improvement in comparison to chemotherapy.5,6
Immunotherapy has made a breakthrough in cancer treatment, in many different types of tumors. However, many patients still fail to respond to immune checkpoint therapies, as only around 20% to 40% of the patients will initially respond to those treatments, and some responders will eventually acquire resistance to the treatment and relapse after a period of response.7 Thus, there is a need to gain knowledge on the mechanism of action of these approaches and overcome resistance.
Response to immune checkpoint inhibitors seems to be conditioned by the infiltration of tumors by activated T cells, evidenced by an ongoing anti-tumor immune response.8-10 Tumors with a T-cell-inflamed microenvironment, also referred to as “hot tumors”, are characterized by a high CD8+ T cell infiltrate, the expression of chemokines involved in T-cell recruitment,8 and a type-I interferon (IFN) signature, and are associated with improved patient survival and better responses to immunotherapies.8,9,11 Otherwise, tumors that lack infiltration of activated T cells in the tumor microenvironment, defined as “cold” tumors, have been described as presenting low or no clinical response to immunotherapies.12
Efforts are being made to better understand the mechanisms leading to T-cell exclusion or T-cell inactivation in the tumor microenvironment. Hence, understanding the tumor-intrinsic molecular bases, such as oncogenic pathways, dictating the presence or absence of tumor-infiltrating T cells (TILs), should provide a new rationale for improved anti-tumor immune therapies.
The PI3K/AKT/mTOR pathway is involved in different cellular functions and regulates cell cycle, cell growth, metabolism, proliferation, and survival that are all hallmarks of cancer cells implicated in initial growth and extension of cancer.12 Genetic alterations, leading to overactivation of the PI3K/AKT/mTOR pathway are frequent events in many types of cancers.13
The phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene is an oncogene frequently implicated in the overactivation of the PI3K/AKT/mTOR pathway, somatic PIK3CA mutations leading to an increase of kinase activity of phosphoinositide 3-kinase (PI3Ks). It has been found that recurrent somatic mutations of PIK3CA, known as hotspot mutations, are frequently involved in overactivation of the PI3K/AKT/mTOR pathway in tumors. These hotspot-activating point mutations are frequently detected either on exon 9 (encoding for the helical domain of the protein, E542K, and E545K) or on exon 20 (encoding for the kinase domain of the protein, H1047R and H1047L) (COSMIC database). Also, frequent genetic alterations leading to the loss of phosphatase and tensin homolog (PTEN, a tumor suppressor that inhibits PI3K), can result in the overactivation of the PI3K/AKT/mTOR pathway.
Interestingly, Parsa and colleagues described that in human glioma the expression of PD-L1 is increased post-transcriptionally after the deletion of PTEN and the activation of the PI3K/AKT/mTOR pathway.14 It has already been shown in a melanoma model that the loss of PTEN and the consequent activation of the PI3K/AKT/mTOR pathway promotes resistance to T cell-mediated immunotherapy.15
These molecular events do not explain all the cases of non-T-cell-infiltrated melanomas, and the particular cases of other types of tumors remain to be studied so as to better understand resistance mechanisms to anti-immune checkpoint inhibitors. Along these lines, Sweis and colleagues showed that β-Catenin, PPAR-γ, and FGFR3 pathways were activated in non-T-cell-inflamed bladder tumors, correlating those tumor-intrinsic oncogenic pathways with T-cell exclusion, and thus identifying those pathways as potential targetable pathways of tumor-intrinsic immunotherapy resistance.16
Therefore, in our study, we assessed the correlation between genetic alterations of selected druggable tumor-intrinsic oncogenic pathways and immune cell infiltration in bladder cancer, more specifically in MIBC, so as to provide a new rationale for improved anti-tumor immune therapies in this setting.
Results
Mutation of PIK3CA correlates with non-T-cell-inflamed MIBCs
We first evaluated whether bladder tumors could be classified as “T-cell-inflamed” or “non-T-cell-inflamed” by evaluating their immune gene expression profile using quantitative PCR (qRT-PCR). For that, we first defined an immune gene signature, of 57 genes that comprised: i) genes commonly used to identify major immune cell populations described to be present in the human tumor microenvironment17 (i.e. FOXP3 to identify Tregs, see Table 1 for the complete list), ii) a selection of immunomodulatory genes comprising druggable immune checkpoints (in clinical use or under evaluation) and including at least one member of the most studied families,18 and iii) major histocompatibility complex (MHC) genes (HLA genes) and IFN genes, which have been associated with tumor resistance to immunotherapies.19,20
Table 1.
List of the 57 genes selected for the immune gene signature.
| ID | GeneID | GeneSymbol | GeneName |
|---|---|---|---|
| Immune cell population genes | |||
| CD2 | 914 | CD2 | CD2 molecule |
| CD3E | 916 | CD3E | CD3e molecule |
| CD4 | 920 | CD4 | CD4 molecule |
| CD8A | 925 | CD8A | CD8a molecule |
| CTLA4 | 1493 | CTLA4 | Cytotoxic T-lymphocyte associated protein 4 |
| FOXP3 | 50943 | FOXP3 | Forkhead box P3 |
| XCR1 | 2829 | XCR1 | Chemokine (C motif) receptor 1 |
| MERTK | 10461 | MERTK | MER proto-oncogene, tyrosine kinase |
| PTPRC | 5788 | PTPRC CD45 | Protein tyrosine phosphatase, receptor type C |
| MS4A1 | 931 | MS4A1 CD20 | Membrane spanning 4-domains A1 |
| NCAM1 | 4684 | NCAM1 CD56 | Neural cell adhesion molecule 1 |
| PDGFRB | 5159 | PDGFRB | Platelet derived growth factor receptor beta |
| PECAM1 | 5175 | PECAM1 | Platelet/endothelial cell adhesion molecule 1 |
| T cell activation genes | |||
| PRF1 | 5551 | PRF1 | Perforin 1 |
| GZMA | 3001 | GZMA | Granzyme A |
| GZMB | 3002 | GZMB | Granzyme B |
| Checkpoint T cell genes | |||
| CD28 | 940 | CD28 | CD28 molecule |
| ENTPD1 | 953 | ENTPD1 CD39 | Ectonucleoside triphosphate diphosphohydrolase 1 |
| NT5E | 4907 | NT5E CD73 | 5ʹ-nucleotidase ecto |
| CD96 | 10225 | CD96 | CD96 molecule, TIGIT family |
| TIGIT | 201633 | TIGIT | T-cell immunoreceptor with Ig and ITIM domains |
| CD226 | 10666 | CD226 | CD226 molecule, TIGIT family |
| TNFRSF14 | 8764 | TNFRSF14 | Tumor necrosis factor receptor superfamily member 14 |
| TNFRSF18 | 8784 | GITR | Tumor necrosis factor receptor superfamily member 18 |
| TNFRSF4 | 7293 | OX-40, CD134 | Tumor necrosis factor receptor superfamily member 4 |
| TNFRSF7 | 939 | CD27 | CD27 molecule |
| TNFRSF9 | 3604 | CD137, 4-1BB | Tumor necrosis factor receptor superfamily member 9 |
| HAVCR2 | 84868 | HAVCR2, Tim-3 | Hepatitis A virus cellular receptor 2 |
| ICOS | 29851 | ICOS | Inducible T-cell co-stimulator |
| LAG3 | 3902 | LAG3 | Lymphocyte activating 3 |
| PDCD1 | 5133 | PDCD1 | Programmed cell death 1 |
| Checkpoint tumor cell genes | |||
| IDO1 | 3620 | IDO1 | Indoleamine 2,3-dioxygenase 1 |
| CD80 | 941 | CD80, B7-1 | CD80 molecule |
| CD86 | 942 | CD86, B7-2 | CD86 molecule |
| CD276 | 80381 | CD276, B7H3 | CD276 molecule |
| LGALS9 | 3965 | LGALS9 | Lectin, galactoside-binding, soluble, 9 |
| CD274 | 29126 | CD274, PDL1 | CD274 molecule |
| PDCD1LG2 | 80380 | PDCD1LG2 | Programmed cell death 1 ligand 2 |
| ICOSLG | 23308 | ICOSLG | Inducible T-cell co-stimulator ligand |
| PVR | 5817 | PVR, CD155 (Tigit ligand) | Poliovirus receptor |
| PVRIG | 79037 | PVRIG, CD112R | Poliovirus receptor related immunoglobulin domain containing |
| TNFSF4 | 7292 | OX40L | Tumor necrosis factor superfamily member 4 |
| Inferferon signature genes | |||
| CXCL10 | 3627 | CXCL10 | C-X-C motif chemokine ligand 10 |
| IFI27 | 3429 | IFI27 | Interferon, alpha-inducible protein 27 |
| IFI44L | 10964 | IFI44L | Interferon induced protein 44 like |
| IFI6 | 2537 | IFI6 | Interferon, alpha-inducible protein 6 |
| IFIT1 | 3434 | IFIT1 | Interferon induced protein with tetratricopeptide repeats 1 |
| IRF8 | 3394 | IRF8 | Interferon regulatory factor 8 |
| MX1 | 4599 | MX1 | MX dynamin like GTPase 1 |
| OAS1 | 4938 | OAS1 | 2ʹ-5ʹ-oligoadenylate synthetase 1 |
| RSAD2 | 91543 | RSAD2 | Radical S-adenosyl methionine domain containing 2 |
| G1P2 | 9636 | ISG15 | ISG15 ubiquitin-like modifier |
| Major histocompatibility complex genes | |||
| HLA-A | 3105 | HLA-A | Major histocompatibility complex, class I, A |
| HLA-B U3/L3 | 3106 | HLA-B | Major histocompatibility complex, class I, B |
| HLA-C U2/L2 | 3107 | HLA-C | Major histocompatibility complex, class I, C |
| HLA-DRA | 3122 | HLA-DRA | Major histocompatibility complex, class II, DR alpha |
| HLA-DRB | 3123 | HLA-DRB1 | Major histocompatibility complex, class II, DR beta 1 |
We first applied the 57-immune gene signature to a cohort of 98 human bladder cancer samples for which we also analyzed the mutational status of PIK3CA, BRAF, RAS, and FGFR3 genes, which are implicated in the activation of the respective oncogenic pathways (Supplementary Figure 1). Using unsupervised hierarchical clustering, bladder cancers were segregated into “high” or “low” immune-infiltrated (also referred to as “hot” or “cold” tumors17,21) based on the level of expression of the immune gene signature. The high level of expression of the immune gene signature reflects the presence of a reactive immune infiltrate in the tumor microenvironment. Along these lines, we have previously confirmed that qRT-PCR mRNA and protein immunohistochemistry expression were strongly associated with immune markers in MIBC patients.22 We observed that, as previously described,16 urothelial bladder cancers showed different levels of immune infiltration depending on the histological group type: NMIBCs showed a significantly lower expression of the immune gene signature than MIBCs (Fisher’s exact test, p < 0.001). We also found that tumors bearing a PIK3CA mutation or a FGFR3 mutation were significantly associated with a lower expression of the immune gene signature in comparison to their wild type counterparts (Fisher’s exact test, p < 0.05 and p < 0.001, respectively). RAS-mutated tumors did not segregate into hot or cold tumors, and there was only one BRAF mutation found in this cohort of bladder tumor patients, in accordance with the low frequency of this oncogenic mutation in bladder tumors.
Given that NMIBCs showed very low levels of expression of the immune gene signature, we then focused on the MIBC subgroup. Among the 56 MIBC samples, we observed that tumors bearing a PIK3CA mutation showed a significantly lower expression of the immune gene signature compared to PIK3CA-unmutated ones (Fisher’s exact test, p = 0.04, Figure 1a). However, when we reduced the analysis to only MIBC samples, there were no significant associations between the expression of the immune gene signature and FGFR3 mutational status (Fisher’s exact test, not significant (NS)) or RAS mutational status (Fisher’s exact test, NS, Figure 1a).
Figure 1.
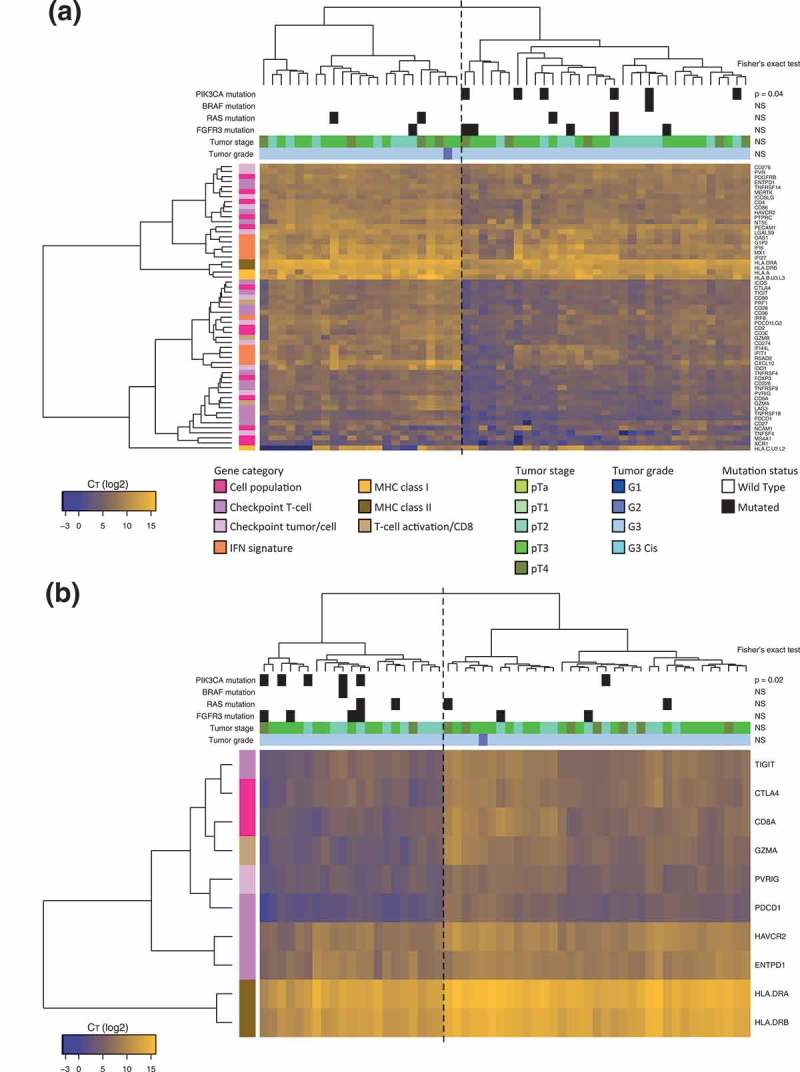
Heatmaps displaying unsupervised clustering of MIBCs into “high” or “low” immune-infiltrated tumors based on immune gene expression and displaying PIK3CA mutational status.
a. Hierarchical clustering heatmap of 56 MIBCs according to the qRT-PCR expression level of 57 immune genes. Dotted line delimitates clusters of “high” or “low” immune-infiltrated tumors, based on the level of expression of immune genes. Presence or absence of activating mutations of PIK3CA, BRAF, RAS, and FGFR3 oncogenes is indicated, along with tumor stage and tumor grade for each sample. MIBCs bearing a PIK3CA mutation show a significantly lower expression of the immune gene signature than wild type tumors (Fisher’s exact test, p < 0.05). Gradient represents the log2 CT value for each gene (yellow = high expression, blue-violet = low expression, dark blue = no expression).b. Hierarchical clustering heatmap of 56 MIBCs according to the qRT-PCR expression level of the 10 most statistically significant differentially expressed genes between wild type and PIK3CA-mutated samples (according to Mann–Whitney Wilcoxon test). Dotted line delimitates clusters of “high” or “low” immune-infiltrated tumors, based on the level of expression of immune genes. PIK3CA-mutated tumors segregate from the PIK3CA-wild type tumors (p < 0.05, Fisher exact test) and fall in the “low” immune-infiltrated tumor cluster. Gradient represents the log2 CT value for each gene (yellow = high expression, blue-violet = low expression, dark blue = no expression).
We then defined a minimal immune gene signature comprising the 10 most statistically significant differentially expressed genes between wild type and PIK3CA-mutated samples (according to Mann–Whitney Wilcoxon test) (Figure 1b). The 10 genes were TIGIT, CTLA4, CD8A, GZMA, PVRIG, PDCD1, HAVCR2, ENTPD1, HLA-DRA, HLA-DRB, which reflect an activated T-cell infiltrate in the tumor. Using this 10-gene signature, the PIK3CA-mutated tumors segregated from the PIK3CA-wild type tumors (p = 0.02, Fisher exact test) and fell in the “cold” tumor cluster. Furthermore, this signature effectively discriminated high- versus low- immune-infiltrated MIBC tumors (p < 0.0001, Fisher exact test).
Overall, our results suggest that mutations in the PIK3CA gene, leading to the activation of the PI3K pathway, are associated with a reduced immune infiltration of the tumor stroma of MIBCs.
Therapeutic inhibition of PI3K pathway inhibits tumor growth in a humanized mice model
To confirm the correlation between the PIK3CA gene activating mutation and the level of tumor T-cell-infiltrate in MIBCs, we set up a humanized mouse model allowing to directly assess the effect of a clinical-grade PI3K inhibitor on human tumor cells and human immune cells in vivo.
We used NOD scid gamma (NSG) recipient mice, which are highly immunodeficient, as they have no B, T, and NK cells, and consequently allow the engraftment of tumor and immune cells of human origin.23,24 Mice were subcutaneously injected with cells of either the VMCUB1 cell line, a PIK3CA-mutated human bladder cancer cell line, or as a control, the HT1376 cell line, a PIK3CA-wild type human bladder cancer cell line. When the tumors were palpable, mice were grafted with human peripheral blood mononuclear cells (PBMCs) isolated from blood samples of human healthy donors. In this humanized model, the majority of human immune cells that reconstitute the mice are T cells derived from the injected PBMCs.24 Three days later, mice were assigned to no treatment or treatment with a class IA-PI3K inhibitor, BKM120 (Figure 2a). Individual and mean tumor growth curves of each cell line are shown in Figure 2b and c, respectively. In the PIK3CA-mutated VMCUB1 group, tumors in mice treated with BKM120 grew at a significantly slower rate, in comparison to the untreated mice (Mann–Whitney test, P < 0.0005, Figure 2b and c). In the PIK3CA-wild type HT1376 group, no significant effect of BKM120 on tumor growth was observed when compared to the untreated mice (Figure 2b and c).
Figure 2.
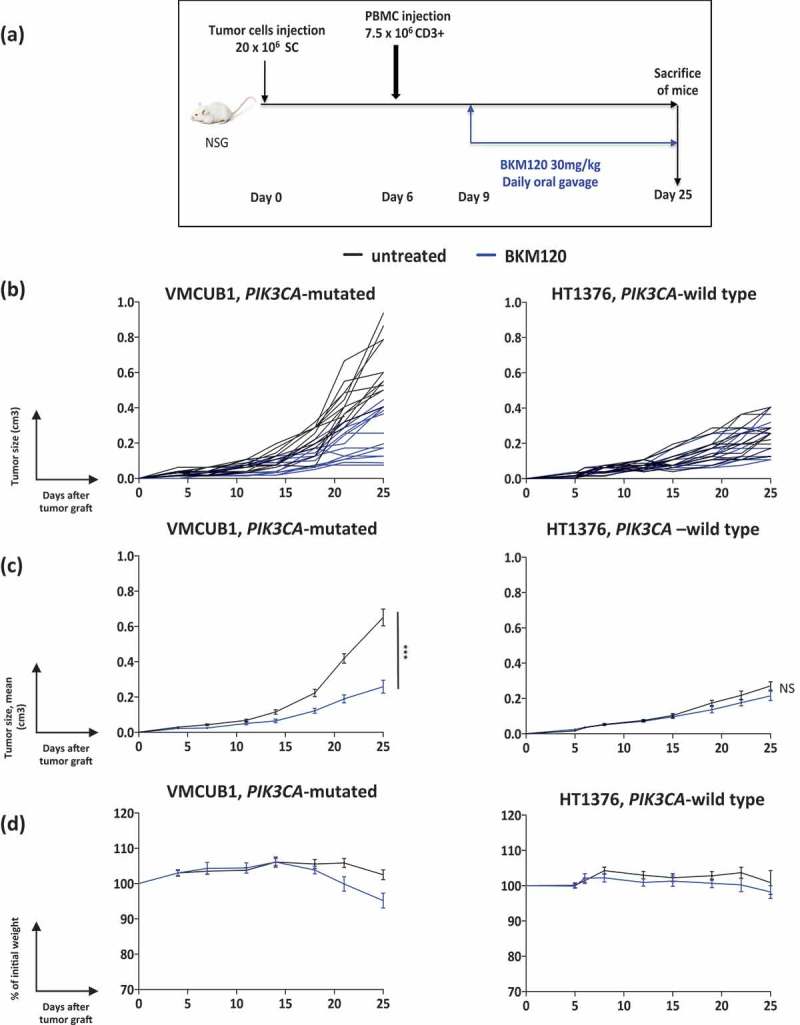
Effect of BKM120 treatment on the growth of VMCUB1, PIK3CA-mutated and HT1376, PIK3CA-wild type human bladder tumors in humanized mice.
a. Experimental protocol: NSG mice were s.c. grafted with the indicated tumor cell line, and when tumors were palpable, they were i.v. injected with PBMCs. Three days later, mice were randomized to the “untreated” (N = 14) or “BKM120-treated” (N = 14) groups. BKM120 was administered daily at a dose of 30mg/kg by oral gavage.b-c. Growth kinetics (b: individual curves; c: mean curves + standard error of the mean) of VMCUB1 (left panel) and HT1376 (right panel) tumors; untreated (black lines), BKM120-treated (blue lines). Tumors were measured twice per week.d. GvHD was followed by the loss of body weight, shown as a percentage of initial weight (mean curves). Results shown are the pool of two independent experiments with similar results. Statistical significance was calculated using the Mann–Whitney test. NS, not significant; ***, P < 0.0005.
The main caveat of this model is that the injected human PBMCs react against mice xeno-antigens, invariably leading to xeno-graft-versus-host-disease (GvHD), which induces progressive body weight loss and death of the mice.24-26 To evaluate GvHD development, we registered mice body weight along the length of the experiment. As shown in Figure 2d, no weight loss higher than 20% (what is considered as a hallmark of clinical GvHD) was observed until day 25 in any of the two models. Consequently, similar to the results already published using PBMC injection in NSG mice,24,27,28 the therapeutic observational window in this model is around 3–4 weeks after PBMCs injection, before evident signs of GvHD.
As BKM120 inhibited tumor growth, we used qRT-PCR to evaluate a panel of genes involved in cell cycling/division that could explain the direct drug effect on tumor cell proliferation. We assessed the expression profile of the proliferation genes ETV4, ETV5, DUSP1, which are downstream in the MAPK pathway, and the CA9 gene which is downstream in the PI3K/AKT/mTOR pathway29 (Supplementary Figure 2). These genes were significantly downregulated in the PIK3CA-mutated VMCUB1 humanized mice after BKM120 treatment, indicating an anti-proliferative effect and an efficient inhibition of the PI3K oncogenic pathway under BKM120 treatment. Although with this technique we cannot dissociate whether the anti-proliferative effect is occurring in the tumor or in other cells present in the microenvironment, the fact that the activating PIK3CA mutation is only present on the tumor cells suggests that variation on the proliferative genes is mainly occurring in the tumor. Altogether these data show that the class IA-PI3K inhibitor BKM120 significantly inhibited the growth of the PIK3CA-mutated VMCUB1 human bladder cancer cell line in a humanized mouse model.
Blocking PI3K signaling increases tumor-immune infiltrate
We then examined the changes that occurred in the intra-tumoral immune infiltrate upon inhibition of the PI3K pathway by BKM120. Total immune infiltration (HuCD45+ cells) was quantified by FACS in tumor samples harvested at a sacrifice of mice at day 25 (Figure 3a). We observed that in the PIK3CA-mutated VMCUB1 group, BKM120 treatment significantly increased the proportion of HuCD45+ cells in the tumor (p < 0.001), as well as the proportion of CD3+ T cells (p < 0.05), in comparison to untreated mice. In tumors harvested from mice of the PIK3CA-wild type HT1376 group, there were no significant changes in the percentage of total immune cells or CD3+ T cells under BKM120 treatment. We then evaluated the distribution of different immune cell subpopulations in the tumor microenvironment. In the PIK3CA-mutated VMCUB1 group and in the PIK3CA-wild type HT1376 group, there were no statistically significant differences between the untreated and treated groups regarding the composition of immune T cells (including CD8+ cytotoxic, CD4+ FOXP3- conventional and CD4+ FOXP3+ regulatory T cells), B and NK cells (Figure 3b). Immunohistochemistry analysis of the tumor samples confirmed the presence of the transferred human hematopoietic cells in the tumor bed. Representative stainings are shown in Figure 3c, depicting the anatomic distribution of tumor-infiltrating HuCD45+ cells, which were found not only at the periphery, but also interspaced among the tumor cells. Overall, these results suggest that at the dose used, BKM120 reduces the growth of PIK3CA-mutated tumors, yet it does not exert a sizeable toxic effect on T cells.
Figure 3.
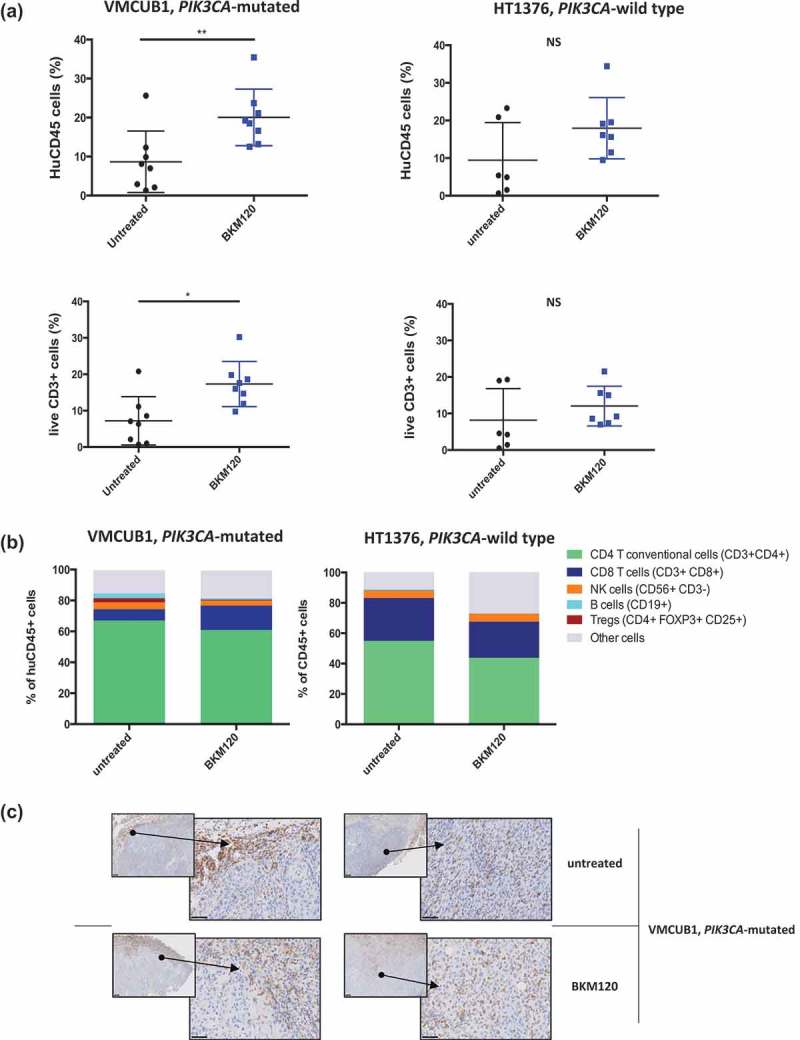
Effect of BKM120 treatment on the tumor immune infiltrate of VMCUB1, PIK3CA-mutated, and HT1376, PIK3CA-wild type human bladder tumors in humanized mice.
Humanized mice were treated as in Figure 2 and tumors were analyzed at day 25.a. FACS analysis showing the proportion of total HuCD45+ and CD3+ T cells infiltrating the tumor, among live cells. Left panels: PIK3CA-mutated VMCUB1 tumors (N = 8 in each group); right panels: PIK3CA-wild type HT1376 tumors (N = 6 in the untreated group and N = 7 in the BKM120 group).b. Proportion of the indicated immune cell subsets among tumor-infiltrating HuCD45+ cells. Results shown are from one representative experiment out of two with similar results. Statistical significance was calculated using the Mann–Whitney test. NS, not significant; *, P < 0.05; **, P < 0.01.C. VMCUB1 PIK3CA-mutated tumors from untreated and BKM120-treated mice were characterized by IHC for human infiltrate (HuCD45). Representative IHC stainings of two mice per indicated group are shown with two different magnifications: smaller square, x50; larger square, x400.
We further assessed the impact of BKM120 treatment on the immune gene expression profile using qRT-PCR analysis, of tumor samples from the humanized mouse model samples. Supplementary Figure 3 shows the effect of BKM120 treatment on the expression of 19 evaluated immune genes (which we selected among the most differentially expressed in Figure 1a). As it could be expected, we observed no or minimal expression of genes corresponding to human immune cell populations that are usually not found (or present at very low levels) in the tumor infiltrate of this humanized model, including neutrophils (FUT4), macrophages (MERTK), dendritic cells (XCR1), Tregs (FOXP3), NK cells (NCAM1) and B cells (MS4A1). Figure 4 depicts individual mRNA expression of the genes that were statistically significantly modified by BKM120 treatment of VMCUB1 PIK3CA-mutated tumor-bearing mice, including PTPRC (HuCD45) (p < 0.005), CD3E (CD3) (p < 0.005), CD8A (CD8) (p < 0.05) GZMA (granzyme A) (p < 0.005), and HLA-DRA (HLA-DRA) (p < 0.05). These genes were not significantly affected in the PIK3CA-wild type HT1376 samples. In PIK3CA-mutated VMCUB1 bearing humanized mice, we also observed a BKM120-mediated increase of the expression of immune checkpoint genes including TIGIT (p < 0.005), CTLA-4 (p < 0.05), PDCD1 (PD-1) (P < 0.005), HAVCR2 (TIM-3)(p < 0.0005) and ENTPD1 (CD39) (p < 0.0005) (data not shown).
Figure 4.
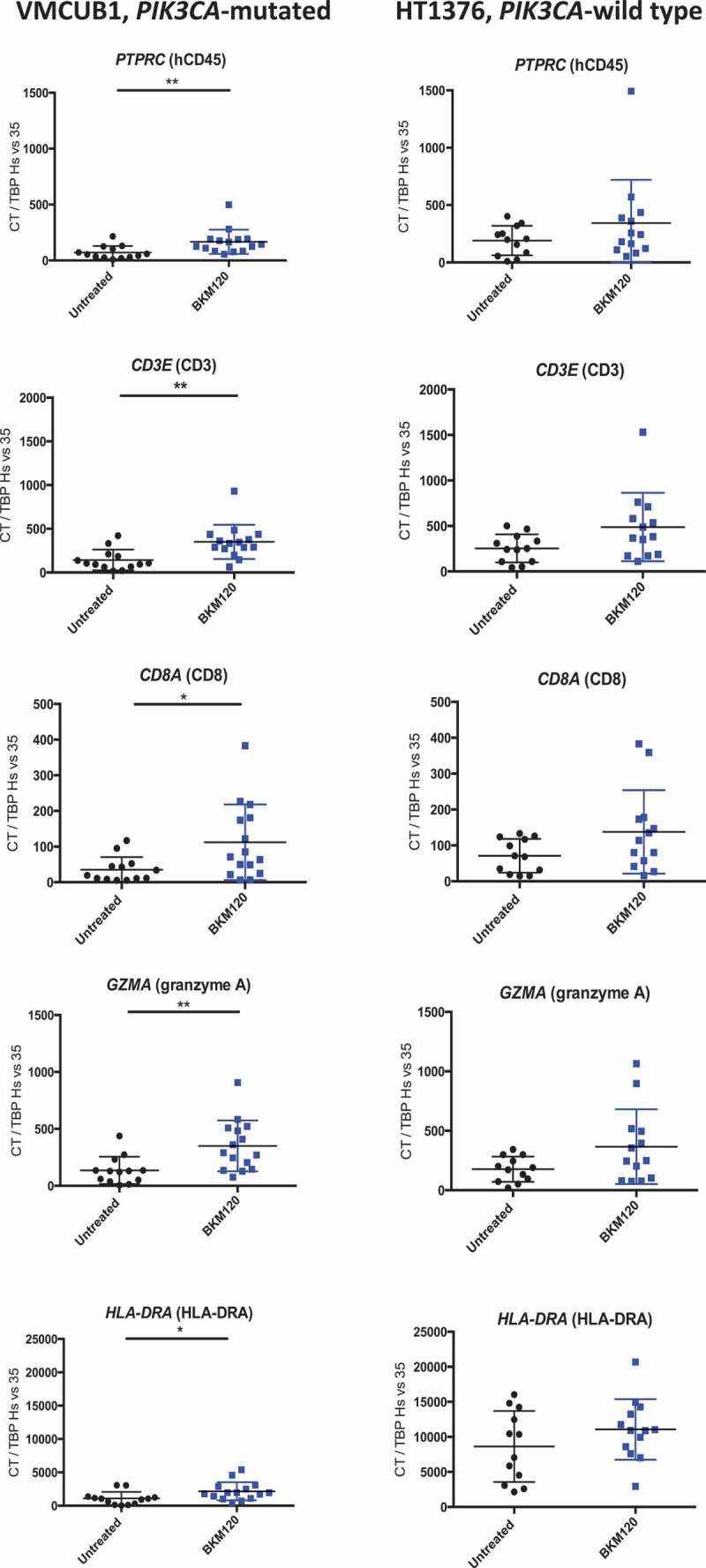
Immune gene expression changes induced by BKM120 treatment on PIK3CA-mutated VMCUB1 and PIK3CA-wild type HT1376 human bladder tumors in humanized mice.
NSG humanized mice were treated with BKM120 or untreated, as detailed in Figure 2 and tumors were analyzed at day 25. Shown is the mRNA expression quantified by qRT-PCR of the indicated genes in PIK3CA-mutated VMCUB1 tumors (left panels) and PIK3CA-wild type HT1376 tumors (right panels), untreated (black dots) or BKM120-treated (blue dots). PIK3CA-mutated VMCUB1 tumors: N = 13 in the untreated group and N = 15 in the BKM120 group. PIK3CA-wild type HT1376 tumors: N = 12 in the untreated group and N = 13 in the BKM120 group. Statistical significance was calculated using the Mann–Whitney test; * p < 0.05, ** p < 0.01.
To gain insight into the mechanism underlying the efficacy of BKM120 on the control of PIK3CA-mutated VMCUB1 tumor growth, we used qRT-PCR to evaluate a panel of genes involved in leukocyte attraction and migration (that could explain the increased T-cell infiltration) (Table 2), including 36 genes coding for chemokines responsible for immune cell (effector or suppressive subsets) migration to the tumor, for molecules involved in angiogenesis and for several adhesion molecules. We compared the genes’ expression level in untreated versus BKM120 treated mice. Figure 5 shows those genes that were statistically differentially expressed upon treatment (Figure 5a shows fold-change expression, and Figure 5b and Supplementary Figure 4 show quantification of individual genes). Transcripts up-regulated by BKM120 treatment included CXCL13 (known to recruit TFH cells),30 CCL2, CCL3, CCL5 (known to recruit monocytes and macrophages) and CXCL9, CXCL10, CXCL11 (known to recruit CXCR3-expressing immune cells Th1, CD8+ T cells, and NK cells).31 Transcripts down-regulated by BKM120 treatment included ITGB3, ITGB5, ITGB8, which code for adhesion molecules described to promote tumor invasion or tumor escape from immune control.32,33 These results suggest that BKM120 treatment re-shapes the landscape of chemokines and integrins present in the tumor microenvironment, promoting tumor infiltration by immune cells.
Table 2.
List of the evaluated 36 genes coding for human chemokines and integrins.
| ID | GeneID | GeneSymbol | GeneName |
|---|---|---|---|
| Chemokines genes | |||
| CCL2 | 6347 | CCL2 | C-C motif chemokine ligand 2 |
| CCL3 | 6348 | CCL3 | C-C motif chemokine ligand 3 |
| CCL4 | 6351 | CCL4 | C-C motif chemokine ligand 4 |
| CCL5 | 6352 | CCL5 | C-C motif chemokine ligand 5 |
| CCL8 | 6355 | CCL8 | C-C motif chemokine ligand 8 |
| CCL21 | 6366 | CCL21 | C-C motif chemokine ligand 21 |
| CSF2 | 1437 | CSF2 | Colony stimulating factor 2 |
| CX3CL1 | 6376 | CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CXCL1 | 2919 | CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL2 | 2920 | CXCL2 | C-X-C motif chemokine ligand 2 |
| CXCL3 | 2921 | CXCL3 | C-X-C motif chemokine ligand 3 |
| CXCL5 | 6374 | CXCL5 | C-X-C motif chemokine ligand 5 |
| CXCL6 | 6372 | CXCL6 | C-X-C motif chemokine ligand 6 |
| CXCL9 | 4283 | CXCL9 | C-X-C motif chemokine ligand 9 |
| CXCL10 | 3627 | CXCL10 | C-X-C motif chemokine ligand 10 |
| CXCL11 | 6373 | CXCL11 | C-X-C motif chemokine ligand 11 |
| CXCL12 | 6387 | CXCL12 | C-X-C motif chemokine ligand 12 |
| CXCL13 | 10563 | CXCL13 | C-X-C motif chemokine ligand 13 |
| Adhesion molecules genes | |||
| ITGA1 | 3672 | ITGA1 | Integrin subunit alpha 1/CD49a/VLA1 |
| ITGA2 | 3673 | ITGA2 | Integrin subunit alpha 2 |
| ITGA4 | 3676 | ITGA4 | Integrin subunit alpha 4 |
| ITGA5 | 3678 | ITGA5 | Integrin subunit alpha 5 |
| ITGA6 | 3655 | ITGA6 | Integrin subunit alpha 6 |
| ITGAV | 3685 | ITGAV | Integrin subunit alpha V |
| ITGB1 | 3688 | ITGB1 | Integrin subunit beta 1 |
| ITGB3 | 3690 | ITGB3 | Integrin subunit beta 3 |
| ITGB4 | 3691 | ITGB4 | Integrin subunit beta 4 |
| ITGB5 | 3693 | ITGB5 | Integrin subunit beta 5 |
| ITGB8 | 3696 | ITGB8 | Integrin subunit beta 8 |
| ICAM1 | 3383 | ICAM1 | Intercellular adhesion molecule 1 |
| ITGAM | 3684 | ITGAM | Integrin subunit alpha M |
| SELE | 6401 | SELE | Selectin E |
| VCAM1 | 7412 | VCAM1 | Vascular cell adhesion molecule 1 |
| Angiogenesis involved genes | |||
| VEGF | 7422 | VEGFA | Vascular endothelial growth factor A |
| VEGFB | 7423 | VEGFB | Vascular endothelial growth factor B |
| VEGFC | 7424 | VEGFC | Vascular endothelial growth factor C |
Figure 5.
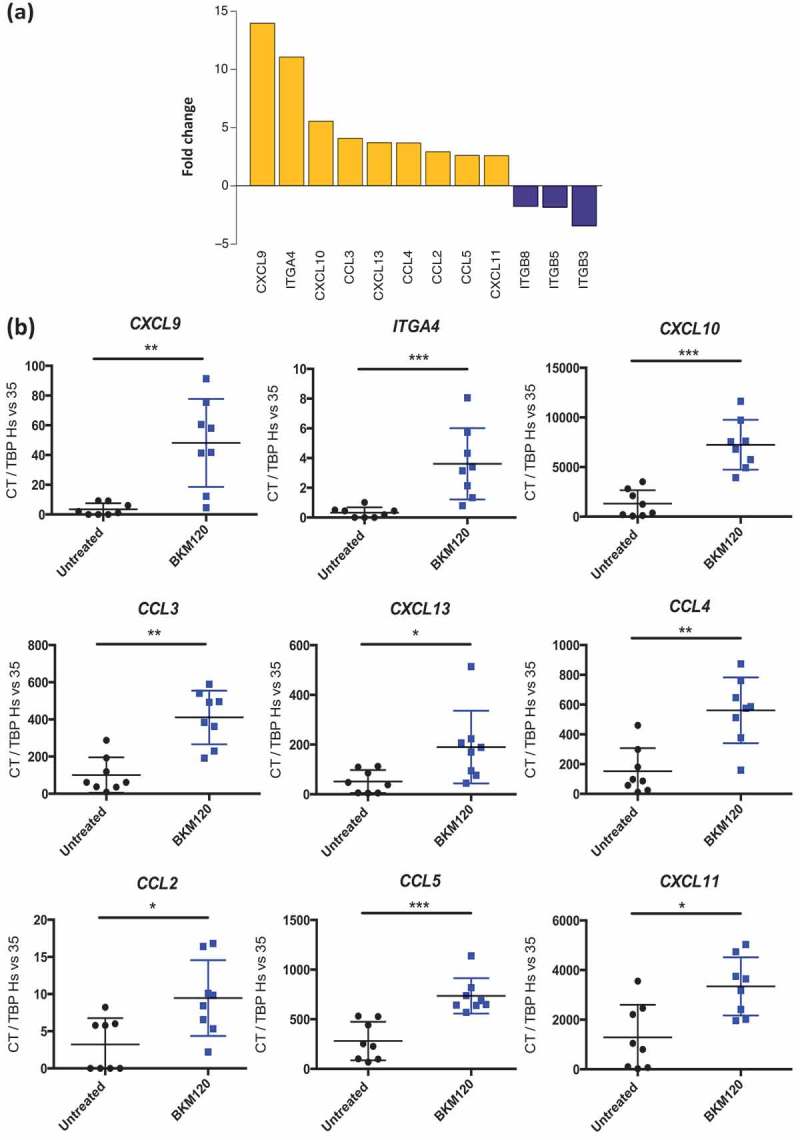
Chemokines and adhesion molecules gene expression changes induced by BKM120 treatment on PIK3CA-mutated VMCUB1 human bladder tumors in humanized mice.
NSG humanized mice grafted with VMCUB1 PIK3CA-mutated tumors were treated with BKM120 or untreated, as detailed in Figure 2 and tumors were analyzed at day 25. mRNA expression of selected chemokines and adhesion molecules was quantified by qRT-PCR. a. Waterfall plot displaying fold-change expression, between BKM120-treated versus non-treated samples, of genes showing a significant statistical difference among 36 genes tested. The genes upregulated in BKM120-treated tumors are in yellow, and the genes downregulated are in blue. b. Shown is the mRNA expression of indicated genes (shown in A) for individual mice (see also Supplementary Figure 4). N = 8 per group. Statistical significance was calculated using the Mann–Whitney test; * p < 0.05, ** p < 0.01, *** p < 0.0005.
Increased tumor-immune infiltration induced by PI3K-signaling blockade renders tumors sensitive to anti-PD-1
Recent studies indicate that sufficient tumor infiltration by T cells is essential for response to immunotherapy.12,18 Consequently, increasing tumor-infiltrating T cells, as observed in BKM120 treatment of PIK3CA-mutated VMCUB1 tumors (Figure 3 and Supplementary Figure 3), should improve the therapeutic efficacy of PD-1 checkpoint blockade therapy. To test this hypothesis, we evaluated the effect of combining BKM120 with an anti-PD-1 antibody used in the clinic (nivolumab) on tumor growth in the humanized mice (Figure 6). Mice bearing PIK3CA-mutated VMCUB1 tumors and grafted with human PBMCs were assigned to the following groups: untreated, BKM120 alone, nivolumab alone or the combination of both (Figure 6a). Individual curves and mean tumor growth curves are shown in Figure 6b and c, respectively. It can be observed that the administration of nivolumab alone did not significantly affect tumor growth, BKM120 treatment alone significantly delayed tumor growth as previously observed in Figure 2b and c (One-way ANOVA and Kruskal–Wallis test P< 0.01), and both drugs combined showed an additional effect (One-way ANOVA and Kruskal–Wallis test P< 0.0001). We verified that no body weight loss higher than 20% (clinical definition of GvHD) occurred in any of the groups, during the first 25 days of the experiment (Figure 6d).
Figure 6.
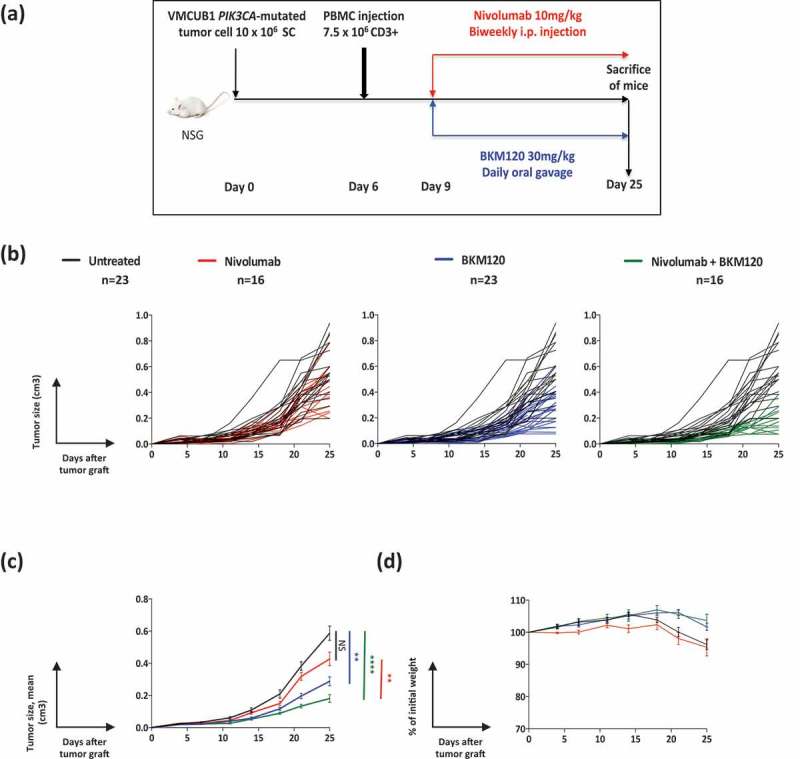
Nivolumab synergizes with BKM120 to reduce the growth of VMCUB1, PIK3CA-mutated human bladder tumors in humanized mice.
a. Experimental protocol: NSG mice were s.c. grafted with VMCUB1, PIK3CA-mutated tumor cells, and when tumors were palpable, they were i.v. injected with PBMCs. Three days later, mice were randomized to the “untreated” (N = 23), “BKM120-treated” (N = 23), “nivolumab-treated” (N = 16) or combined “nivolumab-plus-BKM120-treated” (N = 16) groups. BKM120 was administered daily at a dose of 30mg/kg by oral gavage and nivolumab was administered two times per week at a dose of 10mg/kg, i.p.b-c. Tumor growth kinetics (b: individual curves; c: mean curves + standard error of the mean) of VMCUB1, PIK3CA-mutated tumors untreated (black line), treated with nivolumab (red line), BKM120 (blue line) or combination of both (green line). Tumors were measured twice per week.d. GvHD was followed by the loss of body weight shown as a percentage of initial weight (mean curves). Results shown are the pool of three independent experiments with similar results. Statistical significance was calculated using One-way ANOVA and Kruskal–Wallis test. **, P< 0.01, ****, P< 0.0001.
These results indicate that treatment of the PIK3CA-mutated VMCUB1 tumor with a targeted inhibitor rendered this PD-1 resistant tumor sensitive to immunotherapy, and therefore demonstrate the potential benefit of combining a targeted therapy with an immunotherapy.
To understand the requirement of immune cells for the observed synergistic effect of the combinatory treatment, we performed a similar experiment as in Figure 6 in mice non-grafted with human PBMC (Supplementary Figure 5). We observed that BKM120 was efficient in inhibiting tumor growth even in the absence of human immune cells, but the additional effect of combining nivolumab with BKM120 strictly required the presence of human PBMCs, confirming that anti-PD-1 is acting through T cells.
Discussion
Sensitivity of tumors to therapies targeting immune checkpoints seems to be conditioned by the quality and quantity of the tumor immune infiltrate.
We defined a qRT-PCR-based immune gene signature that allows weighing in a semi-quantitative way the immune infiltrate of solid tumors and that could discriminate high T-cell infiltrated bladder tumors from low/non-T-cell infiltrated tumors. This signature, which can be reduced to 10 genes, represents a low-cost and rapid technique that could be adapted and further validated in the clinical setting to evaluate tumor immune infiltration, so as to help in the clinical management of cancer patients.
Moreover, our approach indicates that the combination of this signature with the mutational status of oncogenic genes represents a simple way to identify tumoral oncogenic pathways responsible for the exclusion of the immune infiltrate out of the tumor microenvironment. The identified druggable mutations could represent a potential target for combination with anti-checkpoint antibodies, allowing that T cells re-invigorated by the immunotherapy effectively access the tumor bed.
We found that in a cohort comprising 98 NMIBC and MIBC human samples, tumors bearing activating mutations in PIK3CA or FGFR3 genes were associated with lower expression of the immune gene signature, compared to tumors bearing the wild-type genes, what could reflect the presence of a less active tumor immune infiltrate. Our findings confirm and extend a previous report associating FGFR3 pathway activation in non-T-cell-inflamed MIBCs, as well as a lower immune gene expression in NMIBCs compared to MIBCs.16 When we focused on MIBC tumor sub-type, we observed that MIBCs from patients bearing PIK3CA gene mutations but not BRAF, RAS or FGFR3 mutations were still significantly associated to non-T-cell-infiltrated tumors, compared to unmutated ones, which, to our knowledge, has not been reported before. These results indicate that the identified molecular events do not explain all the cases of infiltrated or non-infiltrated tumors. On the contrary, our work indicates that the influence of each oncogenic pathway on the immune infiltrate is tumor-type dependent and even more, tumor sub-type dependent, as was the case for the muscle-invasive and non-invasive bladder tumors in our studied series. Another point that could explain the fact that FGFR3 pathway was not associated with immune gene expression patterns in MIBC anymore is that FGFR3 mutations are more frequent in NMIBC than MIBC.34
To confirm this association between the tumor T-cell infiltrate in MIBCs and mutations of the PIK3CA gene, we set up a humanized mouse model allowing direct assessment of the effect of a clinical-grade PI3K inhibitor on human tumor cells and human immune cells in vivo. We observed that BKM120, a pan class 1A-PI3K inhibitor, significantly inhibited the growth of a human bladder cancer cell line bearing a druggable PIK3CA mutation, associated with an increase in tumor infiltration by an immune cell (HuCD45+) and more specifically CD3+ T cells.
Different primary and adaptive acquired mechanisms of resistance to immune checkpoint inhibitors have been described. Among them, the absence of tumor recognition by T cells, due to the lack of immunogenic tumor antigens (i.e. low mutational burden, absence of cancer-testis antigen), or to the development by the tumor of mechanisms that blunt antigen presentation (i.e. alterations in proteasome subunits or transporters associated with antigen processing or deletion of the beta-2-microblobulin or the MHC molecules themselves).12,19,35 Also, tumor-intrinsic activated oncogenic pathways have been associated with poor responses to immunotherapy. Along these lines, the loss of PTEN and consequent activation of the PI3K/AKT/mTOR pathway has been associated with a post-transcriptional increase of PD-L1 expression in gliomas,14 and to decreased T-cell infiltration at tumor sites in melanoma in part due to the increase in the expression of immunosuppressive cytokines.15 Additionally, tumor-intrinsic active WNT/β-catenin signaling pathway has also been described to induce T-cell exclusion from the tumor tissue mediated by decreased production of chemokines.36 Moreover, a panel of chemokines (CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10) was described to be expressed in metastatic melanomas rich in infiltrating T-cells.8 These results are in accordance with the upregulated chemokine transcripts that we detected after inhibition of the PI3K pathway. Furthermore, we observed that adhesion molecules’ expression (ITGB3, ITGB5, and ITGB8) was downregulated after BKM120 administration, which could further contribute to the increase in tumor infiltration by immune cells, as previously described33,37).
The anti-tumoral immune response can also be inhibited by immunosuppressive cells, such as Tregs and tumor-associated macrophages. Indeed, inhibition of PI3K pathway could also have an inhibitory effect on myeloid-derived suppressor cells (MSDCs), which could restore the activity of T cells, without increasing the quantity of T cells present in the tumor microenvironment, but leading to an increase of activated T cells. This has been shown for anti-angiogenic treatments which seem to act, at least in part, by reducing the number of MSDCs in tumor immune infiltrate, and consequently promoting the activation of T cells. This hypothesis has been investigated recently in a head and neck squamous cell carcinoma model, in which inhibition of PI3K pathway with a PI3K inhibitor could functionally inhibit MDSCs, leading to enhanced responses to PD-L1 blockade. Moreover, in this model, the combination therapy of this PI3K inhibitor with anti-PD-L1 antibody induced CD8+ T cell-dependent primary tumor growth delay.38 However, in the humanized mouse model that we used for our study, the human myeloid cells do not efficiently reconstitute the immunodeficient mice, hampering the evaluation of the therapy’s effect on human myeloid cells.
Finally, we observed that inhibition of the PI3K pathway effectively increased immune infiltration of PIK3CA-mutated bladder tumors, thus transforming “cold” tumors into “hot” tumors, which is thought to represent a key step in obtaining significant responses to immunotherapies. We describe that BKM120 induced an increase of the expression of several immune cell population genes such as PTPRC (HuCD45), CD3E (CD3), CD8A (CD8) or GZMA (granzyme A) reflecting an increasing active immune infiltrate, along with genes encoding for immune checkpoints such as TIGIT, CTLA-4, PD-1, TIM-3, or CD39, suggesting potential for tumor sensitivity to immune checkpoint blockade, and identifying other potential immune checkpoint targets other than anti-CTLA-4 and anti-PD-1/PD-L1 antibodies. Furthermore, our data showed that the combination of a PI3K inhibitor and nivolumab treatment rendered an anti-PD-1 resistant tumor sensitive to the immunotherapy.
Our results suggest a relevant rationale for combination strategies of PI3K inhibitors with immune checkpoint inhibitors. Actually, several combination strategies of immune checkpoint inhibitors with either molecularly targeted therapies, chemotherapy, radiotherapy, or other immunotherapies in development are evaluated, to try to overcome primary resistance to immune checkpoint inhibitors in various tumor types.39 Strategies evaluating PI3K inhibitors in combination with immune checkpoint inhibitors are already being assessed in early clinical trials.40
Further studies will be needed to better decipher the mechanisms of immune resistance, and also to define the most efficient combination strategies, with the most efficient timing and dosage of targeted and immune therapies.
Materials and methods
Classification of urothelial bladder cancer subtypes by immune gene expression profile and presence of mutations in oncogenic pathways
Patients and samples
Patients included 17 women (17.3%) and 81 men (82.7%). Pathological staging included 42 NMIBC, 18 low-grade (42.9%) and 24 high-grade (57.1%) NMIBC, and 56 high-grade MIBC. Clinical and histological characteristics of patients along with survival are summarized in Tables 3 and 4.
Table 3.
Clinicopathological characteristics and survival of 56 MIBC patients.
| Whole population |
Disease-free survival |
Overall survival |
|||
|---|---|---|---|---|---|
| Number of patients (%) | Number of events (%)a | p-value* | Number of events (%)b | p-value* | |
| Total population | 56 (100.0) | 36 (64.3) | 34 (60.7) | ||
| Age (years) | |||||
| ≥60 | 40 (71.4) | 30 (83.3) | 0.0082 | 30 (88.2) | 0.0005 |
| <60 | 16 (28.6) | 6 (16.7) | 4 (11.8) | ||
| Sex | |||||
| Male | 42 (75.0) | 24 (66.7) | 0.053 (NS) | 26 (76.5) | 0.75 (NS) |
| Female | 14 (25.0) | 12 (33.3) | 8 (23.5) | ||
| Smoking statusc | |||||
| Non-smoker | 9 (18.0) | 7 (23.3) | 0.41 (NS) | 7 (24.1) | 0.34 (NS) |
| Smoker | 41 (82.0) | 23 (76.7) | 22 (75.9) | ||
| History of NMIBC | |||||
| No | 35 (62.5) | 21 (58.3) | 0.39 (NS) | 20 (58.8) | 0.48 (NS) |
| Yes | 21 (37.5) | 15 (41.7) | 14 (41.2) | ||
| Associated pTis | |||||
| No | 50 (89.3) | 34 (94.4) | 0.17 (NS) | 32 (94.1) | 0.20 (NS) |
| Yes | 6 (10.7) | 2 (5.6) | 2 (5.9) | ||
| Tumor stage | |||||
| T2 | 19 (33.9) | 11 (30.6) | 0.47 (NS) | 8 (23.5) | 0.041 |
| ≥T3 | 37 (66.1) | 25 (69.4) | 26 (76.5) | ||
| Lymph node statusd | |||||
| N- | 35 (63.6) | 18 (51.4) | 0.013 | 17 (50.0) | 0.0075 |
| N+ | 20 (36.4) | 17 (48.6) | 17 (50.0) | ||
| FGFR3 status | |||||
| Mutated | 6 (10.7) | 4 (11.1) | >0.99 (NS) | 4 (11.8) | >0.99 (NS) |
| Not mutated | 50 (89.3) | 32 (88.9) | 30 (88.2) | ||
| PIK3CA status | |||||
| Mutated | 6 (10.7) | 2 (5.6) | 0.17 (NS) | 2 (5.9) | 0.20 (NS) |
| Not mutated | 50 (89.3) | 34 (94.4) | 32 (94.1) | ||
*Chi2 test, Chi2 test with Yates’ correction or Fisher test if appropriate
NS: not significant
afirst recurrence (local or metastatic)
bdeath
cdata available for 50 patients
ddata available for 55 patients
Table 4.
Clinicopathological characteristics and survival of 42 NMIBC patients.
| Whole population |
No recurrence |
Recurrence |
Muscle-invasive progression | |||
|---|---|---|---|---|---|---|
| Number of patients (%) | Number (%) | Number (%) | p-value* | Number (%) | p-value** | |
| Total population | 42 (100) | 13 (31.0) | 21 (50.0) | 8 (19.0) | ||
| Age (years) | ||||||
| ≥60 | 33 (78.6) | 9 (69.2) | 16 (76.2) | 0.96 (NS) | 8 (100.0) | 0.17 (NS) |
| <60 | 9 (21.4) | 4 (30.8) | 5 (23.8) | 0 (0.0) | ||
| Sex | ||||||
| Male | 39 (92.9) | 12 (92.3) | 19 (90.5) | >0.99 (NS) | 8 (100.0) | >0.99 (NS) |
| Female | 3 (7.1) | 1 (7.7) | 2 (9.5) | 0 (0.0) | ||
| Smoking status | ||||||
| Non-smoker | 17 (40.5) | 3 (23.1) | 11 (52.4) | 0.092 (NS) | 3 (37.5) | 0.83 (NS) |
| Smoker | 25 (59.5) | 10 (76.9) | 10 (47.6) | 5 (62.5) | ||
| History of NMIBC | ||||||
| No | 22 (52.4) | 10 (76.9) | 9 (42.9) | 0.052 (NS) | 3 (37.5) | 0.59 (NS) |
| Yes | 20 (47.6) | 3 (23.1) | 12 (57.1) | 5 (62.5) | ||
| Associated pTis | ||||||
| No | 40 (95.2) | 13 (100.0) | 21 (100.0) | >0.99 (NS) | 6 (75.0) | 0.033 |
| Yes | 2 (4.8) | 0 (0.0) | 0 (0.0) | 2 (25.0) | ||
| Grade | ||||||
| Low grade | 18 (42.9) | 8 (61.5) | 9 (42.9) | 0.29 (NS) | 1 (12.5) | 0.13 (NS) |
| High grade | 24 (57.1) | 5 (38.5) | 12 (57.1) | 7 (87.5) | ||
| Tumor stage a | ||||||
| Ta | 28 (68.3) | 9 (69.2) | 17 (81.0) | 0.71 (NS) | 2 (28.6) | 0.024 |
| T1 | 13 (31.7) | 4 (30.8) | 4 (19.0) | 5 (71.4) | ||
*Chi2 test, Chi2 test with Yates’ correction or Fisher test if appropriate (recurrence versus no recurrence)
**Chi2 test, Chi2 test with Yates’ correction or Fisher test if appropriate (muscle-invasive progression versus others)
NS: not significant
adata available for 41 patients
Patients included in this study had undergone transurethral bladder resection or radical cystectomy in Cochin hospital between January 2002 and January 2006. Data were obtained from the patient’s medical records. All patients signed an informed consent. This study received approval from an institutional review board and was conducted according to the principles outlined in the Declaration of Helsinki.
Immediately after surgery, tumor samples were frozen in liquid nitrogen and stored at −80℃ (for RNA extraction) and fixed in formaldehyde.
qRT-PCR analysis of the immune gene signature in human bladder tumor samples
The theoretical basis, RNA extraction, cDNA synthesis, design of primers and PCR reaction conditions have been previously described in detail.41 Briefly, quantitative values were obtained from the threshold cycle (Ct) number at which the increase in the signal associated with the exponential growth of PCR products began to be detected. One endogenous RNA control gene was chosen, namely TBP (Gen-Bank Accession No. NM_003194), which encodes TATA box-binding protein. Each sample was normalized based on its TBP content. Results, expressed as N-fold differences in target gene expression relative to the TBP gene and termed Ntarget, were determined as Ntarget = 2Ct sample, where the Ct value of the sample was determined by subtracting the average Ct value of the target gene from the average Ct value of the TBP gene. For all genes, a Ct value below 38 was considered as quantifiable and the mRNA values were normalized such that a Ct value of 38 was equal to 0.125, i.e. smallest quantifiable amount of mRNA. A Ct value above 38 was considered as not quantifiable. Positive controls for all genes were obtained by performing an RNA pool control, which was prepared by mixing identical amounts of RNA from various human normal and tumoral tissues.
To generate the heatmaps we used the “heatmap.3 function in the R software”. Hierarchical clusterings were produced using as input the log2 expression values of normalized Ct, using Ward’s method on euclidean distances. To solve the error caused by log transformation of values of 0, we have added 0.001 to all values before log transformation. Values in the heatmap are expressed as log2 (normalized Ct), where (as explained above), a normalized Ct value of 38 is equal to 0.125 (then, log2 (0.125) = −3). This means that in the heatmap all values lower than −3 correspond to non-quantifiable expression of a gene. Fisher’s exact test was applied to verify if identified clusterings permit to discriminate wild type and mutated samples for each oncogenic pathway according to the immune gene signature.
Mann-Whitney-Wilcoxon test was applied between wild type and mutated gene sample populations on each gene to identify the genes that were statistically differentially expressed between both populations.
Evaluation of the effect of targeted therapy inhibition of PI3K pathway on tumor-immune infiltration in vivo
Humanized mouse model
NOD-scid-IL2rγ-/- (NSG) mice were grafted with human tumor cell line and then reconstituted with human peripheral blood mononuclear cells (PBMC) from blood samples of healthy donors.
PBMC were isolated using Ficoll and the quantification of CD3+ cells was done by Fluorescence-activated cell sorting (FACS) and then the total PBMC were immediately stored in liquid nitrogen.
Human bladder cancer cell lines, either VMCUB1, a PIK3CA-mutated human bladder cell line, or HT1376, a PIK3CA-wild type human bladder cancer cell line, were injected ectopically in the right flank of each NSG mouse subcutaneously with 10 or 20 × 106 of cancer cells/mouse.
When the tumors were palpable (above 3x3mm), we injected intravenously (IV), by retro-orbital injection, the amount of PBMC that corresponds to 7.5 × 106 human CD3+ T cells/mouse.
Tumor growth was measured twice a week using a caliper to determine the tumor size, calculated as ((length x width2)/2). Mice were sacrificed before the tumor reached 2 cm3. Mice were weighted at the same time points, to follow graft versus host disease (GvHD), and following ethical guidance, were sacrificed upon a loss of 20% of body weight, considered as the clinical diagnosis of GvHD.
Starting 3 days after PBMC injection, BKM120 (MedChemExpress), a pan-PI3K inhibitor, was administered at a dose of 30 mg/kg per day, via oral gavage every day. Nivolumab (OPDIVO, BMS) is also administered (i.p.) 3 days after PBMC injection at a dose of 10mg/kg twice a week.
At the experimental endpoint, mice were sacrificed; the tumors were collected and processed individually. Half of each tumor sample was used for FACS staining, and the other half was divided in two, one part was used for the histochemistry analysis, and the other was kept in liquid nitrogen directly after dissection for future qPCR analysis to assess our immune gene signature on tumor samples from the humanized mouse model.
Flow cytometry
Half of the tumor was processed for FACS analysis. The tumor was cut into small pieces and mixed with 2 ml of CO2 independent medium (Gibco® Life technologie) containing 30 μl Dnase (10mg/ml, Roche®), and 60 μl liberase LT (5mg/ml, Roche®) per sample. The tumor sample was processed on a Gentlemacs dissociator (Miltenyi Biotec, San Diego, CA) according to the manufacturer’s instructions, then the dissociated tumor sample was filtered through a cell strainer (BD Biosciences), washed with PBS, then incubated with the antibodies (Abs) for FACS staining.
For the phenotypic analysis of human immune cell populations, tumor cells were stained for surface markers with LIVE/DEAD Fixable Aqua (Life TechnologiesTM), hCD27-BV605 clone O323 (Biolegend), hCD3-BV650 clone OKT3 (Biolegend), hCD4-BV785 clone OKT4 (Biolegend), hPD-1-BV711 clone EH12.2H7 (Biolegend), hCD8-PECF594 clone RPA-T8 (BD Biosciences), hCD56-PE-Cy5 clone N901 (BD Biosciences), hCD45-APC Cy7 clone 2D1 (BD Biosciences), hCD19-Alexa 700 clone HIB19 (BD Biosciences), hCD45RA-PECy5 clone HI100 (eBiosciences), hCD25-PE (BD Biosciences), hTCRgd-FITC clone 11F2 (BD Biosciences). Then, for intracellular staining, cells were fixed and permeabilized with fixation/permeabilization solution (eBiosciences) according to the manufacturer’s instructions and stained with FOXP3-efluor 450 clone 236A/E7 (eBioscience).
Samples were then analyzed on a Fortessa flow cytometer (BD Biosciences). FACS data were analyzed with FlowJo Version v10.0.8 (TreeStar).
Immunohistochemistry
Paraffin-embedded tumors from mice untreated or treated with BKM120 were stained for human CD45 cells. Sequential sections of 3μm were cut, deparaffinized, and rehydrated through a series of xylene and ethanol washes. The samples were then exposed to citrate buffer pH 6 (Dako) for 20 min in a microwave (antigen retrieval) followed by an incubation of 10 min with 3% hydrogen peroxidase in deionized water (blocking of endogenous peroxidase), and washed with water and PBS. The samples were then incubated with anti-human CD45 Ab (2B11 and PD7/26 clones, Dako) followed by immunodetection with the biotin-conjugated anti-mouse antibody. Afterward, the samples were incubated with peroxidase-labeled streptavidin and followed by chromogenic reaction with DAB counterstaining with hematoxylin. All the staining process was performed using a Dako autostainer.
Funding Statement
This work was supported by Institut Curie (PIC3i-2015), Institut National de la Santé et de la Recherche Médicale INSERM. Association pour la Recherche sur le Cancer [ARC, PJA 20131200444]; SIRIC [Site de Recherche Intégrée sur le Cancer, INCa-DGOS-Inserm_12554]; Labex DCBIOL [Agence Nationale de la Recherche, ANR-10-IDEX-0001-02 PSL] and [ANR-11-LABX0043].
Acknowledgments
We thank the mouse facility, flow cytometry core, and the Pathex platform from Institut Curie.
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
Authors’ contributions
B.E., DLR. P., B. I. and. P. E. designed the research. B.E., DLR. P., V.S., D. M., C. S., and C.W. performed experiments and analyzed data. R.A. and R.W. performed bioinformatic analysis. K.C., R.F. and Y.A. provided reagents and contributed to experimental design. P.G., B.L., and D.D contributed with patient data and samples, B. I., A. Y., R.F and P. E. critically revised the manuscript. B.E., DLR. P. and. P.E. wrote the manuscript. P.E. conceived and designed the project and wrote the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A.. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Sylvester RJ, van der Meijden APM, Oosterlinck W, Witjes JA, Bouffioux C, Denis L, Newling DWW, Kurth K.. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. Eur Urol. 2006;49(3):466–477. discussion 475-477. doi: 10.1016/j.eururo.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 3.von der Maase H, Hansen SW, Roberts JT, Dogliotti L, Oliver T, Moore MJ, Bodrogi I, Albers P, Knuth A, Lippert CM, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18(17):3068–3077. doi: 10.1200/JCO.2000.18.17.3068. [DOI] [PubMed] [Google Scholar]
- 4.Bellmunt J, Fougeray R, Rosenberg JE, von der Maase H, Schutz FA, Salhi Y, Culine S, Choueiri TK. Long-term survival results of a randomized phase III trial of vinflunine plus best supportive care versus best supportive care alone in advanced urothelial carcinoma patients after failure of platinum-based chemotherapy. Ann Oncol. 2013;24(6):1466–1472. doi: 10.1093/annonc/mdt007. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji -R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirilovsky A, Marliot F, El Sissy C, Haicheur N, Galon J, Pagès F. Rational bases for the use of the immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. Int Immunol. 2016;28(8):373–382. doi: 10.1093/intimm/dxw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 12.Ramos RN, Piaggio E, Romano E. Mechanisms of resistance to immune checkpoint antibodies. Handb Exp Pharmacol. 2017. March 18. doi: 10.1007/164_2017_11. [DOI] [PubMed] [Google Scholar]
- 13.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 15.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF. Molecular drivers of the non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol Res. 2016;4(7):563–568. doi: 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 18.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019. January 4. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 19.Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Maβen S, Horn S, Moll I, Maltaner R, Horn PA, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res. 2014;20(24):6593–6604. doi: 10.1158/1078-0432.CCR-14-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5(200):200ra116–200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Goux C, Vacher S, Pignot G, Sibony M, Barry Delongchamps N, Terris B, Piaggio E, Zerbib M, Damotte D, Bieche I. mRNA expression levels of genes involved in antitumor immunity: identification of a 3-gene signature associated with prognosis of muscle-invasive bladder cancer. Oncoimmunology. 2017;6(11):e1358330. doi: 10.1080/2162402X.2017.1358330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7(2):118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 24.De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39(9):748–763. doi: 10.1016/j.it.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Pérol L, Martin GH, Maury S, Cohen JL, Piaggio E. Potential limitations of IL-2 administration for the treatment of experimental acute graft-versus-host disease. Immunol Lett. 2014;162(2Pt B):173–184. doi: 10.1016/j.imlet.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 26.Naserian S, Leclerc M, Thiolat A, Pilon C, Le Bret C, Belkacemi Y, Maury S, Charlotte F, Cohen JL. Simple, reproducible, and efficient clinical grading system for murine models of acute graft-versus-host disease. Front Immunol. 2018;9:10. doi: 10.3389/fimmu.2018.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 28.King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157(1):104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L, et al. KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell. 2017;168(5):817–829.e15. doi: 10.1016/j.cell.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Leval L, Rickman DS, Thielen C, de Reynies A, Huang Y-L, Delsol G, Lamant L, Leroy K, Brière J, Molina T, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11):4952–4963. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- 31.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, Anderson RL. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006;8(2):R20. doi: 10.1186/bcr1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, Ito S, Yanagisawa H, Hashimoto M, Ma R, et al. Integrin αvβ8-expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight. 2018;3(20). doi: 10.1172/jci.insight.122591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kompier LC, Lurkin I, van der Aa MNM, van Rhijn BWG, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One. 2010;5(11):e13821. doi: 10.1371/journal.pone.0013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 37.Chae YK, Choi WM, Bae WH, Anker J, Davis AA, Agte S, Iams WT, Cruz M, Matsangou M, Giles FJ. Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci Rep. 2018;8(1):1023. doi: 10.1038/s41598-018-19454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, Silvin C, Van Waes C, Allen C. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kδ/γ. Cancer Res. 2017;77(10):2607–2619. doi: 10.1158/0008-5472.CAN-16-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan RJ, Hong D, Tolcher A, Patnaik A, Shapiro G, Chmielowski B, Ribas A, Brail LH, Roberts J, Lee L, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol. 2018;36(suppl; abstr 3013):3013. doi: 10.1200/JCO.2018.36.15_suppl.3013. [DOI] [Google Scholar]
- 41.Bièche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R. Prognostic value of ERBB family mRNA expression in breast carcinomas. Int J Cancer. 2003;106(5):758–765. doi: 10.1002/ijc.11273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.