

Summary
The determining factor of spinal muscular atrophy (SMA), the most common motor neuron degenerative disease of childhood, is the survival motor neuron (SMN) protein. SMN and its Gemin associates form a complex that is indispensible for the biogenesis of small nuclear ribonucleoproteins (snRNPs), which constitute the building blocks of spliceosomes. It is as yet unclear whether a decreased capacity of SMN in snRNP assembly, and, hence, transcriptome abnormalities, account for the specific neuromuscular phenotype in SMA. Across metazoa, the SMN‐Gemins complex concentrates in multiple nuclear gems that frequently neighbour or overlap Cajal bodies. The number of gems has long been known to be a faithful indicator of SMN levels, which are linked to SMA severity. Intriguingly, a flurry of recent studies have revealed that depletion of this nuclear structure is also a signature feature of amyotrophic lateral sclerosis (ALS), the most common adult‐onset motor neuron disease. This review discusses such a surprising crossover in addition to highlighting the most recent work on the intricate world of spliceosome building, which seems to be at the heart of motor neuron physiology and survival.
Keywords: Amyotrophic lateral sclerosis, Cajal body, Gems, SMN‐Gemins complex, Spinal muscular atrophy
Introduction
The correct synthesis, handling and processing of messenger RNAs (mRNAs) is fundamental for the manufacturing of functional proteins, without which, life in its most basic or complex form is simply impossible. Splicing or the pruning away of protein noncoding sequences (introns) and the fusion of coding segments (exons) is particularly important in transforming a pre‐mRNA just transcribed from DNA into a mature mRNA that can be readily translated. The machine responsible for orchestrating this ongoing process is the spliceosome, which, at its core, is essentially a coalition of small nuclear ribonucleoproteins (snRNPs). The intricate world revolving around spliceosome building would have remained obscure had a catastrophic neurodegenerative disease not been discovered in which snRNP assembly was found to be “out of tune” 1, 2, 3. Characterized by loss of lower motor neurons as well as progressive muscle weakness and wasting, spinal muscular atrophy (SMA) is a motor neuron disorder (MND) that is recessively inherited, and as there is currently no cure, it remains the most common genetic cause of infant death 4. In the majority of cases (>97%), SMA has been pinned on low levels of the ubiquitously expressed survival motor neuron (SMN) protein as a result of homozygous deletions or, less frequently, mutations in the SMN1 gene, and a lack of full compensation by a duplicate gene, SMN2 5. Copy number polymorphisms of SMN2 and, thereby, SMN protein levels, are coupled with the degree of phenotype intensity in SMA 2, 6, 7.
Although the biology of the SMN protein has been intensely explored throughout more than two decades since its discovery as the factor determining SMA, three fundamental truths remain. First, SMN is oligomeric in nature 8, 9, 10, 11 and stably associates with a set of core Gemin proteins to form the SMN‐Gemins complex, the size and membership of which is dependent on organism complexity 12. Second, SMN and its Gemin associates are indispensable for the biogenesis of snRNPs 13, 14, 15, 16, and in case of SMN, a tight correlation exists between the degree of its deficiency and snRNP assembly capability 3, 17, 18. Third, across metazoa, the SMN‐Gemins complex concentrates in multiple nuclear puncta named gems or Gemini of Cajal bodies (CBs) in view of them frequently neighbouring or overlapping CBs 19, 20. The number of gems has long been known to be a faithful indicator of SMN levels and, hence, SMA severity 2, 14, 21. Intriguingly, a flurry of recent studies have revealed that depletion of this nuclear structure, which is symptomatic of low SMN levels, is also a hallmark feature of the most common adult‐onset MND, amyotrophic lateral sclerosis (ALS) 22, 23, 24, 25, 26, 27, 28. These emerging findings raise the question of whether spliceosome building is at the heart of motor neuron physiology and survival. Should this be the case, therapeutics aimed at modulating this pathway most likely through augmentation of SMN levels may be universally beneficial in MND. In this account, I review the most recent work on spliceosomal snRNP assembly including the surprising discovery that the less famed Gemin associates of SMN have a key stake in this process and, in this context, their loss of function in vivo disrupts normal motor function in a similar manner to reduced levels of SMN. Importantly, I highlight why SMN has been and, still remains more than ever, the best door to successfully gain insights into how building blocks of the spliceosome are manufactured and why this process might selectively impact the motor unit, when it gets “out of key”.
Recent Developments in Spliceosome Building
Molecular Architecture of the snRNP Assemblyosome
In vertebrates and, hence, humans, the SMN‐Gemins complex is an elaborate nine‐membered union of diverse proteins that largely do not share sequences or domains (Figure 1). SMN, seven Gemin proteins (Gemin2‐Gemin8) 16, 29, 30, 31, 32, 33, 34, 35, 36, and Unrip 37, 38 complete its membership. Not all metazoan organisms are endowed with a sizable complex, thus the fruit fly Drosophila melanogaster possesses a minimalistic SMN‐Gemins complex counting only SMN, Gemin2, Gemin3 and Gemin5 amongst its members 12, 39, 40, 41, 42. In addition to suggesting that the SMN‐Gemins complex gained complexity in evolution, the simplicity of the fly version is an alluring feature considering that Drosophila has a well‐earned reputation as a superb model organism of neurodegenerative disease (reviewed in 43). Although they come together to establish the snRNP assemblyosome, SMN‐Gemins complex members have additional unrelated functions and select components including Gemin3 and Gemin5 even participate in alternative multi‐subunit complexes with a function in an snRNP‐independent activity that is nonetheless RNP‐centric (reviewed 12).
Figure 1.
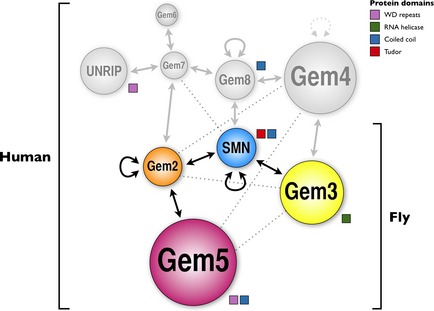
Survival motor neuron (SMN)‐Gemins complex members: united in diversity. Interactions between SMN‐Gemins complex members verified in one and more than one experimental system are depicted in solid and dashed arrows, respectively. Circular arrows indicate self‐association. Protein domains in the Simple Modular Architecture Research Tool (SMART) database are also indicated for each member. Only the coiled coil domain is shared by more than one complex member. The fly version of the SMN‐Gemins complex is minimalistic and composed of only SMN, Gemin2, Gemin3 and Gemin5. Addition of other components with evolution increased the complexity of the SMN‐Gemins complex to the present nine‐membered version present in vertebrates including humans (adapted from 12).
The protein–protein interactions within the vertebrate SMN‐Gemins complex are quite intricate, however, a consensus interaction map was recently drafted that establishes a troika formed of SMN, Gemin2 and Gemin8 at the backbone of the human SMN‐Gemins complex that is capable of recruiting the remaining members in blocks. In this respect, SMN associates to Gemin2, Gemin3, and Gemin8. Gemin2 and Gemin3, in turn, interact with Gemin5 and Gemin4, respectively, and Gemin8 interacts with Gemins 4 and 7 11. UNRIP and Gemin6 are recruited by Gemin7, which is also capable of associating with Gemin2 11, 44. Interestingly, in addition to SMN, only Gemin2 and Gemin8 were reported to self‐associate 9, 10, 11, 45 with the Gemin2–Gemin2 interaction stabilizing SMN oligomers 45. Furthermore, Gemin5 and UNRIP are viewed as partial rather than full members in view of their weak and cytoplasmic‐restricted association with the human SMN‐Gemins complex 11, 37, 38, 46, 47.
Leading Roles for the Gemins in snRNP Assembly
snRNPs consist of one or two short noncoding RNA molecules (small nuclear RNAs or snRNAs) bound to a set of seven Smith (Sm) or Sm‐like (Lsm) proteins, and a unique set of snRNP‐specific proteins 48. In partnership with numerous non‐snRNP splicing factors, U1, U2, U4/U6 and U5 snRNPs form the major spliceosome, which is responsible for splicing the majority of pre‐mRNA introns, whereas U11, U12, U4atac/U6atac and U5 snRNPs constitute the less abundant minor spliceosome, that is specialized in processing a rare group of introns 49. In contrast to Lsm‐class U6 and U6atac snRNPs, which are synthesized entirely in the nucleus 50, for reasons as yet unclear, the core structure of the remaining Sm‐class snRNPs is assembled in the cytoplasm. Several studies of late have only scratched the surface of the intricacies, intrigues and vicissitudes defining this key segmented phase in vivo, which is ATP‐dependent, requires the participation of the entire SMN‐Gemins complex 11, 13, 14, 15, 16, 44, 45, and essentially involves the uploading of a heptameric Sm D1/D2/E/F/G/D3/B ring‐shaped core domain onto the “Sm site”, a conserved uridine‐rich sequence motif intrinsic to snRNAs.
snRNAs that are expelled from the nucleus following transcription are thought to be swiftly identified by SMN‐Gemins complex‐independent Gemin5 protein through the stringent recognition of a code formed of sequences and structural motifs 15. Gemin5 charged with snRNAs via its N‐terminal WD‐repeat domain 51, then docks into the SMN‐Gemins complex, most probably proximate to Gemin2, to deliver its cargo for Sm core assembly 52. The majority of Sm proteins are recognized by Gemin2, which wraps itself around a crescent‐shaped pentamer formed of Sm D1/D2/E/F/G to contact each of the five Sm proteins. Importantly, the N‐terminal tail of Gemin2 reaches into the snRNA‐binding pocket on the Sm pentamer to block their inclination for promiscuous RNA binding, presumably until the delivery of the bona fide RNA substrates, snRNAs 53 (Figure 2). Gemins 6 and 7 form a heterodimer that resembles Sm core protein dimers even though they both lack significant sequence similarity with Sm proteins. In this context, the Gemin6/Gemin7 dimer is thought to serve as a surrogate for the SmB‐SmD3 dimer around which the Sm pentamer is arranged, eventually facilitating the pentamer's association with the Gemin2 arm of the SMN‐Gemins complex 54. Unrip may be important for the exchange of the Gemin6/Gemin7 heterodimer by the Sm D3/B particle, which should ensure Sm ring closure within the SMN‐Gemins complex in preparation for uploading onto snRNAs 44. The chaperoning of RNA and, eventually, RNP molecules as well as ATP breakdown during the assembly reaction, are probably entrenched jobs of the DEAD‐box RNA helicase Gemin3 30, 36, 55 assisted by its probable cofactor, Gemin4 31.
Figure 2.
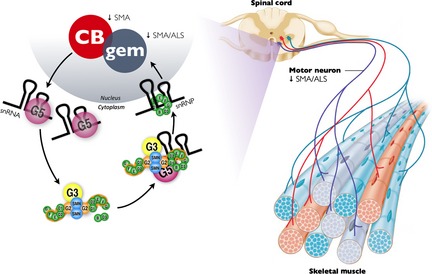
Cajal bodies, gems, spliceosome building and motor neuron degeneration. Newly transcribed snRNAs are exported to the cytoplasm where they are captured by Gemin5, and transferred to the survival motor neuron (SMN)‐Gemins complex for assembly of a ring‐shaped heptameric Sm protein core assembly. For simplicity, the SMN‐Gemins complex depicted here is the Drosophila version. Gemin2 is the factor that recognizes and captures the majority of Sm proteins by wrapping itself around an Sm D1/D2/E/F/G pentamer. snRNP assembly is energetically demanding, and ATP hydrolysis is probably a function of DEAD‐box RNA helicase Gemin3, which might also act as an RNA or RNP chaperone during the assembly reaction. Following Sm ring closure, snRNPs are imported into the nucleus, and prior to functioning in pre‐mRNA splicing, they undergo maturation in the Cajal body (CB). The function of gems is as yet unclear but they associate with CBs and their loss is a signature marker of both spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS) pathology.
It is unexpected that SMN does not feature in the crucial steps of snRNP assembly. Does this imply that it is not so important as previously thought for this essential housekeeping activity? Or, in view of substantial evidence (reviewed in 56, 57), does SMN burden itself with a less‐defined non‐canonical function, likely linked to axonal mRNP trafficking? It is the opinion of many that SMN's function in snRNP assembly is the most decisive of all SMN‐Gemins complex members, as its oligomers probably form a sturdy scaffold on which the Gemin builders operate in several concurrent assembly reactions following “due diligence” of the assembly blocks. This activity might be supported by other SMN‐Gemins complex components especially those such as Gemin8, which are known to have a propensity to oligomerize 11. Although extensive in vitro studies reported on the possibility that the protein arginine methyltransferase 5 (PRMT5) complex sequesters and modifies select Sm proteins post‐translationally, an action which is believed to enhance their interactions with the SMN‐Gemins complex (reviewed in 58), in vivo experiments concluded that this action is probably not indispensable for snRNP assembly 59. Proper Sm core assembly is not only required for the in vivo stability and function of snRNPs but it is also a prerequisite for subsequent steps in the biogenesis of snRNPs including cap hypermethylation, 3′ terminal trimming, and eventual import into the nucleus. In the latter compartment, snRNPs undergo maturation in CBs prior to their participation in pre‐mRNA splicing including nucleotide modification guided by small CB‐specific RNPs (scaRNPs), incorporation of several snRNP‐specific proteins as well as coupling of individual snRNPs into di‐ and tri‐snRNPs 12, 60, 61. Following several rounds of action, snRNPs are known to repeatedly return to CBs where they are regenerated or recycled 62.
The Link Between Spliceosome Building and Motor Function
Confirming their participation in a crucial housekeeping function, early lethality is the phenotypic endpoint of knockouts or strong global knockdowns of SMN‐Gemins complex members analyzed thus far in a variety of model organisms 42, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72. However, SMA is the result of insufficient levels but not complete loss of the SMN protein, and replicating this situation in worm, fly, zebrafish and mouse gives rise to phenotypes that closely resemble SMA's signature pathology 5, 41, 63, 65, 73, 74, 75, 76, 77. Considering their intimate liaison with SMN as well as their emerging key roles in spliceosome building, why are the Gemin genes not linked to SMA or similar motor conditions? The answer probably lies in the unique genetics of SMN in humans, hence, the absence of an SMN2‐like pseudogene means that deleterious mutations in any of the Gemin genes is incompatible with life. Gemins (and Unrip) are also probably not redundant or interchangeable, therefore, they are responsible for exclusive activities during snRNP biogenesis. In this context, building on elegant biochemical and structural studies that support this direction 15, 52, 53, my laboratory demonstrated that organisms that have reduced levels of Gemins selectively in the motor unit develop motor deficits that are similar to those observed on attenuation of SMN 42, 64. In other words, if one considers the in vivo state, inadequate levels of any one member is sufficient to arrest the SMN‐Gemins complex in a function that is critical for normal motor behaviour.
How can a decreased capacity of the SMN‐Gemins complex account for the selective neuromuscular phenotype in SMA? Though still controversial, substantial evidence have been garnered to support one school of thought asserting that reduced snRNP biosynthesis and, consequently, defective splicing is selectively disruptive to the motor unit. To this end, early studies have demonstrated a significant phenotypic rescuing effect on restoration of normal snRNP levels in zebrafish and mouse SMA models 78, 79. More recently, in corroboration, studies on symptomatic severe SMA animal models uncovered an negative impact on splicing and expression levels of minor‐intron‐containing genes that are essential for motor‐sensory circuit function 80, 81, due to a previously described preferential reduction in the snRNPs that constitute the minor spliceosome 17, 18, 82. The link between snRNP assembly and selective neuromuscular degeneration is, however, refuted by studies demonstrating that the role of SMN in snRNP assembly can be uncoupled from motor or axonal defects 83, 84 and, recent reports have even presented evidence against a minor‐intron‐dependent aetiology for SMA 85, 86. Nevertheless, in a technically challenging study, which in contrast to others 18, 87, involved the sequencing of RNA from microdissected motor neurons of presymptomatic SMA mice, the Dreyfuss’ laboratory has yet again led the way by successfully identifying specific transcriptome abnormalities that link SMN deficiency to the distinct motor neuron pathology in SMA 86. Overall, the evidence is substantial to tip the balance in favour of a vital role for SMN and, likely the SMN‐Gemins complex, in preventing transcriptome irregularities.
MND Crossover
Gems and Cajal Bodies
What is the function of Gems? We are still in the dark when it comes to answering this question although great progress has been made on the Cajal body (CB), gems’ twin nuclear structure. More than a century ago, making use of the silver staining technique, Spanish neuroscientist Santiago Ramón y Cajal described a nuclear structure that is generally smaller than nucleoli, but with which it is sometimes associated (hence, the original name cuerpo accesorio or accessory body), in the neurons, including motor neurons, of many different vertebrates 88. Although hosting an expanding list of proteins and RNAs or RNPs, the CB is usually identified by the presence of scaRNAs and its signature protein coilin (reviewed in 60, 61). CBs are the sites of several crucial steps in the maturation and regeneration of the RNA‐processing machinery so it was quite surprising that loss of CBs through genetic depletion of coilin is not associated with lethality in Drosophila 89. Furthermore, in this genetic background and in coilin‐ or SMN‐deficient cells, scaRNA levels are normal and snRNAs are properly modified 90, 91, 92. The story does not repeat itself in vertebrates, in that, besides the obvious CB dispersal phenotype 93, coilin knockout mice show reduced viability, fertility and fecundity 94, and in zebrafish, coilin depletion during embryogenesis leads to deficits in snRNP biogenesis, disrupted pre‐mRNA splicing and consequently, reduced cell proliferation followed by developmental arrest 95. These findings suggest that by concentrating several components in discrete nuclear compartments, CBs act as catalysts that accelerate reactions including those linked to snRNP biogenesis and this is especially important in highly metabolically active cells 61, 96.
Gems are snRNP‐free concentrates of SMN‐Gemins complexes minus Unrip that in their majority occur as distinct nuclear structures in foetal tissues but increasingly colocalize with CBs in later developmental stages and adulthood 97, 98. Coilin, which binds to SMN, has been reported to mediate the recruitment of SMN‐Gemins complexes, and, hence, gems, to CBs 99. It is highly likely that gems are storage depots for supplementary SMN‐Gemins complexes that are recruited to CBs most probably for the regeneration of worn snRNPs and, in agreement, the upregulation of either SMN or its Gemin associates is known to induce gem formation or promote an increase in gem numbers 20, 26, 42, 47, 100. In addition, whereas canonical CBs and gems are lost on loss of SMN or Gemins, knockdown of snRNA export or cap modification factors only disrupted CBs 13, 101, 102, 103, 104, hence, dissociating gem stability from that of CBs as well as indicating that CB integrity is dependent on ongoing snRNP biogenesis (Figure 2). Truth must be told. Gems are not the only membrane‐free cellular structures in which the SMN‐Gemins complex concentrates, hence, in the cytoplasmic compartment, it accumulates with snRNPs and nuclear import factors in U bodies 40, 105, 106, which probably are the factories where the actual assembly reactions take place. In cultured neurons, the SMN‐Gemins complex also localizes to large stationary and small actively transported snRNP‐independent granules in neuronal processes, possibly underscoring a tissue‐specific role in mRNP trafficking 107, 108, 109, 110. Similar to gems, crosstalk with compartment‐specific organelles is also observed for the cytoplasmic SMN‐Gemins complex‐rich bodies 40, 103, 104, 105, and both nuclear and cytoplasmic bodies respond to metabolic changes in the cell 19, 111.
Underpinnings of Gem Depletion in ALS
In both ALS and SMA, lower motor neurons in the anterior horn of the spinal cord are exceedingly vulnerable to injury, raising the question of whether both disorders share a common pathogenesis. ALS, which also affects upper motor neurons in the cerebral cortex, is mostly sporadic and, in this respect, only a minority of cases (5–10%) are inherited. Until recently, the field was fixated on the pathogenic processes resulting from dominant mutations in copper/zinc superoxide dismutase 1 (SOD1) gene, which account for ~20% of the familial ALS forms. These mutations appear to confer a toxic gain of function to the protein rather than loss of the metalloenzymatic activity that converts the highly toxic superoxide anion to molecular oxygen and hydrogen peroxide. A game changer was the identification of mutations in an additional diverse number of genes, including a subset that is intricately linked to RNA processing. Amongst these, RNA‐binding proteins TAR DNA‐binding protein 43 (TDP‐43), and fused in sarcoma (FUS) are both nuclear proteins and shuttle between the nuclear and the cytoplasmic compartment as part of their cellular activities. Furthermore, under pathogenic conditions, both proteins are missing from their normal nuclear location and found bundled in cytoplasmic inclusions found in both neuronal and glial cells. This behaviour suggests that loss of normal nuclear function or, alternatively, gain of toxic function by aggregates play significant roles in ALS pathogenesis 112, 113, 114, 115.
In the midst of the experimental frenzy surrounding the characterization of newly identified ALS‐linked genes, Turner and co‐workers were the first to audaciously probe for evidence favouring an involvement of SMN in the pathogenesis of ALS. To this end, although they found that TDP‐43 distribution, expression or biochemistry was unaltered in spinal cords of affected SMA mice 116, transgenic mutant SOD1 mice were characterized by low SMN (and Gemin2) protein levels, but not mRNA, and a further reduction was found to accelerate the phenotypic severity associated with this model 117. Importantly, replicating the murine situation, SMN protein levels are also significantly reduced (~50%) in the spinal cords of sporadic ALS patients 26, in agreement with earlier evidence demonstrating an association between lower SMN2 copy numbers and increased severity of sporadic ALS 118. Loss of total SMN protein, reflected in the depletion of motor neuron gems initiates before disease onset and is progressive, linked to the evolution of the disease 27, 28. Interestingly, despite the loss of gems, CB numbers are preserved in motor neurons of mutant SOD1 mice 27, 117. This phenotype indicates that mutant SOD1 interferes with the shuttling of the SMN‐Gemins complex to CBs in motor neurons, at least in part, by disrupting the interaction between SMN and coilin. In this regard, SMN and its associate, Gemin2, were found depleted from the nucleus and enriched within the cytoplasm, a behaviour that is also followed by both endogenous and mutant SOD1 26, 28, 117.
This mechanism underpinning gem depletion is cell‐autonomous as mutant SOD1‐expressing astrocytes, which are known to influence the neurodegenerative process (reviewed in 113), do not contribute to loss of gems in wild‐type motor neurons 27. Recently, in the reverse experiment, both the Turner and Monani labs have showed that neuronal overexpression of SMN, and, hence, elevated gem numbers, significantly improved neuromuscular function as well as motor neuron survival in mutant SOD1 mice 26, 27. Furthermore, upregulated SMN protein levels in spinal motor neurons conferred greater resistance to the degenerative effects of mutant SOD1‐expressing astrocytes 27, reaffirming SMN's neuroprotective facet. It is noteworthy that overexpression of wild‐type SOD1 protein does not alter SMN levels or ameliorate the neuromuscular and survival phenotype in a severe SMA model, an observation suggesting that SMN functions downstream of SOD1, and in this respect, phenotype modulation can only be one‐way 117.
The plot got really exciting when the total number of gems was found to be altered in spinal motor neurons of mice by knockout of endogenous TDP‐43 or overexpression of human TDP‐43 22. Strikingly still, gem numbers are dramatically reduced in cells that are either transfected with mutant TDP‐43 or FUS, are derived from patients with ALS or have disrupted levels of either protein 23, 24, 25. Despite normal levels of SMN and Gemins on FUS (but not TDP‐43 25) knockdown, gems were efficiently restored on upregulation of SMN. This result indicates that the requirement for FUS in gem formation is not due to an effect on protein levels of SMN‐Gemins complex members, and similar to SOD1, SMN seems to act downstream of FUS 23. TDP‐43 is known to associate directly with FUS 119, and the latter acts downstream of TDP‐43 in a pathway essential for normal survival and motor function 120, 121. Both TDP‐43 and FUS were also reported to localize to gems 24. Additionally, biochemical experiments reveal that both TDP‐43 and FUS interact with the SMN‐Gemins complex 23, 24, 122, 123. In case of FUS, the association with SMN is mediated through not only a direct binding between the two but also U1 snRNP, a finding that might hint at a form of collaboration on spliceosome building 23.
Downstream Consequences: snRNP Disruptions and Axonal Defects
Considering that the locale and/or levels of the SMN‐Gemins complex are disturbed in ALS, one begs the question of how this scenario influences downstream pathways including snRNP biogenesis. In this regard, studies have provided conflicting evidence. Tsuiji et al. 24 report that snRNA levels are up‐regulated in cells depleted of TDP‐43. This phenotype is also present in spinal cords of sporadic ALS patients, where snRNPs were shown to accumulate extensively in motor neuron nuclei. However, findings by Ishihara et al. 25 are indicative of an alteration in the snRNA repertoire of TDP‐43‐depleted cells or symptomatic ALS tissues, a phenotype that is reminiscent of that reported in symptomatic SMA mice 17, 18. Furthermore, in a recent study, cytoplasmic mis‐localization of FUS, in cells expressing mutant FUS, was found to cause partial accumulation of snRNPs to the cytoplasm 122. Interestingly, crossover does not seem to be limited to processes underpinning spliceosome building but also extends to noncanonical functions associated with SMN, especially those confined to neuronal processes, and in this regard, it is well known that paucity of SMN gives rise to axonal defects (reviewed in 56). In this regard, FUS mutants were found to disturb the axonal localization of SMN. Importantly, overexpression of SMN rescued the axonal defects induced by mutant FUS, raising the question of whether such defects arise as a result of SMN sequestration by mutant FUS 123. Further research on this theme is warranted and can potentially give a clearer picture.
Conclusion
It is presently conceivable to deduce that RNA metabolism is crucial to motor neuron physiology and survival. Clues have been exposed in a melange of studies including those that report the identification of MND‐linked mutations in genes encoding proteins with crucial roles in RNA processing and the in‐depth investigations that they triggered 124, 125, 126. Combine these with studies that link splicing factors or snRNPs to neurodegeneration (127, 128). The reason for the selective vulnerability of motor neurons is still opaque though it is worth noting that accumulating evidence is suggesting that MND is more far‐reaching and, hence, it may not be a cell‐autonomous disease of motor neurons (reviewed in 129, 130). Gems are quite prominent in motor neurons 2, 21, 97, 98, and though they are probably not essential for cell survival given their absence in certain cell types 97, is it plausible that they work in conjunction with CBs to expedite the final steps of the snRNP production line and/or the regeneration of snRNPs? Investigations that address this open question can potentially impact our understanding of the molecular nature underpinning motor neuron degeneration.
Aiming at identifying compounds that augment cellular SMN, gem counts have already been successfully exploited either as an integral component of image‐based screens 131 or as a confirmatory assay 100, 132. The unexpected finding that gem depletion is a signature feature of ALS pathology, in addition to SMA (Figure 2), consolidates the use of this cellular phenotype to screen for novel pharmacologic modifiers that are universally applicable in MND. Importantly, ALS patients might also benefit from the various SMA‐directed therapeutics in the pipeline. Principally relevant are those that boost SMN protein levels including viral and non‐viral mediated gene delivery, oligonucleotide‐mediated SMN2‐directed splicing modification as well as small molecules that spur SMN expression (reviewed in 133). The coming together of two major motor neuron degenerative disorders heralds a bright outlook for treatment strategies targeting their eradication.
Conflict of Interest
The author declares no conflict of interest.
Acknowledgments
The author is thankful to members of his laboratory and colleagues for stimulating discussions. Research on MND in the author's laboratory is supported by the University of Malta Research Fund and the Malta Council for Science & Technology through the National Research & Innovation Programme 2012 (R&I‐2012‐066). The authors thank EU COST Action CM1103 “Structure‐based drug design for diagnosis and treatment of neurological diseases: dissecting and modulating complex function in the monoaminergic systems of the brain.”
References
- 1. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 2. Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997;16:265–269. [DOI] [PubMed] [Google Scholar]
- 3. Wan L, Battle DJ, Yong J, et al. The survival of motor neurons protein determines the capacity for snRNP assembly: Biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 2005;25:5543–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kolb SJ, Kissel JT. Spinal muscular atrophy: A timely review. Arch Neurol 2011;68:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burghes AH, Beattie CE. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 2009;10:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wirth B, Brichta L, Schrank B, et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 2006;119:422–428. [DOI] [PubMed] [Google Scholar]
- 7. Prior TW, Swoboda KJ, Scott HD, et al. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A 2004;130:307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA 1999;96:11167–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lorson CL, Strasswimmer J, Yao JM, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 1998;19:63–66. [DOI] [PubMed] [Google Scholar]
- 10. Young PJ, Man NT, Lorson CL, et al. The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self‐association and SIP1 binding. Hum Mol Genet 2000;9:2869–2877. [DOI] [PubMed] [Google Scholar]
- 11. Otter S, Grimmler M, Neuenkirchen N, et al. A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J Biol Chem 2007;282:5825–5833. [DOI] [PubMed] [Google Scholar]
- 12. Cauchi RJ. SMN and Gemins: ‘we are family’.. or are we? Insights into the partnership between Gemins and the spinal muscular atrophy disease protein SMN. BioEssays 2010;32:1077–1089. [DOI] [PubMed] [Google Scholar]
- 13. Shpargel KB, Matera G. Gemin proteins are required for efficient assembly of Sm‐class ribonucleoproteins. Proc Natl Acad USA 2005;102:17372–17377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng W, Gubitz AK, Wan L, et al. Gemins modulate the expression and activity of the SMN complex. Hum Mol Genet 2005;14:1605–1611. [DOI] [PubMed] [Google Scholar]
- 15. Battle DJ, Lau CK, Wan L, et al. The Gemin5 protein of the SMN complex identifies snRNAs. Mol Cell 2006;23:273–279. [DOI] [PubMed] [Google Scholar]
- 16. Carissimi C, Saieva L, Baccon J, et al. Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J Biol Chem 2006;281:8126–8134. [DOI] [PubMed] [Google Scholar]
- 17. Gabanella F, Butchbach ME, Saieva L, et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007;2:e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue‐specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008;133:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 20. Cauchi RJ. Gem formation upon constitutive Gemin3 overexpression in Drosophila. Cell Biol Int 2011;35:1233–1238. [DOI] [PubMed] [Google Scholar]
- 21. Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997;6:1205–1214. [DOI] [PubMed] [Google Scholar]
- 22. Shan X, Chiang PM, Price DL, et al. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP‐43 transgenic mice. Proc Natl Acad Sci USA 2010;107:16325–16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamazaki T, Chen S, Yu Y, et al. FUS‐SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2012;2:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsuiji H, Iguchi Y, Furuya A, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 2013;5:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishihara T, Ariizumi Y, Shiga A, et al. Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum Mol Genet 2013;22:4136–4147. [DOI] [PubMed] [Google Scholar]
- 26. Turner BJ, Alfazema N, Sheean RK, et al. Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol Aging 2013;35:906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kariya S, Re DB, Jacquier A, et al. Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum Mol Genet 2012;21:3421–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gertz B, Wong M, Martin LJ. Nuclear localization of human SOD1 and mutant SOD1‐specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice. J Neuropathol Exp Neurol 2012;71:162–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Q, Fischer U, Wang F, et al. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997;90:1013–1021. [DOI] [PubMed] [Google Scholar]
- 30. Charroux B, Pellizzoni L, Perkinson RA, et al. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of Gems. J Cell Biol 1999;147:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charroux B, Pellizzoni L, Perkinson RA, et al. Gemin4: A novel component of the SMN complex that is found in both gems and nucleoli. J Cell Biol 2000;148:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gubitz AK, Mourelatos Z, Abel L, et al. Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J Biol Chem 2002;277:5631–5636. [DOI] [PubMed] [Google Scholar]
- 33. Pellizzoni L, Baccon J, Rappsilber J, et al. Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J Biol Chem 2002;277:7540–7545. [DOI] [PubMed] [Google Scholar]
- 34. Baccon J, Pellizzoni L, Rappsilber J, et al. Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J Biol Chem 2002;277:31957–31962. [DOI] [PubMed] [Google Scholar]
- 35. Carissimi C, Saieva L, Gabanella F, et al. Gemin8 is required for the architecture and function of the survival motor neuron complex. J Biol Chem 2006;281:37009–37016. [DOI] [PubMed] [Google Scholar]
- 36. Campbell L, Unter KMD, Mohaghegh P, et al. Direct interaction of Smn with dp103, a putative RNA helicase: Role for Smn in transcription regulation? Hum Mol Genet 2000;9:1093–1100. [DOI] [PubMed] [Google Scholar]
- 37. Carissimi C, Baccon J, Straccia M, et al. Unrip is a component of SMN complexes active in snRNP assembly. FEBS Lett 2005;579:2348–2354. [DOI] [PubMed] [Google Scholar]
- 38. Grimmler M, Otter S, Peter C, et al. Unrip, a factor implicated in cap‐independent translation, associates with the cytosolic SMN complex and influences its intracellular localization. Hum Mol Genet 2005;14:3099–3111. [DOI] [PubMed] [Google Scholar]
- 39. Kroiss M, Schultz J, Wiesner J, et al. Evolution of an RNP assembly system: A minimal SMN complex facilitates formation of UsnRNPs in Drosophila melanogaster . Proc Natl Acad Sci USA 2008;105:10045–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cauchi RJ, Sanchez‐Pulido L, Liu JL. Drosophila SMN complex proteins Gemin2, Gemin3, and Gemin5 are components of U bodies. Exp Cell Res 2010;316:2354–2364. [DOI] [PubMed] [Google Scholar]
- 41. Grice SJ, Sleigh JN, Liu JL, et al. Invertebrate models of spinal muscular atrophy: Insights into mechanisms and potential therapeutics. BioEssays 2011;33:956–965. [DOI] [PubMed] [Google Scholar]
- 42. Borg R, Cauchi RJ. The Gemin Associates of Survival Motor Neuron are Required for Motor Function in Drosophila. PLoS ONE 2013;8:e83878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cauchi RJ, van den Heuvel M. The fly as a model for neurodegenerative diseases: Is it worth the jump? Neurodegener Dis 2006;3:338–356. [DOI] [PubMed] [Google Scholar]
- 44. Ogawa C, Usui K, Ito F, et al. Role of survival motor neuron complex components in small nuclear ribonucleoprotein assembly. J Biol Chem 2009;284:14609–14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ogawa C, Usui K, Aoki M, et al. Gemin2 plays an important role in stabilizing the survival of motor neuron complex. J Biol Chem 2007;282:11122–11134. [DOI] [PubMed] [Google Scholar]
- 46. Battle DJ, Kasim M, Wang J, et al. SMN‐independent subunits of the SMN complex. Identification of a small nuclear ribonucleoprotein assembly intermediate. J Biol Chem 2007;282:27953–27959. [DOI] [PubMed] [Google Scholar]
- 47. le Hao T, Fuller HR, le Lam T, et al. Absence of gemin5 from SMN complexes in nuclear Cajal bodies. BMC Cell Biol 2007;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matera AG, Terns RM, Terns MP. Non‐coding RNAs: Lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol 2007;8:209–220. [DOI] [PubMed] [Google Scholar]
- 49. Patel AA, Steitz JA. Splicing double: Insights from the second spliceosome. Nat Rev Mol Cell Biol 2003;4:960–970. [DOI] [PubMed] [Google Scholar]
- 50. Mroczek S, Dziembowski A. U6 RNA biogenesis and disease association. Wiley Interdiscip Rev RNA 2013;4:581–592. [DOI] [PubMed] [Google Scholar]
- 51. Lau CK, Bachorik JL, Dreyfuss G. Gemin5‐snRNA interaction reveals an RNA binding function for WD repeat domains. Nat Struct Mol Biol 2009;16:486–491. [DOI] [PubMed] [Google Scholar]
- 52. Yong J, Kasim M, Bachorik JL, et al. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell 2010;38:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang R, So BR, Li P, et al. Structure of a key intermediate of the SMN complex reveals Gemin2's crucial function in snRNP assembly. Cell 2011;146:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma Y, Dostie J, Dreyfuss G, et al. The Gemin6‐Gemin7 heterodimer from the survival of motor neurons complex has an Sm protein‐like structure. Structure 2005;13:883–892. [DOI] [PubMed] [Google Scholar]
- 55. Yan X, Mouillet JF, Ou Q, et al. A novel domain within the DEAD‐box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol 2003;23:414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: The role of SMN in axonal mRNA regulation. Brain Res 2012;1462:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Briese M, Esmaeili B, Sattelle DB. Is spinal muscular atrophy the result of defects in motor neuron processes? BioEssays 2005;27:946–957. [DOI] [PubMed] [Google Scholar]
- 58. Fischer U, Englbrecht C, Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA 2011;2:718–731. [DOI] [PubMed] [Google Scholar]
- 59. Gonsalvez GB, Praveen K, Hicks AJ, et al. Sm protein methylation is dispensable for snRNP assembly in Drosophila melanogaster . RNA 2008;14:878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nizami Z, Deryusheva S, Gall JG. The Cajal Body and Histone Locus Body. Cold Spring Harb Perspect Biol 2010;2:a000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Machyna M, Heyn P, Neugebauer KM. Cajal bodies: Where form meets function. Wiley Interdiscip Rev RNA 2013;4:17–34. [DOI] [PubMed] [Google Scholar]
- 62. Stanek D, Pridalova‐Hnilicova J, Novotny I, et al. Spliceosomal small nuclear ribonucleoprotein particles repeatedly cycle through Cajal bodies. Mol Biol Cell 2008;19:2534–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Briese M, Esmaeili B, Fraboulet S, et al. Deletion of smn‐1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet 2009;18:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cauchi RJ, Davies KE, Liu JL. A motor function for the DEAD‐box RNA helicase, Gemin3, in Drosophila. PLoS Genet 2008;4:e1000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chan YB, Miguel‐Aliaga I, Franks C, et al. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 2003;12:1367–1376. [DOI] [PubMed] [Google Scholar]
- 66. Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 1997;94:9920–9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mouillet JF, Yan X, Ou Q, et al. DEAD‐box protein‐103 (DP103, Ddx20) is essential for early embryonic development and modulates ovarian morphology and function. Endocrinology 2008;149:2168–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jablonka S, Holtmann B, Meister G, et al. Gene targeting of Gemin2 in mice reveals a correlation between defects in the biogenesis of U snRNPs and motoneuron cell death. Proc Natl Acad Sci USA 2002;99:10126–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gates J, Lam G, Ortiz JA, et al. rigor mortis encodes a novel nuclear receptor interacting protein required for ecdysone signaling during Drosophila larval development. Development 2004;131:25–36. [DOI] [PubMed] [Google Scholar]
- 70. Hannus S, Buhler D, Romano M, et al. The Schizosaccharomyces pombe protein Yab8p and a novel factor, Yip1p, share structural and functional similarity with the spinal muscular atrophy‐associated proteins SMN and SIP1. Hum Mol Genet 2000;9:663–674. [DOI] [PubMed] [Google Scholar]
- 71. Owen N, Doe CL, Mellor J, et al. Characterization of the Schizosaccharomyces pombe orthologue of the human survival motor neuron (SMN) protein. Hum Mol Genet 2000;9:675–684. [DOI] [PubMed] [Google Scholar]
- 72. Shpargel KB, Praveen K, Rajendra TK, et al. Gemin3 is an essential gene required for larval motor function and pupation in Drosophila. Mol Biol Cell 2009;20:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rajendra TK, Gonsalvez GB, Walker MP, et al. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol 2007;176:831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chang HC, Dimlich DN, Yokokura T, et al. Modeling spinal muscular atrophy in Drosophila. PLoS ONE 2008;3:e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sleigh JN, Buckingham SD, Esmaeili B, et al. A novel Caenorhabditis elegans allele, smn‐1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre‐approved compounds. Hum Mol Genet 2011;20:245–260. [DOI] [PubMed] [Google Scholar]
- 76. Sleigh JN, Gillingwater TH, Talbot K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis Model Mech 2011;4:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Beattie CE, Carrel TL, McWhorter ML. Fishing for a mechanism: Using zebrafish to understand spinal muscular atrophy. J Child Neurol 2007;22:995–1003. [DOI] [PubMed] [Google Scholar]
- 78. Winkler C, Eggert C, Gradl D, et al. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 2005;19:2320–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Workman E, Saieva L, Carrel TL, et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet 2009;18:2215–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lotti F, Imlach WL, Saieva L, et al. An SMN‐dependent U12 splicing event essential for motor circuit function. Cell 2012;151:440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Imlach WL, Beck ES, Choi BJ, et al. SMN is required for sensory‐motor circuit function in Drosophila. Cell 2012;151:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Boulisfane N, Choleza M, Rage F, et al. Impaired minor tri‐snRNP assembly generates differential splicing defects of U12‐type introns in lymphoblasts derived from a type I SMA patient. Hum Mol Genet 2011;20:641–648. [DOI] [PubMed] [Google Scholar]
- 83. Carrel TL, McWhorter ML, Workman E, et al. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 2006;26:11014–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Praveen K, Wen Y, Matera AG. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep 2012;1:624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Garcia EL, Lu Z, Meers MP, et al. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron‐containing genes. RNA 2013;19:1510–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang Z, Pinto AM, Wan L, et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci USA 2013;110:19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Baumer D, Lee S, Nicholson G, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 2009;5:e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gall JG. The centennial of the Cajal body. Nat Rev Mol Cell Biol 2003;4:975–980. [DOI] [PubMed] [Google Scholar]
- 89. Liu JL, Wu Z, Nizami Z, et al. Coilin is essential for Cajal body organization in Drosophila melanogaster . Mol Biol Cell 2009;20:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Deryusheva S, Gall JG. Small Cajal body‐specific RNAs of Drosophila function in the absence of Cajal bodies. Mol Biol Cell 2009;20:5250–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Deryusheva S, Choleza M, Barbarossa A, et al. Post‐transcriptional modification of spliceosomal RNAs is normal in SMN‐deficient cells. RNA 2012;18:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jady BE, Darzacq X, Tucker KE, et al. Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J 2003;22:1878–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tucker KE, Berciano MT, Jacobs EY, et al. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol 2001;154:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Walker MP, Tian L, Matera AG. Reduced viability, fertility and fecundity in mice lacking the cajal body marker protein, coilin. PLoS ONE 2009;4:e6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Strzelecka M, Trowitzsch S, Weber G, et al. Coilin‐dependent snRNP assembly is essential for zebrafish embryogenesis. Nat Struct Mol Biol 2010;17:403–409. [DOI] [PubMed] [Google Scholar]
- 96. Matera AG, Izaguire‐Sierra M, Praveen K, et al. Nuclear bodies: Random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev Cell 2009;17:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Young PJ, Le TT, thi Man N, et al. The relationship between SMN, the spinal muscular atrophy protein, and nuclear coiled bodies in differentiated tissues and cultured cells. Exp Cell Res 2000;256:365–374. [DOI] [PubMed] [Google Scholar]
- 98. Young PJ, Le TT, Dunckley M, et al. Nuclear gems and Cajal (coiled) bodies in fetal tissues: Nucleolar distribution of the spinal muscular atrophy protein, SMN. Exp Cell Res 2001;265:252–261. [DOI] [PubMed] [Google Scholar]
- 99. Hebert MD, Szymczyk PW, Shpargel KB, et al. Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev 2001;15:2720–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jarecki J, Chen X, Bernardino A, et al. Diverse small‐molecule modulators of SMN expression found by high‐throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet 2005;14:2003–2018. [DOI] [PubMed] [Google Scholar]
- 101. Lemm I, Girard C, Kuhn AN, et al. Ongoing U snRNP biogenesis is required for the integrity of Cajal bodies. Mol Biol Cell 2006;17:3221–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Girard C, Neel H, Bertrand E, et al. Depletion of SMN by RNA interference in HeLa cells induces defects in Cajal body formation. Nucleic Acids Res 2006;34:2925–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee L, Davies SE, Liu JL. The spinal muscular atrophy protein SMN affects Drosophila germline nuclear organization through the U body‐P body pathway. Dev Biol 2009;332:142–155. [DOI] [PubMed] [Google Scholar]
- 104. Cauchi RJ. Conserved requirement for DEAD‐box RNA helicase Gemin3 in Drosophila oogenesis. BMC Res Notes 2012;5:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu JL, Gall JG. U bodies are cytoplasmic structures that contain uridine‐rich small nuclear ribonucleoproteins and associate with P bodies. Proc Natl Acad Sci USA 2007;104:11655–11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Natalizio AH, Matera AG. Identification and characterization of Drosophila Snurportin reveals a role for the import receptor Moleskin/importin‐7 in snRNP biogenesis. Mol Biol Cell 2013;24:2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang H, Xing L, Rossoll W, et al. Multiprotein complexes of the survival of motor neuron protein SMN with gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci 2006;26:8622–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Todd AG, Morse R, Shaw DJ, et al. Analysis of SMN‐neurite granules: Core Cajal body components are absent from SMN‐cytoplasmic complexes. Biochem Biophys Res Commun 2010;397:479–485. [DOI] [PubMed] [Google Scholar]
- 109. Todd AG, Morse R, Shaw DJ, et al. SMN, Gemin2 and Gemin3 associate with beta‐actin mRNA in the cytoplasm of neuronal cells in vitro. J Mol Biol 2010;401:681–689. [DOI] [PubMed] [Google Scholar]
- 110. Todd AG, Shaw DJ, Morse R, et al. SMN and the Gemin proteins form sub‐complexes that localise to both stationary and dynamic neurite granules. Biochem Biophys Res Commun 2010;394:211–216. [DOI] [PubMed] [Google Scholar]
- 111. Buckingham M, Liu JL. U bodies respond to nutrient stress in Drosophila. Exp Cell Res 2011;317:2835–2844. [DOI] [PubMed] [Google Scholar]
- 112. Andersen PM, Al‐Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat Rev Neurol 2011;7:603–615. [DOI] [PubMed] [Google Scholar]
- 113. Ferraiuolo L, Kirby J, Grierson AJ, et al. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:616–630. [DOI] [PubMed] [Google Scholar]
- 114. Lagier‐Tourenne C, Polymenidou M, Cleveland DW. TDP‐43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010;19:R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sheean RK, Turner BJ. Genetics of motor neuron disorders: From gene diversity to common cellular conspirators in selective neuronal killing In: Cauchi RJ, editor. Drosophila melanogaster models of motor neuron disease. New York: Nova Biomedical, 2013;1–34. [Google Scholar]
- 116. Turner BJ, Baumer D, Parkinson NJ, et al. TDP‐43 expression in mouse models of amyotrophic lateral sclerosis and spinal muscular atrophy. BMC Neurosci 2008;9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Turner BJ, Parkinson NJ, Davies KE, et al. Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol Dis 2009;34:511–517. [DOI] [PubMed] [Google Scholar]
- 118. Veldink JH, Kalmijn S, Van der Hout AH, et al. SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 2005;65:820–825. [DOI] [PubMed] [Google Scholar]
- 119. Ling SC, Albuquerque CP, Han JS, et al. ALS‐associated mutations in TDP‐43 increase its stability and promote TDP‐43 complexes with FUS/TLS. Proc Natl Acad Sci USA 2010;107:13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang JW, Brent JR, Tomlinson A, et al. The ALS‐associated proteins FUS and TDP‐43 function together to affect Drosophila locomotion and life span. J Clin Invest 2011;121:4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kabashi E, Bercier V, Lissouba A, et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 2011;7:e1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gerbino V, Carri MT, Cozzolino M, et al. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol Dis 2013;55:120–128. [DOI] [PubMed] [Google Scholar]
- 123. Groen EJ, Fumoto K, Blokhuis AM, et al. ALS‐associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 2013;22:3690–3704. [DOI] [PubMed] [Google Scholar]
- 124. Baumer D, Ansorge O, Almeida M, et al. The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev Mol Med 2010;12:e21. [DOI] [PubMed] [Google Scholar]
- 125. Kolb SJ, Sutton S, Schoenberg DR. RNA processing defects associated with diseases of the motor neuron. Muscle Nerve 2010;41:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lemmens R, Moore MJ, Al‐Chalabi A, et al. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci 2010;33:249–258. [DOI] [PubMed] [Google Scholar]
- 127. Jia Y, Mu JC, Ackerman SL. Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration. Cell 2012;148:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhao C, Bellur DL, Lu S, et al. Autosomal‐dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet 2009;85:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hamilton G, Gillingwater TH. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol Med 2013;19:40–50. [DOI] [PubMed] [Google Scholar]
- 130. Ilieva H, Polymenidou M, Cleveland DW. Non‐cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009;187:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Makhortova NR, Hayhurst M, Cerqueira A, et al. A screen for regulators of survival of motor neuron protein levels. Nat Chem Biol 2011;7:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mattis VB, Rai R, Wang J, et al. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet 2006;120:589–601. [DOI] [PubMed] [Google Scholar]
- 133. Van Meerbeke JP, Sumner CJ. Progress and promise: The current status of spinal muscular atrophy therapeutics. Discov Med 2011;12:291–305. [PubMed] [Google Scholar]