

Cryptococcus neoformans is a model organism for the study of fungal infections. This encapsulated fungus is found worldwide, particularly in soil samples contaminated by bird excreta (1). Despite frequent exposure, immunologically intact persons rarely get disease, with an annual incidence estimated at only 0.001%. However, the incidence rises astronomically in patients with impaired T-cell function. In persons with AIDS, C. neoformans is one of the five most common causes of life-threatening opportunistic infections. Meningoencephalitis is the most common clinical presentation of cryptococcosis, although any organ system can be affected. C. neoformans is a facultative intracellular parasite of macrophages and it seems that the capacity to survive both inside and outside the phagocyte is critical for the organism's virulence (2). In this issue of PNAS, Steenbergen et al. (3) provide tantalizing data regarding the evolution and maintenance of virulence in C. neoformans. The authors examined the interactions of C. neoformans with the free-living soil amoeba, Acanthamoeba castellanii. They found that the fungus was phagocytosed by the amoebae. Once inside, the fungus efficiently replicated, eventually killing the amoebae. The process was remarkably similar to that seen in mammalian macrophages (2). Thus, with both macrophages and amoebae, after phagocytosis the fungi get localized into membrane-bound vacuoles. Fungal replication ensues, accompanied by disruption of the vacuolar membrane, intracellular accumulation of capsular polysaccharide, and cell lysis.
Nearly a quarter of a century ago, Bunting et al. (4) published their observation that free-living amoebae could ingest C. neoformans. The whimsical title of the manuscript, Cryptococcus neoformans: Gastronomic Delight of a soil Ameba, reflected the ability of the studied amoeba, Acanthamoeba polyphaga, not only to phagocytose C. neoformans, but also to kill the vast majority of ingested fungi. In contrast, the findings of Steenbergen et al. (3) suggest that this “gastronomic delight” is actually a “poison pill” that kills its host. More experimental work will be needed to reconcile these disparate findings. One possible explanation is fungal strain-related differences. In support of this concept, one of the three wild-type strains tested by Steenbergen et al. did not replicate well inside the amoebae whereas the other two strains did. Moreover, different species of Acanthamoeba were used in the two studies raising the possibility that the fungicidal capacity of amoebae varies among species. Alternatively, akin to the situation with macrophages, perhaps amoebae can exist in either an “activated” or “inactivated” state with respect to fungicidal capacity.
Of the >100,000 fungal species that exist on our planet, fewer than 20 regularly cause serious infections in humans (5). Overwhelmingly, those afflicted have severely compromised immune systems, such as AIDS or neutropenia. Thus, innate and acquired immunity normally forms very effective host defenses against fungi. This is true not only in humans, but in all other members of the animal kingdom studied thus far. In fact, there is remarkable conservation in the pathways leading to antifungal defenses. Accordingly, in Drosophila, successful defenses following challenge with the mold Aspergillus fumigatus require activation of Toll and signal transduction pathways that are homologous to the mammalian toll-like receptors and NF-κB nuclear translocation pathways activated after fungal stimulation (6, 7). Nevertheless, fungi have well defined virulence factors that greatly enhance pathogenicity (5). This begs the question that if host defenses are so effective against fungi so as to make lethal mycoses rare, why did these virulence factors evolve and what is the selective pressure to maintain these factors?
The major virulence factor of C. neoformans is its polysaccharide capsule (Fig. 1; refs. 5 and 8). C. neoformans is the only fungus of medical importance to possess a capsule. Deletion of genes necessary for capsule formation results in organisms that are avirulent in murine models of cryptococcosis. Moreover, reinsertion of the genes restores virulence (9). The major component of capsule is a high molecular weight polysaccharide, glucuronoxylomannan, which both coats the fungus and is shed. In patients with cryptococcosis, shed glucuronoxylomannan accumulates locally in the surrounding tissue, particularly macrophages, but also circulates in the blood and cerebrospinal fluid. A myriad of mechanisms has been proposed to account for the virulence properties of the capsule including inhibition of leukocyte migration and phagocytosis, complement consumption, dysregulation of cytokine responses, and suppression of lymphocytic function (5, 8). Interestingly, the capsule seems to enable the fungus to avoid phagocytosis and to thrive once inside the macrophage (2). Other well defined virulence factors include laccase (a phenoloxidase that enables the organism to make melanin) and phospholipase. The genes encoding both of these enzymes have been knocked out in C. neoformans, and in each case, the loss of enzymatic function results in organisms that are hypovirulent in mouse models (10, 11). Melanin, which deposits in the cell wall of the fungus, is thought to protect the fungus against oxidative damage by phagocytes, although other mechanisms have been postulated (5, 8). Phospholipase may enhance virulence by helping the fungus penetrate phospholipid-rich host barriers such as membranes and pulmonary surfactant (11). The work by Steenbergen et al. (3) provides provocative evidence that the interaction of C. neoformans with amoebae could drive the evolution and maintenance of these three cryptococcal virulence factors. C. neoformans strains lacking capsule were efficiently killed by the amoebae whereas the parental encapsulated strain replicated ≈70-fold over a 48-h period inside the amoebae. Although it was not killed, the phospholipase mutant did not replicate within the amoebae. The results with laccase-deficient mutants were less clear cut. The amoebae had roughly equivalent anticryptococcal activity against the wild-type and laccase mutant strains. Moreover, growth of wild-type strains in medium containing the laccase substrate, L-dopa, which results in fungi that are heavily melanized, had no significant effect on subsequent fungal growth in the amoebae. However, a melanized acapsular strain did grow within the amoebae, in stark contrast to the unmelanized acapsular fungi that were killed. This latter observation is of uncertain biological relevance as naturally occurring strains of C. neoformans are encapsulated. Moreover, it is unknown whether substrates for laccase exist in amoebae. In humans, catecholamines, including the neurotransmitter dopamine, serve as substrates for laccase, a factor that has been invoked to explain the neurotropism of C. neoformans (12).
The results of Steenbergen et al. support the hypothesis that amoebae are a replication niche of the fungus.
Figure 1.
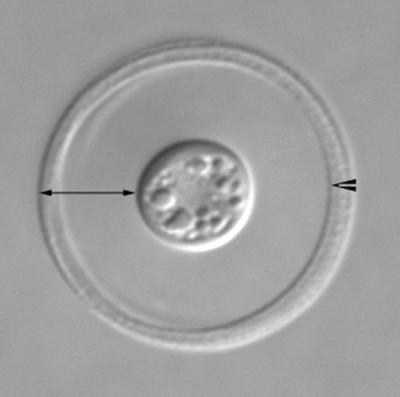
Photomicrograph of C. neoformans acquired with differential interference contrast (DIC) optics. The fungus has been incubated with anticapsular Abs that demarcate the rim of the capsule (arrowhead). The full width of the capsule is indicated by the double-headed arrow. (Courtesy of T. Kozel and P. Thorklidson, University of Nevada School of Medicine, Reno, NV.)
Free-living amoebae adhere to soil particles and under favorable conditions their numbers are estimated to exceed one million cells per gram of soil (13). The small size and elasticity of these protozoa enable them to feed on bacteria and fungi located in small pores in the soil (13). The molecular mechanisms by which amoebae and macrophages ingest microbes bear striking similarities, and cell biologists have used amoebae, particularly Dictyostelium discoideum, as models to study phagocytosis (14). It is perhaps not surprising then that the strategies used by pathogens to parasitize macrophages overlap with those enabling survival within amoebae. The demonstration by Steenbergen et al. (3) that virulence factors empowering C. neoformans to survive within amoebae coincide with those that facilitate intracellular survival in macrophages has precedence in the bacterial kingdom. Best studied in this regard has been the Gram-negative pathogen Legionella pneumophila. At least 13 protozoan species can be infected by L. pneumophila and there is a clear link between bacterial multiplication and the presence of amoebae in water (15). Virulence factors that promote L. pneumophila survival and replication in macrophages have a remarkable overlap with those required for growth in amoebae. For example, in one study, 89 transposon mutants were isolated that exhibited defects in cytotoxicity, intracellular survival, and replication within both the human macrophage-like cell line, U937, and A. polyphaga (16). Moreover, inoculation of mice with a mixture of L. pneumophila and amoebae results in more severe disease compared with mice infected with bacteria alone. Finally, a more virulent and antibiotic-resistant phenotype emerges after growth of L. pneumophila in amoebae compared with in broth. The opportunist pathogen, Mycobacterium avium, also demonstrates increased virulence after passage in amoebae (17). Amoebae containing L. pneumophila and M. avium are frequent contaminants of potable water supplies and pulmonary exposure can occur after inhalation of aerosolized bacteria inside of amoebae. A potential role of protozoans as reservoirs for other human bacterial pathogens, including Listeria monocytogenes, Chlamydia pneumoniae, and Burkholderia cepacia, has also been suggested (18–20).
It would be interesting to examine whether C. neoformans demonstrates increased virulence after passage in amoebae, especially as the fungus has been shown to undergo phenotypic switching (21). Moreover, signature-tagged mutagenesis could be used to identify fungal genes that favor intracellular replication in amoebae with the eventual goal of discovering virulence factors that enhance pathogenicity in mammalian systems. Our understanding of how C. neoformans succeeds in the environment remains rudimentary. Clearly, establishing an ecological niche is complex and most successful organisms will use multiple means to do so. C. neoformans seems to be no exception. The results of Steenbergen et al. (3) support the hypothesis that amoebae are a replication niche of the fungus. Definitive proof, although, will likely require, at a minimum, meticulous study of soil (and perhaps water) samples. Ultimately, too, it will be important to determine whether aerosolization of C. neoformans contained within amoebae occurs naturally.
Based on the finding that C. neoformans can be isolated from the excreta of virtually all species of birds studied, it has long been thought that birds are important in propagating C. neoformans (1, 22). Interestingly, the birds themselves do not get infected, perhaps because their high body temperature is inhospitable to the growth of the fungus. The properties that enable C. neoformans to colonize birds are not known. One wonders whether some of the virulence factors that enable C. neoformans to parasitize macrophages and amoebae could also promote colonization of birds. One such candidate virulence factor is urease. Urease-negative mutants of C. neoformans are hypovirulent in mouse models of cryptococcosis (23). In bird excreta, the primary role of urease may be to enable C. neoformans to convert urea to the usable nitrogen source ammonia. There is reason too to suspect that besides amoebae and birds, other environmental reservoirs of C. neoformans exist that help maintain the organism's virulence factors. C. neoformans has been isolated from decaying wood hollows (24). Decaying wood contains large amounts of the aromatic polymer lignin. Laccases, which are present in wood rot fungi, use lignins as substrates. Thus, it has been hypothesized that cryptococcal laccase helps the organism establish an ecological niche in rotting wood (24). Melanin, the end product of laccase activity, may also protect the organism from environmental stresses such as ionizing radiation (12).
Although rare reports have epidemiologically linked human cryptococcosis to bird exposure, in the vast majority of cases, the source of the fungus is never determined (1, 22). From a medical standpoint, a greater understanding of the ecological niches of C. neoformans is important because it could lead to commonsense recommendations so that immunocompromised persons at high risk of infection can minimize exposure to this opportunistic fungus. In this regard, the report by Steenbergen et al. (3) could be an important step in this direction.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AI37532 and R01 AI25780. S.M.L. is a recipient of a Burroughs Wellcome Fund Scholar Award in Pathogenic Mycology.
Footnotes
See companion article on page 15245.
References
- 1.Levitz S M. Rev Infect Dis. 1991;13:1163–1169. doi: 10.1093/clinids/13.6.1163. [DOI] [PubMed] [Google Scholar]
- 2.Feldmesser M, Tucker S, Casadevall A. Trends Microbiol. 2001;9:273–278. doi: 10.1016/s0966-842x(01)02035-2. [DOI] [PubMed] [Google Scholar]
- 3.Steenbergen J N, Shuman H A, Casadevall A. Proc Natl Acad Sci USA. 2001;98:15245–15250. doi: 10.1073/pnas.261418798. . (First Published December 11, 2001; 10.1073/pnas.261418798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunting L A, Neilson J B, Bulmer G S. Sabouraudia. 1979;17:225–232. doi: 10.1080/00362177985380341. [DOI] [PubMed] [Google Scholar]
- 5.Hogan L H, Klein B S, Levitz S M. Clin Microbiol Rev. 1996;9:469–488. doi: 10.1128/cmr.9.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemaitre B, Nicolas E, Michaut L, Reichhart J M, Hoffmann J A. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 7.Shoham S, Huang C, Chen J M, Golenbock D T, Levitz S M. J Immunol. 2001;166:4620–4626. doi: 10.4049/jimmunol.166.7.4620. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan K L, Murphy J W. Emerg Infect Dis. 1998;4:71–83. doi: 10.3201/eid0401.980109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Y C, Kwon-Chung K J. Mol Cell Biol. 1994;14:4912–4919. doi: 10.1128/mcb.14.7.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salas S D, Bennett J E, Kwon-Chung K J, Perfect J R, Williamson P R. J Exp Med. 1996;184:377–386. doi: 10.1084/jem.184.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox G M, McDade H C, Chen S C, Tucker S C, Gottfredsson M, Wright L C, Sorrell T C, Leidich S D, Casadevall A, Ghannoum M A, Perfect J R. Mol Microbiol. 2001;39:166–175. doi: 10.1046/j.1365-2958.2001.02236.x. [DOI] [PubMed] [Google Scholar]
- 12.Casadevall A, Rosas A L, Nosanchuk J D. Curr Opin Microbiol. 2000;3:354–358. doi: 10.1016/s1369-5274(00)00103-x. [DOI] [PubMed] [Google Scholar]
- 13.Ekelund F, Ronn R. FEMS Microbiol Rev. 1994;15:321–353. doi: 10.1111/j.1574-6976.1994.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 14.Janssen K P, Schleicher M. Biochim Biophys Acta. 2001;1525:228–233. doi: 10.1016/s0304-4165(01)00108-8. [DOI] [PubMed] [Google Scholar]
- 15.Swanson M S, Hammer B K. Annu Rev Microbiol. 2000;54:567–613. doi: 10.1146/annurev.micro.54.1.567. [DOI] [PubMed] [Google Scholar]
- 16.Gao L Y, Harb O S, Abu Kwaik Y. Infect Immun. 1997;65:4738–4746. doi: 10.1128/iai.65.11.4738-4746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cirillo J D, Falkow S, Tompkins L S, Bermudez L E. Infect Immun. 1997;65:3759–3767. doi: 10.1128/iai.65.9.3759-3767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marolda C L, Hauroder B, John M A, Michel R, Valvano M A. Microbiology. 1999;145:1509–1517. doi: 10.1099/13500872-145-7-1509. [DOI] [PubMed] [Google Scholar]
- 19.Essig A, Heinemann M, Simnacher U, Marre R. Appl Environ Microbiol. 1997;63:1396–1399. doi: 10.1128/aem.63.4.1396-1399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ly T M, Muller H E. J Med Microbiol. 1990;33:51–54. doi: 10.1099/00222615-33-1-51. [DOI] [PubMed] [Google Scholar]
- 21.Goldman D L, Fries B C, Franzot S P, Montella L, Casadevall A. Proc Natl Acad Sci USA. 1998;95:14967–14972. doi: 10.1073/pnas.95.25.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nosanchuk J D, Shoham S, Fries B C, Shapiro D S, Levitz S M, Casadevall A. Ann Intern Med. 2000;132:205–208. doi: 10.7326/0003-4819-132-3-200002010-00006. [DOI] [PubMed] [Google Scholar]
- 23.Cox G M, Mukherjee J, Cole G T, Casadevall A, Perfect J R. Infect Immun. 2000;68:443–448. doi: 10.1128/iai.68.2.443-448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazera M S, Salmito Cavalcanti M A, Londero A T, Trilles L, Nishikawa M M, Wanke B. Med Mycol. 2000;38:379–383. doi: 10.1080/mmy.38.5.379.383. [DOI] [PubMed] [Google Scholar]