

Summary
Vitamin D has been demonstrated to influence multiple aspects of amyotrophic lateral sclerosis (ALS) pathology. Both human and rodent central nervous systems express the vitamin D receptor (VDR) and/or its enzymatic machinery needed to fully activate the hormone. Clinical research suggests that vitamin D treatment can improve compromised human muscular ability and increase muscle size, supported by loss of motor function and muscle mass in animals following VDR knockout, as well as increased muscle protein synthesis and ATP production following vitamin D supplementation. Vitamin D has also been shown to reduce the expression of biomarkers associated with oxidative stress and inflammation in patients with multiple sclerosis, rheumatoid arthritis, congestive heart failure, Parkinson's disease and Alzheimer's disease; diseases that share common pathophysiologies with ALS. Furthermore, vitamin D treatment greatly attenuates hypoxic brain damage in vivo and reduces neuronal lethality of glutamate insult in vitro; a hallmark trait of ALS glutamate excitotoxicity. We have recently shown that high‐dose vitamin D 3 supplementation improved, whereas vitamin D 3 restriction worsened, functional capacity in the G93A mouse model of ALS. In sum, evidence demonstrates that vitamin D, unlike the antiglutamatergic agent Riluzole, affects multiple aspects of ALS pathophysiology and could provide a greater cumulative effect.
Keywords: Amyotrophic lateral sclerosis, Apoptosis, Calcidiol, Calcitriol, D3, G93A mice, Excitotoxicity, Inflammation, Neurodegenerative disease, Neuromuscular disease, Motor neuron death, Oxidative stress, Vitamin D
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS; Figure 1), also known as “Lou Gehrig's disease,” is a fatal neurodegenerative disease of the motor cortex, brain stem and spinal cord; responsible for the destruction of upper and lower motor neurons causing paralysis 1. Currently, at least 26 mutant genes are known to cause ALS in humans (Table 1). Of the known genetic defects, the most studied of these is mutant Cu/Zn superoxide dismutase (SOD1, comprising approximately 20% of all known inherited mutations 2). Most ALS patients will begin to experience symptoms usually manifesting as weakness in the limbs, progressing to affect manual dexterity and gait, eventually losing most voluntary control 1. Death is eventually caused due to respiratory failure with a median survival rate of 3–5 years after the onset of symptoms 3. The only generally accepted treatment for the disease is the administration of the antiglutamate drug Riluzole, which is by far the most prescribed therapy for ALS 4. Daily 100 mg oral consumption of the drug is reported to prolong the median survival of patients by approximately 2–3 month and increase the likelihood of survival in the first year by 9% 5.
Figure 1.
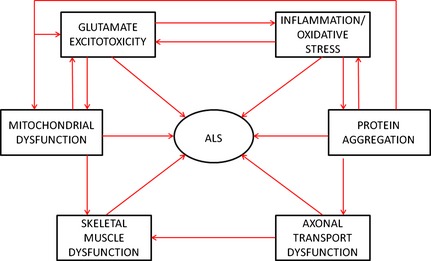
Schematic outlining the multifaceted nature of ALS pathology. ALS, amyotrophic lateral sclerosis.
Table 1.
Known mutated genes known to cause amyotrophic lateral sclerosis
| Gene protein product | Normal protein function | References |
|---|---|---|
| Superoxide dismutase 1 | Antioxidant | 2, 150 |
| Alsin | GTPase regulation | 151 |
| ALS3 | Unknown | 152 |
| ALS7 | Unknown | 153 |
| Senataxin | RNA processing | 154 |
| Vesicle‐associated membrane protein/synaptobrevin‐associated membrane protein B | Intracellular vesicle trafficking | 155, 156 |
| Angiogenin | Angiogenic regulation | 157, 158 |
| TAR DNA binding protein‐43 | Transcriptional regulation | 159 |
| Fused in sarcoma | Transcriptional regulation | 160, 161 |
| Dynactin p150 subunit | Axonal transport | 162 |
| Spatacsin | Axonal transport | 163 |
| Ubiquilin 2 | Protein degredation | 164 |
| SIGMAR 1 | Receptor | 156 |
| C9orf72 | Unknown | 166 |
| Peripherin | Neurofilament subunit | 167 |
| Valosin‐containing protein | Intracellular vesicle trafficking | 168 |
| Ewing sarcoma breakpoint region 1 | RNA processing | 169 |
| Optineurin | RNA processing | 170 |
| Ataxin 2 | RNA processing | 171 |
| Neurofilament heavy chain | Cell structure | 172 |
| Charged multivesicular body protein 2b | Intracellular vesicle trafficking | 173 |
| Phosphatidylinositol 3,5‐bisphosphate 5‐phosphatase | Intracellular vesicle trafficking | 174 |
| D‐amino acid oxidase | Protein metabolism | 175 |
| Profilin 1 | Cell structure | 176 |
| Sequestosome | Protein metabolism | 177 |
| TATA‐binding protein associated factor 15 | RNA binding protein | 178 |
Rationale for Vitamin D as a Therapeutic in ALS
Amyotrophic lateral sclerosis shares pathophysiological similarities with various diseases such as congestive heart failure, rheumatoid arthritis (RA), multiple sclerosis (MS), Alzheimer's disease (AD), and Parkinson's disease (PD). These similarities include oxidative stress, inflammation, neurodegeneration, mitochondrial dysregulation, and apoptosis 6, 7, 8, 9, 10, 11. Evidence suggests that vitamin D ameliorates these pathophysiologies in animal disease models and human patients 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, and may therefore be able to attenuate the sequelae of ALS (Figure 2). Recent studies have shown that high‐dose vitamin D3 (D3) supplementation improves paw grip endurance and motor performance in the G93A mouse model of ALS 21, 22. In contrast, D3 restriction hastens the decline in paw grip endurance and motor performance postdisease onset in the same mouse model 23. Indeed, a very recent ALS clinical study concluded that D3 supplementation reduced the decline in the revised amyotrophic lateral sclerosis functional rating scale (ALSFRS‐R) score versus non‐supplemented patients 24. These findings are supported by a retrospective study, which also found that patients with vitamin D deficiency (serum calcidiol > 25 nM) had a 6 fold higher rate of death compared to patients with high vitamin D status (serum calcidiol > 75 nM) 25. In sum, there is substantial support for vitamin D as a potential therapeutic in ALS.
Figure 2.
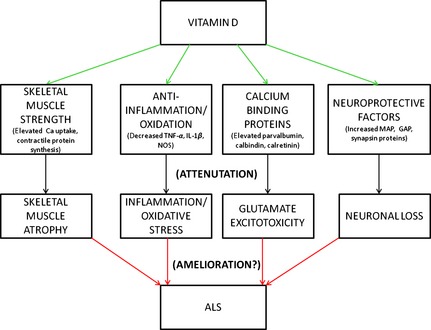
Schematic of the potential amyotrophic lateral sclerosis (ALS) pathophysiologies modulated by vitamin D and the possible subsequent mitigation of ALS. (TNF‐α, tumor necrosis factor‐ α; IL‐1β, interleukin‐1β; NOS, nitric oxide synthase; MAP, microtubule‐associated protein; GAP, growth‐associated protein).
Vitamin D as Related to ALS Pathology
Human Vitamin D Studies Related to ALS
Inflammation and Oxidative Stress
The chronic, feed‐forward cycle of glial cell activation leading to inflammatory cytokine generation, microglial proliferation, and neurotoxicity in ALS constructs an event referred to as “neuroinlammation” 26. Tumour necrosis factor‐alpha (TNF‐α), a potent inflammatory cytokine, induces apoptosis and contributes to oxidative stress by activating microglia 26. This important cytokine is found in elevated amounts in G93A mouse spinal cords 27 and human ALS patient serum 28, 29. Following administration of thalidomide and lenalidomide (agents used to treat some cancers that also inhibit TNF‐α expression) starting at 30 day, G93A mice exhibited significant improvements in multiple outcome measures when compared with saline‐treated controls including: improvement in motor performance between 98 and 155 day of age, attenuation in weight loss from 70 day of age, and extention of mean survival by 12–18.5% 30. Interleukin‐1β (IL‐1β) is also involved in inflammatory‐mediated damage in ALS where it stimulates the destruction of cellular proteins via transcription of apoptotic enzymes including caspase 1 and caspase 3 31. In line with this, human ALS spinal cords exhibit high concentrations of IL‐1β 32. Matrix metalloproteinases (MMP) are capable of degrading all components of the extra cellular matrix 33 and MMP‐9, activated by TNF‐α and IL‐1β 34, is also implicated in ALS pathology as well as other related neurologic conditions such as AD, PD, stroke, and spinal cord trauma 35, 36, 37, 38, 39, 40, 41.
In 174 patients with kidney disease, serum 1,25(OH)2D3 negatively correlated with urinary MCP‐1 (a marker of inflammation; r = −0.342), renal scarring (r = −0.546), and macrophage infiltration (r = −0.537) 42. Healthy male mononuclear cells insulted with lipopolysaccharide (LPS) exhibited a 64% and 59% reduction in TNF‐α and IL‐1β, respectively, in tandem with an increase in median blood 25(OH)D3 from 43 nM in winter to 89 nM in summer 43. Healthy Canadian natives supplemented with 1000 IU D3/day for 8 months exhibited a 16 fold decrease in IL‐1β mRNA in cultured macrophages exposed to tuberculosis lipoprotein versus baseline 42. In a randomized, double‐blind, placebo‐controlled trial in patients with congestive heart failure (characterized by a reduced cardiac ejection fraction, cardiac hypertrophy, and increased pro‐inflammatory cytokines, particularly TNF‐α 7), 2000 IU D3/day for 9 months increased median expression of the anti‐inflammatory IL‐10 by 43% versus baseline 7. Median TNF‐α levels increased 12% in the placebo group, but did not change significantly from baseline in the D3 group 7. A more recent randomized, double‐blind, placebo‐controlled trial administered a single dose of 250,000 IU D3 to cystic fibrosis patients (characterised by chronic lung infection and inflammation) and observed a 50% reduction in TNF‐α expresstion versus placebo 45. Peterson et al. 46 found that mean serum TNF‐α concentration was 35% lower with high (0.79 pg/mL) versus low (1.22 pg/mL) UV exposure, and that serum 25(OH)D3 was negatively correlated with serum TNF‐α (r = −0.25) in healthy women. Indeed, a 2010 US prospective database study involving 41,497 men and women (age, 55 ± 21 years) showed that those with serum 25(OH)D3 <37.5 nM had a 45%, 45%, and 78% greater likelihood of developing coronary artery disease, peripheral vascular disease, and stroke, respectively, versus those with levels >75 nM 47. Human patients with rheumatoid arthritis (a chronic inflammatory autoimmune disease) exhibit low serum levels of 25(OH)D3 and 1,25(OH)2D3 48, suggesting that vitamin D plays a role in the disease. In a 3‐month clinical trial involving 19 patients with RA treated with standard antirheumatic drugs, daily oral 2 μg doses of alphacalcidiol (a vitamin D analog) improved clinical measures such as Ritchie Articular Index, and improved biological measures such as lymphocyte proliferation and apoptosis in 89% of the patients. These human studies suggest the possibility that vitamin D supplementation could reduce ALS neuroinflammation and oxidative stress.
Muscle
Amyotrophic lateral sclerosis is characterized by “amyotrophy,” indicative of the muscle wasting due to the denervation of muscle fibers and their degenerating motor neurons 1. “Lateral sclerosis” refers to the hardening of ventral and lateral corticospinal tracts as these areas are progressively replaced by gliosis (the process involving the accumulation and death of neuroglia at sites of damage in the central nervous system resulting in scarring) 1. A phenotypic hallmark of ALS is the atrophy of skeletal muscle fibres which become denervated as their corresponding motor neurons degenerate 49.
The nuclear VDR is involved in regulating a large number of genes (up to 5% of the total human genome 50) and is indeed expressed by human muscle tissue 51. A cross‐sectional study involving 127 Dutch elderly aged >65 years found a modest association between serum 25(OH)D3 and appendicular lean mass (β = 0.012), as well as physical performance (β = 0.020) 52. After an acute bout of intense exercise, serum 25(OH)D3 was inversely correlated with the postexercise muscle weakness experienced by one leg versus the other nonexercised control leg immediately after exercise (r = −0.71), as well as at 48 h (r = −0.67) and 72 h (r = −0.72) postexercise 53. Data from 4100 ambulatory ≥ 60 year‐old adults demonstrated a dose–response relationship between serum 25(OH)D3 levels and the ability to walk 8 ft. and sit‐to‐stand test; whereby those in the highest 25(OH)D3 quintile scored 6% and 4% higher, respectively, versus the lowest quintile 54. Supplementing 75–88 year‐old men and women (serum 25(OH)D3 <50 nM) with 800 IU D3/day in double‐blind, RCTs decreased the risk of falls by 27–72% 55, 56, 57, 58. Further, 63–99 year‐old women supplemented for 3 months with 800 IU D3/day + 1200 mg/day calcium improved their musculoskeletal function (knee flexor and extensor strength, grip strength, and the timed up‐and‐go test) by 4–11% versus baseline, in contrast to the −4% to 1% change in strength demonstrated by the control group supplemented only with calcium 55. This increase in strength probably contributed to the 49% lower rate of falls experienced by the group supplemented with D3 and calcium versus only calcium. In contrast, 251 healthy adults aged 18‐50 years given 1000 IU/day or placebo for 4 months did not improve in the chair‐rising test, hand grip strength, or jump height versus baseline 59. Separately, treating vitamin D‐deficient approximately 70 year‐old women with alphacalcidiol (vitamin D analog with a negligible calcemic effect) for 6 months increased isometric knee extensor strength by 13% and total walking distance traveled over 2 min by 10% as compared to baseline 60. In hemodialysis patients, 1,25(OH)2D3 supplementation increased three repetition‐maximum knee extension, knee extension peak torque, and ankle dorsiflexion by 21–38% versus placebo 61. These gains in strength were concurrent with an 11% and 15% increase in tibialis anterior and thigh cross‐sectional areas, respectively. In support, 21 mobility‐limited women aged ≥ 65 years given 4000 IU/day increased intramyonuclear VDR by 30% and muscle fibre size by 11% 62. Most relevant, vitamin D3 supplementation at 2000 IU/d for 9 months was recently shown to reduce the decline in the ALSFRS‐R score 24. A separate study supported these findings retrospectively by associating low vitamin D status with poor ALS prognosis according to the ALSFRS‐R 63.
Neurodegeneration and Neuroprotection
Excessive glutamate, the main excitatory neurotransmitter in the CNS 64, is an inherent part of ALS pathology. Excessive glutamate release by the presynaptic neuron into the synaptic cleft and/or impaired glutamate removal by EAAT2 transposters located on synapse‐enveloping astrocytes in ALS causes prolonged activation of postsynaptic receptors, resulting in the influx of excessive quantities of sodium and calcium into the cell, inducing free radical production 65. Due to a limited amount of intracellular calcium‐buffering proteins, motor neurons are vulnerable to excessive calcium concentrations 66. Mice that highly express intracellular calcium‐buffering proteins are more resistant to excitotoxicity and exhibit lower concentrations of intracellular calcium following AMPA receptor stimulation 66. Poor clearance of glutamate from the synapse can also, in part, be responsible for excitotoxicity due to a decline in the number of functional astrocytic glutamate reuptake transporters, either due to a decrease in the number of transporters or an increase in dysfunctional/nonfunctioning transporters 64. Increases in intracellular calcium can induce mitochondria to generate free radicals which escape the cell and further compromise the ability of synaptic glutamate reuptake by glutamate transporters 65. The high influx of calcium can also cause neuronal mitochondria to swell, opening the mitochondrial permeability transition pore and releasing proapoptotic factors 67.
Support for the role played by vitamin D in the nervous system is strengthened by the discovery of its machinery in the postmortem human brain 68 and nuclear vitamin D uptake in the spinal cord 69. VDR is very strongly expressed in the CA1 and CA2 regions of the brain, but with a lower amount in the CA3 region of the hippocampus, whereas 1α‐OHase is very strongly and evenly distributed throughout the CA1, CA2, and CA3 regions of the hippocampus 68; confirming previous studies in rats 70, 71. The CA areas of the hippocampus are integral for learning and memory and are involved in AD pathology; an illness characterized by neurodegeneration and progressive loss of memory and cognitive function 72. A decrease in VDR mRNA levels has been detected in human Alzheimer CA1 (34%) and CA2 (31%) pyramidal cells, but not in the temporal cortex or cerebellum (unaffected areas), as compared to controls with Huntington disease 73. A long‐term prospective study involving 498 elderly women demonstrated that women who did not develop AD after 7 year follow‐up consumed 17% more dietary vitamin D at baseline versus women who developed AD 74. However, there was no such difference between nonafflicted women and those who developed other dementias. Furthermore, women in the highest quintile for vitamin D intake at baseline decreased their risk (OR = 0.23) for developing AD versus the lowest quintile at the 7 year follow‐up 74. Similarly, using a group of 858 Italian adults aged 65 years+, the risk for cognitive decline at 6 year follow‐up increased (OR = 1.60) with vitamin D deficiency (<25 nM) versus those who were sufficient (>75 nM) 75.
Parkinson's disease is a common neurodegenerative disease whereby selective death of dopaminergic neurons results in dysfunction characterized by tremors, impaired speech, and general loss of muscle control 76. ALS and PD express some of the same pathophysiology. In PD, as in ALS, neuroinflammation presents as a prominent pathologic feature, characterized by activated microglia and infiltrating T cells at sites of spinal cord motor neuron injury 10, 77. ALS induces activation of microglia and increases their release of proinflammatory cytokines and free radicals such as TNF‐α, IL‐1β, inducible nitric oxide synthase, and 78. A similar event occurs in PD patients with mutations in α‐synuclein, with mutant protein aggregates causing activated‐microglial release of a similar array of damaging biochemical factors 10, 11. There is also evidence to suggest that damage mediated by H2O2 and ˙OH through the nonenzymatic Fenton reaction also occurs in PD 79, 80. Furthermore, as in ALS 81, 82, 83, PD electron transport chain (ETC) complex I activity is reduced, namely, in the substantia nigra of the brain 84, 85, 86. Indeed, humans exposed to 1‐methyl 4‐phenyl 1,2,3,6 tetrahydropyridine develop PD through mechanisms that damage nigrostriatal ETC complex I 87.
Clinical vitamin D studies in human PD patients are scarce, but a 1997 case study 88 treated a hospitalized 50 year‐old man diagnosed with PD with 4000 IU D3/day and 1 g Ca/day (body weight not specified) in addition to regular therapy which alone failed to show any clinical benefits after 3 year. The patient exhibited low serum calcium, phosphorus, and 25(OH)D3 prior to supplementation with D3 + Ca. The patient improved significantly in the following year as evidenced by decreased rigidity and akinesia, with a substantive decrease in his multidrug therapy to only 375 mg levodopa/day. At 1‐year follow‐up, examination revealed absent tremor with only moderate rigidity.
The association of PD with low vitamin D status has been suggested through epidemiologic studies showing a higher prevalence of PD among those living in the more northern latitudes 89, 90, 91, 92.
Vitamin D insufficiency (serum 25(OH)D3 ≤75 nM) has also been observed in PD patients. PD patients have a significantly higher prevalence (55%) of hypovitaminosis D versus healthy controls (36%) and AD patients (41%) 93. A high prevalence of low serum 25(OH)D3 (<50 nM) in the mid‐late summer months was found in patients with severe PD when compared with those with less advanced PD 94. Sato et al. ascertained that PD patients also had lower serum 25(OH)D3 (29.7 nM) as compared with healthy control subjects (83.2 nM). These researchers also found a significant and very strong inverse relationship between vitamin D status and the Unified PD Rating Scale III (r = ‐‐0.91) 95, a scale used to measure progression and severity of illness 96. In a longitudinal study 97, a dose–response relationship was found between vitamin D status and risk for developing PD: those with a concentration of at least 50 nM had a relative risk one‐third (RR = 0.33) of those with <25 nM. Furthermore, genetic VDR polymorphisms are associated with PD risk and age‐at‐disease‐onset 98.
Multiple sclerosis is a demyelinating, neurodegenertative disease of the central nervous system. An association with higher latitude and susceptibility to developing MS is well established, as is the association of a higher later‐in‐life incidence of MS when born in late spring versus a lower incidence when born in late autumn 99. In a prospective study involving American nurses, those who supplemented with at least 400 IU D3/day were at a lower risk for MS versus those with no supplemental inake (RR = 0.59) 100.
In Vivo and In Vitro Animal Vitamin D Studies Related to ALS
Inflammation and Oxidative Stress
Human monocytes exposed to LPS exhibited a dose‐dependent decrease in inflammation as measured by p38 phosphorylation in response to the administration of 15, 30, and 50 ng/mL 25(OH)D3 101. Aged rats (20 months) supplemented with 1.05 μg 1,25(OH)2D3/kg b.wt./day for 21 days exhibited 25% lower IL‐1β and 23% greater IL‐10 expression versus nonsupplemented controls 102. This was observed in tandem with 22% less amyloid‐ β oligomerization and 15% greater neprilysin (amyloid‐ β degrading enzyme) expression. In rodents, injection of type‐2 collagen generates the collagen‐induced arthritis (CIA) model; a model for RA 14. CIA can be prevented by ingestion of 1,25(OH)2D3 in both mice and rats 16, 17. Alternatively, 1,25(OH)2D3 can prevent CIA from progressing from early to more severe stages 14. VDR‐deficient mice cross‐bred with human TNF‐α transgenic mice displayed signs of exacerbated degenerative arthritic disease including accelerated grip strength loss (47%), paw swelling (91%), and synovial bone erosion (106%) versus transgenic mice not deficient in VDR. When compared with wild‐type mice, VDR‐deficient mice exhibited an approximately 20 fold elevated serum level of TNF‐α 103, underscoring the role of VDR ligands in modulating inflammation.
Under normal circumstances, inducible nitric oxide synthase is not expressed by glia, however in diseases involving the CNS such as MS 104, AD 105 and PD 106 its expression in glia is an inherent aspect of pathology. In ALS, nitric oxide production is involved in the conversion of to ONOO−, a process that occurs at a rate 3× faster than the rate at which normal SOD can catalyze the dismutation of the radical to H2O2 107. This event leads to protein nitration and damage to the cytoskeletal structure and enzymes 108 and can contribute to cell death 109. Indeed, nitric oxide synthase levels are found to be elevated in the G93A mouse spinal cord versus control, an event that parallels gliosis and motor neuron loss 110. Since vitamin D has been shown to inhibit nitric oxide synthase in rodents 111, 112, a similar inhibition in ALS animal models or human patients could help mitigate disease pathology.
Rodent experimental allergic encephalomyelitis (EAE, used as a model of human MS) shares common pathophysiology with ALS. Similar to motor neuron destruction in ALS, oligodendrocytes (the myelin‐producing cells of the CNS) are vulnerable to glutamate excitotoxicity 113. Treatment with glutamate receptor antagonists in this model increased oligodendrocyte survival and decreased markers for axonal degeneration 108, 109 which correlated with improved EAE rodent disease score 115. Furthermore, glutamate antagonists in this model reduced ventral horn motor neuron loss; neurons of central importance in ALS pathology 114. It follows that Riluzole (the only established and moderately effective drug‐based treatment for ALS) is also effective in reducing inflammation, demyelenation, axonal damage, and overall disease severity in rodent EAE 110. Vitamin D treatment may follow a similar mechanism, since in vitro administration also protected rodent cortical neurons against glutamate excitotoxicity 117, 118.
In vivo, dietary administration of 20 ng 1,25(OH)2D3 1 day before EAE disease induction (35–56 days of age) fully prevented onset of disease, whereas all control mice fed regular chow became paralyzed in both fore and hind limbs. In the same study, a 300 ng intraparietal injection of 1,25(OH)2D3 at the first sign of symptoms (limp tail) 10 days after myelin basic protein immunization (45–66 days of age) halted the advancement of the disease for the remainder of the observation period (approximately 30 days) 13, whereas the controls developed paralysis in both fore and hind limbs. To test if the protective effect is reversible, 1,25(OH)2D3 was removed from half of the vitamin D‐treated mice at age 63–84 days, thus creating three different groups: (1) mice maintained on 1,25(OH)2D3 throughout the study, (2) mice temporarily provided with, then restricted of, 1,25(OH)2D3, and (3) mice fed a diet devoid of 1,25(OH)2D3 throughout the study. Group 3 experienced more severe signs of disease as compared to the other two groups, however, group 2 eventually caught up with group 3 in disease severity 10 days post‐1,25(OH)2D3 withdrawal. This strongly establishes that 1,25(OH)2D3 supplementation interferes with EAE.
Vitamin D also has direct antioxidant effects in vivo. Injection of 0.6 pmol 1,25(OH)2D3 into rat substantia nigra reduced zinc‐induced lipid peroxidation and dopamine loss by approximately 20% and 33%, respectively, after 7 days versus zinc alone 6. As well, 1,25(OH)2D3 reduced zinc‐induced substantia nigra apoptosis as evidenced by significantly reduced presence of cytosolic cytochrome C. 1,25(OH)2D3 pretreatment for 15 days (i.p. 5 IU/g b.wt./day) in diabetic rats increased the enzyme activity of liver and kidney catalase, glutathione peroxidase, and SOD1 by approximately 2–4.4 fold, while simultaneously decreasing lipid peroxidation as indicated by thiobarbituric acid reactive substances by 40–46% versus controls, indicating a reduction in oxidative stress‐induced damage 119.
Muscle
G93A mice transgenically overexpress the mutant human SOD1 gene and follow the same disease pattern as human ALS clinically and neuropathologically 120, 121, and is thus the most widely used animal model of ALS. We have previously demonstrated that dietary D3 supplementation at 10‐fold (10 IU D3/g feed) the adequate intake (AI, 1 IU D3/g feed) delays the decline in paw grip endurance and motor performance by 7% and 22%, respectively, versus the AI in the transgenic G93A mouse model for ALS 21. In a later blinded study, dietary D3 at 50‐fold the AI (50 IU D3/g feed) delayed the decline in paw grip endurance by 12% versus the AI 22. Alternatively, D3 restriction (0.025 IU D3/g feed) decreased cumulative scores for paw grip endurance and motor performance postdisease onset by 23% and 18%, respectively, versus control 23. Complete analysis of the mouse skeletal muscle tissue to elucidate what molecular aspects are regulated by this vitamin D supplementation will be forthcoming 122, 123, 124, 125. Despite the observed improvements in functional ability in the two supplementation studies, there were no significant differences in age at disease onset, duration of disease progression or lifespan. However, it is of note that the AI mice (the control mice) were likely to be consuming D3 at levels considerably above what is truly adequate 21, 22.
In vitro, mouse skeletal muscle cells treated with 100 nM 1,25(OH)2D3 exhibited increased expression and nuclear translocation of the VDR and decreased cell proliferation versus placebo. 1,25(OH)2D3 treatment also promoted myogenic differentiation by increasing IGF‐II and follistatin expression, while decreasing myostatin expression; the only known biological inhibitor of muscle mass 126. In other studies, cultured skeletal and cardiac muscle cells demonstrate increased calcium uptake following exposure to physiological concentrations of 25(OH)D3 or 1,25(OH)2D3 127, 128. Additionally, 1,25(OH)2D3 treatment at physiological concentrations elevated cell density and fusion in chick skeletal muscle cell culture, indicating a role for vitamin D in muscle cell proliferation and differentiation 129. The improved functional capacity in G93A mice 21, 22, as well as the improved musculoskeletal function and reduction in falls observed in human studies [51, 52, 53, 54, 55, 56, 57,60,61] following D3 supplementation may also be due to muscle‐specific mechanisms involving contractile protein synthesis and energy homeostasis. In D3‐deficient rats, a single oral dose of 400 IU D3 significantly increased muscle leucine incorporation (a measure of muscle protein synthesis) at 7 h compared with untreated controls 130. Intravenous injection of 0.4 μg 25(OH)D3 significantly increased intramuscular leucine concentrations at 4 h, whereas removal of the kidneys [and therefore the ability to renally convert 25(OH)D3 to 1,25(OH)2D3] did not abolish this effect 130, suggesting a direct role of 25(OH)D3 independent of 1,25(OH)2D3 in muscle function. In evidence, rat epitrochlear muscle had greater leucine incorporation and ATP content in a medium containing 50 nM 25(OH)D3 versus untreated muscle. Administration of D3 at 52,000 nM had no measurable effect, indicating that D3's action in skeletal muscle is conditional upon its conversion to 25(OH)D3, and that 25(OH)D3 is the active genomic vitamin D metabolite in skeletal muscle. The authors found no measureable effect of 1.25 nM 1,25(OH)2D3 on muscle amino acid incorporation or ATP content versus untreated muscle 130. In support, hatchling chicks fed a D3‐free diet for 2 week followed by 1 week supplementation with 80 IU/day D3 had greater concentrations of the contractile proteins actin and troponin C compared with chicks maintained on the D3‐free diet 131.
Neurodegeneration and Neuroprotection
In vivo, radiolabelled 1,25(OH)2D3 uptake in mouse spinal cord 3–4 h postinjection (1 or 3.8 ng/g b.wt.) was clearly strongest in the nuclei of large motor neurons of the spinal cord anterior horn, even in animals that received the lower dose 69. This demonstrates the presence of the nuclear VDR particularly in the large motor neurons of the spinal cord, indicating a role for vitamin D in maintaining the health of motor neurons; cells which are destroyed in ALS.
Vitamin D receptor‐knockout mice have significant locomotor and muscular functional impairment but no apparent cognitive dysfunction, in line with human ALS characteristics 132. Recently, in the cuprizone mouse model of MS, high doses of dietary D3 (6.2 and 12.5 IU/g feed) significantly attenuated brain white matter demyelination and microglia activation 133. Rats orally administered 500 IU D3/kg b.wt./day for 12 weeks after surgical peroneal nerve injury exhibited a 71% greater number of axons in the proximal area of injury versus vehicle‐only treated rats. Furthermore, D3‐supplemented rats demonstrated 8% greater proximal and 10% greater distal myelination; assessed using the G‐ratio (defined as the ratio of axon diameter to myelinated fibre outer diameter) 134. Animal cerebral artery ligation involves the over‐release of excitatory amino acids, overinflux of calcium into the cell, oxidative stress, mitochondrial respiratory damage, and programmed cell death 18; pathologic mechanisms shared by ALS. Rats pretreated with 1,25(OH)2D3 for 8 days (i.p., 1 ng/g b.wt./day), but not 4 days, exhibited 2.3‐fold less volume of infarcted brain tissue due to 90 min cerebral artery ligation versus controls 18. This protection can be at least partially explained by the nearly 2‐fold increase in glial‐derived neurotrophic factor (GDNF) endogenous protein expression; a finding confirmed by what has previously been demonstrated in vitro 135. The same group showed that rats lesioned with 6‐hydroxydopamine after being pretreated for 8 days with 1,25(OH)2D3 (1 ng/g b.wt./day) had hypokinesia significantly attenuated 1 month postlesioning. This was evidenced by approximately 35–100% greater locomotor activity versus saline‐treated rats 12. In vitro work showed that 1,25(OH)2D3 pretreatment attenuated H2O2‐induced neuronal cell death by approximately 3.4‐fold versus saline pretreatment. Even in healthy wild‐type rats, 1,25(OH)2D3 administration (i.p., 1 ng/g b.wt./day) for 7 days increased brain GDNF protein expression by 40% versus saline controls 19. Various other studies have demonstrated that 1,25(OH)2D3 acts on cells of the nervous system in vitro to increase synthesis of other neurotrophic factors which promote neuronal survival, growth, development, and maintenance such as, neural growth factor 136, 137, 138, and neurotrophin‐3 139.
Vitamin D may also exert neuroprotective effects through the upregulation of calcium‐binding proteins. Specific groups of motor neurons such as those found in Onuf's nucleus and the oculomotor nerve are resistant to the ALS degeneration observed in other neurons 140. Motor neurons of Onuf's nucleus and the oculomotor nerve are responsible for the bladder/rectal functions and eye movement often preserved in ALS, even in the late stages of the disease 141, 142. Protection in these neurons may be attributed to the greater expression of calbindin and/or parvalbumin versus neurons which are lost early in ALS 135, 142, 144, 145. Spinal cord analysis in G93A mice showed that parvalbumin‐positive anterior horn motor neurons were severely diminished versus controls before the onset of symptoms, whereas calbindin‐positive neurons were mostly preserved 146. During the symptomatic stage, however, parvalbumin and calbindin immunoreactivity was almost completely abolished. In the brain stem, oculomotor and abducens motor neurons which stained parvalbumin‐positive were as well preserved in transgenic mice as in the controls, even at the end‐stage of disease 146. Indeed, G93A mice with enhanced levels of parvalbumin experienced delayed disease onset by 17% and extended survival by 11% versus controls, accompanied by a 33% greater rate of lumbar spinal cord neuronal survival 147.
Vitamin D increases expression of calcium‐binding protein in vivo. Rats fed 20 IU D3/g b.wt./day for 113 days via gastric cannulation exhibited a 50% increased basal ganglia parvalbumin protein expression versus controls (approximately 0.15 IU D3/g b.wt./day), although no significant changes were found in total cortex or total hippocampus 20. These changes occurred despite multiple signs of D3 toxicity in the animals receiving the extremely high dose of D3 20. Separately, rats intracerebroventricularly injected with 1,25(OH)2D3 (80–250 ng) exhibited strong parvalbumin, calbindin, and calretinin protein immunoreactivity in spinal cord motor neurons versus control, with the strongest detection occurring with 100 ng 148.
In addition to the potential for vitamin D in mitigating the sequelae of hyper‐intracellular calcium concentrations in motor neurons in ALS, vitamin D could also mitigate the severity of ALS by attenuating glutamate excitotoxicity‐induced motor neuron death. Chronic 1,25(OH)2D3 treatment of rat cortical neurons provided cellular protection against glutamate excitotoxicity in a dose‐dependent fashion, where 10 and 100 nM 1,25(OH)2D3 allowed for 10% and 30% more neuronal survival, respectively, versus control 117. Furthermore, cells treated with 1,25(OH)2D3 increased the expression of the neuronal markers microtubule associated protein‐2, growth‐associated protein‐43, and synapsin‐1, suggesting a neuroprotective role for 1,25(OH)2D3 117. Separately, 1,25(OH)2D3 at 100 nM protected mouse neocortical and hippocampal neurons from glutamate insult versus controls despite a delay of 6 h after the initiation of an excitotoxic challenge 118.
Limitations
Different diet‐based interventions in rodent models of ALS have yielded varying effects on disease onset, lifespan, and/or functional capacity 149. Unfortunately, successful interventions in rodent models have not translated well to human clinical trials due to poor design, lack of statistical power, as well as the fact that nearly all animal studies commence prior to disease onset whereas clinical trials are initiated at far more advanced stages of disease 149. Thus, it remains to be seen if the beneficial effects of high dose vitamin D supplemtation observed in rodents 21, 22 will translate to their human counterparts.
Future Directions
Future animal research should measure the effect of the D3 supplementation on markers related to mechanisms implicated in ALS pathophysiology. As such, markers of oxidative stress, antioxidant capacity, inflammation, apoptosis, and neuron count should be measured in the brain/spinal cord/skeletal muscle. As well, the quantification of skeletal muscle contractile proteins would be useful to establish the mechanism for the observed improvement in muscle function and motor performance observed following vitamin D supplementation, as well as the decrement in funcational capacity observed following vitamin D restriction, in the high copy G93A transgenic mouse model of ALS 21, 22, 23. Since vitamin D3 toxicity was observed in G93A females 22, protein analysis should be conducted to confirm the absence of 1α‐OHase in skeletal muscle as well as the corresponding 1,25(OH)2D3 and 25(OH)D3 concentrations, which could explain the observed improvement in functional capacity despite overall toxicity. In addition, vitamin D status, calcium concentrations, and intracellular calcium trafficking/buffering capacity should be measured to establish levels at which toxicity is induced in females, to compare these values to those in males, as well as to understand the role of calcium binding proteins in the cytosol, endoplasmic reticulum, and mitochondria in modulating the sequelae of ALS. Also, of importance is the establishment of a dose which is closer to optimal concerning its effects on functional capacity in this model. This should be done using differential doses of D3. Since 50 IU/g feed is likely to be the approximate threshold for D3 toxicity, we would recommend future experimental high vitamin D doses to be lower than this amount. It would also be of value to experimentally explore the effect of 1,25(OH)2D3 and/or noncalcemic vitamin D analogs in this disease model. Most importantly, studies should be conducted to confirm high dose vitamin D safety in ALS patients, as well as to test the efficacy, if any, of vitamin D on the rapid progression of human ALS pathology.
Conclusion
Vitamin D exerts its influence on a wide variety of different physiological processes in both the healthy and diseased states. Vitamin D may be used as an effective therapy in ALS based on the evidence regarding its effect on muscle function, oxidative stress, inflammation, neuroprotection, mitochondrial function, and apoptosis, in vivo in humans and rodents, as well as in vitro. In addition, vitamin D's influence on diseases which share pathophysiological similarities with ALS suggest that vitamin D may also attenuate ALS pathology. This hypothesis warrants testing in randomized, blinded clinical trials. We have previously shown that vitamin D3 at 10 and 50 IU/g feed (approximately 1.7–8.1 IU D3/g b.wt./day) in the G93A mouse attenuated the decline in functional capacity. Furthermore, we have also shown that vitamin D3 restriction (0.004 IU D3/g b.wt./day) worsened functional capacity in the same mouse model. Since it has been shown to effect multiple aspects of ALS pathophysiology, vitamin D is a strong candidate as a therapeutic for ALS.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis 2009;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;364:362. [DOI] [PubMed] [Google Scholar]
- 3. Vucic S, Kiernan MC. Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med 2009;9:255–272. [DOI] [PubMed] [Google Scholar]
- 4. Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1‐mediated familial ALS. Prog Neurobiol 2008;85:94–134. [DOI] [PubMed] [Google Scholar]
- 5. Deng Y, Xu Z, Xu B, et al. The protective effect of riluzole on manganese caused disruption of glutamate‐glutamine cycle in rats. Brain Res 2009;1289:106–117. [DOI] [PubMed] [Google Scholar]
- 6. Lin AM, Chen KB, Chao PL. Antioxidative effect of vitamin D3 on zinc‐induced oxidative stress in CNS. Ann N Y Acad Sci 2005;1053:319–329. [DOI] [PubMed] [Google Scholar]
- 7. Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double‐blind, randomized, placebo‐controlled trial. Am J Clin Nutr 2006;83:754–759. [DOI] [PubMed] [Google Scholar]
- 8. Andjelkovic Z, Vojinovic J, Pejnovic N, et al. Disease modifying and immunomodulatory effects of high dose 1 alpha (OH) D3 in rheumatoid arthritis patients. Clin Exp Rheumatol 1999;17:453–456. [PubMed] [Google Scholar]
- 9. Compston A, Coles A. Multiple sclerosis. Lancet 2002;359:1221–1231. [DOI] [PubMed] [Google Scholar]
- 10. Appel SH, Beers DR, Henkel JS. T cell‐microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol 2010;31:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W, Wang T, Pei Z, et al. Aggregated alpha‐synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 2005;19:533–542. [DOI] [PubMed] [Google Scholar]
- 12. Wang JY, Wu JN, Cherng TL, et al. Vitamin D(3) attenuates 6‐hydroxydopamine‐induced neurotoxicity in rats. Brain Res 2001;904:67–75. [DOI] [PubMed] [Google Scholar]
- 13. Cantorna MT, Hayes CE, DeLuca HF. 1,25‐Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci U S A 1996;93:7861–7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larsson P, Mattsson L, Klareskog L, Johnsson C. A vitamin D analogue (MC 1288) has immunomodulatory properties and suppresses collagen‐induced arthritis (CIA) without causing hypercalcaemia. Clin Exp Immunol 1998;114:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lemire JM, Archer DC. 1,25‐dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest 1991;87:1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cantorna MT, Hayes CE, DeLuca HF. 1,25‐Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J Nutr 1998;128:68–72. [DOI] [PubMed] [Google Scholar]
- 17. Tsuji M, Fujii K, Nakano T. 1 alpha‐hydroxyvitamin D3 inhibits type II collagen‐induced arthritis in rats. FEBS Lett 1994;337:248–250. [DOI] [PubMed] [Google Scholar]
- 18. Wang Y, Chiang YH, Su TP, et al. Vitamin D(3) attenuates cortical infarction induced by middle cerebral arterial ligation in rats. Neuropharmacology 2000;39:873–880. [DOI] [PubMed] [Google Scholar]
- 19. Sanchez B, Lopez‐Martin E, Segura C, Labandeira‐Garcia JL, Perez‐Fernandez R. 1,25‐Dihydroxyvitamin D(3) increases striatal GDNF mRNA and protein expression in adult rats. Brain Res Mol Brain Res 2002;108:143–146. [DOI] [PubMed] [Google Scholar]
- 20. de Viragh PA, Haglid KG, Celio MR. Parvalbumin increases in the caudate putamen of rats with vitamin D hypervitaminosis. Proc Natl Acad Sci U S A 1989;86:3887–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gianforcaro A, Hamadeh MJ. Dietary vitamin D(3) supplementation at 10x the adequate intake improves functional capacity in the G93A transgenic mouse model of ALS, a pilot study. CNS Neurosci Ther 2012;18:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gianforcaro A, Solomon JA, Hamadeh MJ. Vitamin D3 at 50x the adequate intake attenuates functional decline in the G93A mouse model of amyotrophic lateral sclerosis, but is toxic in females. PLoS ONE 2013;7:e30243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Solomon JA, Gianforcaro A, Hamadeh MJ. Vitamin D3 deficiency differentially affects functional and disease outcomes in the G93A mouse model of amyotrophic lateral sclerosis. PLoS ONE 2011;6:e29354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karam C, Barrett MJ, Imperato T, Macgowan DJ, Scelsa S. Vitamin D deficiency and its supplementation in patients with amyotrophic lateral sclerosis. J Clin Neurosci 2013;20:1550–1553. [DOI] [PubMed] [Google Scholar]
- 25. Camu W, Tremblier B, Plassot C, Alphandery S, Salsac C, Pageot N, Juntas‐Morales R, Scamps F, Daures J, Raoul C. Vitamin D confers protection to motoneurons and is a prognostic factor of amyotrophic lateral sclerosis. Neurobiol Aging 2013. doi: 10.1016/j.neurobiolaging.2013.11.005 [DOI] [PubMed] [Google Scholar]
- 26. Hensley K, Mhatre M, Mou S, et al. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A‐SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal 2006;8:2075–2087. [DOI] [PubMed] [Google Scholar]
- 27. Yoshihara T, Ishigaki S, Yamamoto M, et al. Differential expression of inflammation‐ and apoptosis‐related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 2002;80:158–167. [DOI] [PubMed] [Google Scholar]
- 28. Poloni M, Facchetti D, Mai R, et al. Circulating levels of tumour necrosis factor‐alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett 2000;287:211–214. [DOI] [PubMed] [Google Scholar]
- 29. Moreau C, Devos D, Brunaud‐Danel V, et al. Elevated IL‐6 and TNF‐alpha levels in patients with ALS: inflammation or hypoxia? Neurology 2005;65:1958–1960. [DOI] [PubMed] [Google Scholar]
- 30. Kiaei M, Petri S, Kipiani K, et al. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2006;26:2467–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gurney ME. What transgenic mice tell us about neurodegenerative disease. BioEssays 2000;22:297–304. [DOI] [PubMed] [Google Scholar]
- 32. Guegan C, Przedborski S. Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest 2003;111:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol 2007;82:1375–1381. [DOI] [PubMed] [Google Scholar]
- 34. Fang L, Huber‐Abel F, Teuchert M, et al. Linking neuron and skin: matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J Neurol Sci 2009;285:62–66. [DOI] [PubMed] [Google Scholar]
- 35. Lukes A, Mun‐Bryce S, Lukes M, Rosenberg GA. Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol Neurobiol 1999;19:267–284. [DOI] [PubMed] [Google Scholar]
- 36. Beuche W, Yushchenko M, Mader M, Maliszewska M, Felgenhauer K, Weber F. Matrix metalloproteinase‐9 is elevated in serum of patients with amyotrophic lateral sclerosis. NeuroReport 2000;11:3419–3422. [DOI] [PubMed] [Google Scholar]
- 37. Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 1998;21:75–80. [DOI] [PubMed] [Google Scholar]
- 38. Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2001;2:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB‐94. J Cereb Blood Flow Metab 2000;20:1681–1689. [DOI] [PubMed] [Google Scholar]
- 40. Lorenzl S, Albers DS, LeWitt PA, et al. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci 2003;207:71–76. [DOI] [PubMed] [Google Scholar]
- 41. Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol 2009;8:205–216. [DOI] [PubMed] [Google Scholar]
- 42. Zehnder D, Quinkler M, Eardley KS, et al. Reduction of the vitamin D hormonal system in kidney disease is associated with increased renal inflammation. Kidney Int 2008;74:1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khoo AL, Chai LY, Koenen HJ, et al. Regulation of cytokine responses by seasonality of vitamin D status in healthy individuals. Clin Exp Immunol 2011;164:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larcombe L, Orr P, Turner‐Brannen E, Slivinski CR, Nickerson PW, Mookherjee N. Effect of vitamin D supplementation on Mycobacterium tuberculosis‐induced innate immune responses in a Canadian Dene First Nations cohort. PLoS ONE 2012;7:e40692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grossmann RE, Zughaier SM, Liu S, Lyles RH, Tangpricha V. Impact of vitamin D supplementation on markers of inflammation in adults with cystic fibrosis hospitalized for a pulmonary exacerbation. Eur J Clin Nutr 2012;66:1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peterson CA, Heffernan ME. Serum tumor necrosis factor‐alpha concentrations are negatively correlated with serum 25(OH)D concentrations in healthy women. J Inflamm (Lond) 2008;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anderson JL, May HT, Horne BD, et al. Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am J Cardiol 2010;106:963–968. [DOI] [PubMed] [Google Scholar]
- 48. Kroger H, Penttila IM, Alhava EM. Low serum vitamin D metabolites in women with rheumatoid arthritis. Scand J Rheumatol 1993;22:172–177. [DOI] [PubMed] [Google Scholar]
- 49. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med 2001;344:1688–1700. [DOI] [PubMed] [Google Scholar]
- 50. Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 2008;29:726–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bischoff HA, Borchers M, Gudat F, et al. In situ detection of 1,25‐dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem J 2001;33:19–24. [DOI] [PubMed] [Google Scholar]
- 52. Tieland M, Brouwer‐Brolsma EM, Nienaber‐Rousseau C, van Loon LJ, De Groot LC. Low vitamin D status is associated with reduced muscle mass and impaired physical performance in frail elderly people. Eur J Clin Nutr 2013;67:1050–1055. [DOI] [PubMed] [Google Scholar]
- 53. Barker T, Henriksen VT, Martins TB, et al. Higher serum 25‐hydroxyvitamin D concentrations associate with a faster recovery of skeletal muscle strength after muscular injury. Nutrients 2013;5:1253–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bischoff‐Ferrari HA, Dietrich T, Orav EJ, et al. Higher 25‐hydroxyvitamin D concentrations are associated with better lower‐extremity function in both active and inactive persons aged > or =60 y. Am J Clin Nutr 2004;80:752–758. [DOI] [PubMed] [Google Scholar]
- 55. Bischoff HA, Stahelin HB, Dick W, et al. Effects of vitamin D and calcium supplementation on falls: a randomized controlled trial. J Bone Miner Res 2003;18:343–351. [DOI] [PubMed] [Google Scholar]
- 56. Pfeifer M, Begerow B, Minne HW, Suppan K, Fahrleitner‐Pammer A, Dobnig H. Effects of a long‐term vitamin D and calcium supplementation on falls and parameters of muscle function in community‐dwelling older individuals. Osteoporos Int 2009;20:315–322. [DOI] [PubMed] [Google Scholar]
- 57. Broe KE, Chen TC, Weinberg J, Bischoff‐Ferrari HA, Holick MF, Kiel DP. A higher dose of vitamin D reduces the risk of falls in nursing home residents: a randomized, multiple‐dose study. J Am Geriatr Soc 2007;55:234–239. [DOI] [PubMed] [Google Scholar]
- 58. Pfeifer M, Begerow B, Minne HW, Abrams C, Nachtigall D, Hansen C. Effects of a short‐term vitamin D and calcium supplementation on body sway and secondary hyperparathyroidism in elderly women. J Bone Miner Res 2000;15:1113–1118. [DOI] [PubMed] [Google Scholar]
- 59. Knutsen KV, Madar AA, Lagerlov P, Brekke M, Raastad T, Stene LC, et al. Does vitamin D improve muscle strength in adults? A randomized, double‐blind, placebo‐controlled trial among ethnic minorities in Norway. J Clin Endocrinol Metab 2013. doi: 10.1210/jc.2013-2647. [DOI] [PubMed] [Google Scholar]
- 60. Verhaar HJ, Samson MM, Jansen PA, de Vreede PL, Manten JW, Duursma SA. Muscle strength, functional mobility and vitamin D in older women. Aging (Milano) 2000;12:455–460. [DOI] [PubMed] [Google Scholar]
- 61. Gordon PL, Sakkas GK, Doyle JW, Shubert T, Johansen KL. Relationship between vitamin D and muscle size and strength in patients on hemodialysis. J Ren Nutr 2007;17:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ceglia L, Niramitmahapanya S, Morais MD, Rivas DA, Harris SS, Bischoff‐Ferrari H, et al. A randomized study on the effect of vitamin D3 supplementation on skeletal muscle morphology and vitamin D receptor concentration in older women. J Clin Endocrinol Metab 2013. doi: 10.1210/jc.2013-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karam C, Barrett MJ, Imperato T, Macgowan DJ, Scelsa S. Vitamin D deficiency and its supplementation in patients with amyotrophic lateral sclerosis. J Clin Neurosci 2013;20:1550–1553. [DOI] [PubMed] [Google Scholar]
- 64. Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal 2009;11:1587–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goodall EF, Morrison KE. Amyotrophic lateral sclerosis (motor neuron disease): proposed mechanisms and pathways to treatment. Expert Rev Mol Med 2006;8:1–22. [DOI] [PubMed] [Google Scholar]
- 66. Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta 2006;1762:1068–1082. [DOI] [PubMed] [Google Scholar]
- 67. Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic Biol Med 2005;38:687–697. [DOI] [PubMed] [Google Scholar]
- 68. Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1 alpha‐hydroxylase in human brain. J Chem Neuroanat 2005;29:21–30. [DOI] [PubMed] [Google Scholar]
- 69. Stumpf WE, Clark SA, O'Brien LP, Reid FA. 1,25(OH)2 vitamin D3 sites of action in spinal cord and sensory ganglion. Anat Embryol (Berl) 1988;177:307–310. [DOI] [PubMed] [Google Scholar]
- 70. Clemens TL, Garrett KP, Zhou XY, Pike JW, Haussler MR, Dempster DW. Immunocytochemical localization of the 1,25‐dihydroxyvitamin D3 receptor in target cells. Endocrinology 1988;122:1224–1230. [DOI] [PubMed] [Google Scholar]
- 71. Walbert T, Jirikowski GF, Prufer K. Distribution of 1,25‐dihydroxyvitamin D3 receptor immunoreactivity in the limbic system of the rat. Horm Metab Res 2001;33:525–531. [DOI] [PubMed] [Google Scholar]
- 72. Sato Y, Asoh T, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in elderly women with Alzheimer's disease. Bone 1998;23:555–557. [DOI] [PubMed] [Google Scholar]
- 73. Sutherland MK, Somerville MJ, Yoong LK, Bergeron C, Haussler MR, McLachlan DR. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin‐28k mRNA levels. Brain Res Mol Brain Res 1992;13:239–250. [DOI] [PubMed] [Google Scholar]
- 74. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer's disease: a 7‐year follow‐up. J Gerontol A Biol Sci Med Sci 2012;67:1205–1211. [DOI] [PubMed] [Google Scholar]
- 75. Llewellyn DJ, Lang IA, Langa KM, et al. Vitamin D and risk of cognitive decline in elderly persons. Arch Intern Med 2010;170:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 2008;79:368–376. [DOI] [PubMed] [Google Scholar]
- 77. Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 1993;50:30–36. [DOI] [PubMed] [Google Scholar]
- 78. Zhao W, Beers DR, Henkel JS, et al. Extracellular mutant SOD1 induces microglial‐mediated motoneuron injury. Glia 2010;58:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kienzl E, Puchinger L, Jellinger K, Linert W, Stachelberger H, Jameson RF. The role of transition metals in the pathogenesis of Parkinson's disease. J Neurol Sci 1995;134(Suppl):69–78. [DOI] [PubMed] [Google Scholar]
- 80. Sofic E, Riederer P, Heinsen H, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 1988;74:199–205. [DOI] [PubMed] [Google Scholar]
- 81. Ilieva EV, Ayala V, Jove M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007;130(Pt 12):3111–3123. [DOI] [PubMed] [Google Scholar]
- 82. Jung C, Higgins CM, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem 2002;83:535–545. [DOI] [PubMed] [Google Scholar]
- 83. Wiedemann FR, Winkler K, Kuznetsov AV, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci 1998;156:65–72. [DOI] [PubMed] [Google Scholar]
- 84. Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989;1:1269. [DOI] [PubMed] [Google Scholar]
- 85. Schapira AH, Gu M, Taanman JW, et al. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann Neurol 1998;44(3 Suppl 1):S89–S98. [DOI] [PubMed] [Google Scholar]
- 86. Mann VM, Cooper JM, Daniel SE, et al. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol 1994;36:876–881. [DOI] [PubMed] [Google Scholar]
- 87. Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem 1993;61:1191–1206. [DOI] [PubMed] [Google Scholar]
- 88. Derex L, Trouillas P. Reversible parkinsonism, hypophosphoremia, and hypocalcemia under vitamin D therapy. Mov Disord 1997;12:612–613. [DOI] [PubMed] [Google Scholar]
- 89. Lux WE, Kurtzke JF. Is Parkinson's disease acquired? Evidence from a geographic comparison with multiple sclerosis Neurology 1987;37:467–471. [DOI] [PubMed] [Google Scholar]
- 90. Wermuth L, von Weitzel‐Mudersbach P, Jeune B. A two‐fold difference in the age‐adjusted prevalences of Parkinson's disease between the island of Als and the Faroe Islands. Eur J Neurol 2000;7:655–660. [DOI] [PubMed] [Google Scholar]
- 91. Wermuth L, Pakkenberg H, Jeune B. High age‐adjusted prevalence of Parkinson's disease among Inuits in Greenland. Neurology 2002;58:1422–1425. [DOI] [PubMed] [Google Scholar]
- 92. Wermuth L, Bech S, Petersen MS, Joensen P, Weihe P, Grandjean P. Prevalence and incidence of Parkinson's disease in The Faroe Islands. Acta Neurol Scand 2008;118:126–131. [DOI] [PubMed] [Google Scholar]
- 93. Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol 2008;65:1348–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sato Y, Kikuyama M, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in Parkinson's disease. Neurology 1997;49:1273–1278. [DOI] [PubMed] [Google Scholar]
- 95. Sato Y, Honda Y, Iwamoto J, Kanoko T, Satoh K. Abnormal bone and calcium metabolism in immobilized Parkinson's disease patients. Mov Disord 2005;20:1598–1603. [DOI] [PubMed] [Google Scholar]
- 96. Rosenbaum RB. Understanding Parkinson's disease: a personal and professional view. Westport, CT: Praeger, 2006. [Google Scholar]
- 97. Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol 2011;67:808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Butler MW, Burt A, Edwards TL, et al. Vitamin D receptor gene as a candidate gene for Parkinson disease. Ann Hum Genet 2011;75:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dobson R, Giovannoni G, Ramagopalan S. The month of birth effect in multiple sclerosis: systematic review, meta‐analysis and effect of latitude. J Neurol Neurosurg Psychiatry 2013;84:427–432. [DOI] [PubMed] [Google Scholar]
- 100. Munger KL, Zhang SM, O'Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology 2004;62:60–65. [DOI] [PubMed] [Google Scholar]
- 101. Balden R, Selvamani A, Sohrabji F. Vitamin D deficiency exacerbates experimental stroke injury and dysregulates ischemia‐induced inflammation in adult rats. Endocrinology 2012;153:2420–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Briones TL, Darwish H. Vitamin D mitigates age‐related cognitive decline through the modulation of pro‐inflammatory state and decrease in amyloid burden. J Neuroinflammation 2012;9:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zwerina K, Baum W, Axmann R, et al. Vitamin D receptor regulates TNF‐mediated arthritis. Ann Rheum Dis 2011;70:1122–1129. [DOI] [PubMed] [Google Scholar]
- 104. Bagasra O, Michaels FH, Zheng YM, et al. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc Natl Acad Sci U S A 1995;92:12041–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vodovotz Y, Lucia MS, Flanders KC, et al. Inducible nitric oxide synthase in tangle‐bearing neurons of patients with Alzheimer's disease. J Exp Med 1996;184:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hunot S, Boissiere F, Faucheux B, et al. Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience 1996;72:355–363. [DOI] [PubMed] [Google Scholar]
- 107. Beckman JS. Peroxynitrite versus hydroxyl radical: the role of nitric oxide in superoxide‐dependent cerebral injury. Ann N Y Acad Sci 1994;738:69–75. [DOI] [PubMed] [Google Scholar]
- 108. Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat 1996;10:249–258. [DOI] [PubMed] [Google Scholar]
- 109. Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature 1993;364:584. [DOI] [PubMed] [Google Scholar]
- 110. Liang X, Wang Q, Shi J, et al. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol 2008;64:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1,25‐Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res Mol Brain Res 1997;45:255–267. [DOI] [PubMed] [Google Scholar]
- 112. Garcion E, Sindji L, Montero‐Menei C, Andre C, Brachet P, Darcy F. Expression of inducible nitric oxide synthase during rat brain inflammation: regulation by 1,25‐dihydroxyvitamin D3. Glia 1998;22:282–294. [PubMed] [Google Scholar]
- 113. McDonald JW, Levine JM, Qu Y. Multiple classes of the oligodendrocyte lineage are highly vulnerable to excitotoxicity. NeuroReport 1998;9:2757–2762. [DOI] [PubMed] [Google Scholar]
- 114. Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000;6:62–66. [DOI] [PubMed] [Google Scholar]
- 115. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000;6:67–70. [DOI] [PubMed] [Google Scholar]
- 116. Gilgun‐Sherki Y, Panet H, Melamed E, Offen D. Riluzole suppresses experimental autoimmune encephalomyelitis: implications for the treatment of multiple sclerosis. Brain Res 2003;989:196–204. [DOI] [PubMed] [Google Scholar]
- 117. Taniura H, Ito M, Sanada N, et al. Chronic vitamin D3 treatment protects against neurotoxicity by glutamate in association with upregulation of vitamin D receptor mRNA expression in cultured rat cortical neurons. J Neurosci Res 2006;83:1179–1189. [DOI] [PubMed] [Google Scholar]
- 118. Kajta M, Makarewicz D, Zieminska E, et al. Neuroprotection by co‐treatment and post‐treating with calcitriol following the ischemic and excitotoxic insult in vivo and in vitro. Neurochem Int 2009;55:265–274. [DOI] [PubMed] [Google Scholar]
- 119. Hamden K, Carreau S, Jamoussi K, et al. 1Alpha,25 dihydroxyvitamin D3: therapeutic and preventive effects against oxidative stress, hepatic, pancreatic and renal injury in alloxan‐induced diabetes in rats. J Nutr Sci Vitaminol (Tokyo) 2009;55:215–222. [DOI] [PubMed] [Google Scholar]
- 120. Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994;264:1772–1775. [DOI] [PubMed] [Google Scholar]
- 121. Gurney ME. Transgenic‐mouse model of amyotrophic lateral sclerosis. N Engl J Med 1994;331:1721–1722. [DOI] [PubMed] [Google Scholar]
- 122. Parkhomenko EA, Milionis A, Gianforcaro A, Solomon JA, Hamadeh MJ. Dietary vitamin D3 at 50x the adequate intake increases apoptosis in the quadriceps of the female G93A mouse model of amyotrophic lateral sclerosis: a pilot study. FASEB J 2012;26:255.7 (abst.). [Google Scholar]
- 123. Shahsavar S, Taheri‐Shalmani S, Solomon JA, Gianforcaro A, Hamadeh MJ. Dietary D3 supplementation at 50x the AI increases contractile protein content and improves mitochondrial oxidative capacity in the transgenic G93A mouse model of ALS: a pilot study. FASEB J 2013;27:644.2 (abst.). [Google Scholar]
- 124. Taheri‐Shalmani S, Shahsavar S, Gianforcaro A, Solomon JA, Hamadeh MJ. Dietary vitamin D3 supplementation at 50x the adequate intake decreases calbindin d28k and endoplasmic reticulum stress and increases apoptosis, suggesting toxicity, in the female transgenic G93A mouse model of amyotrophic lateral sclerosis. FASEB J 2013;27:644.1 (abst.). [Google Scholar]
- 125. Milionis A, Parkhomenko EA, Solomon JA, Gianforcaro A, Hamadeh MJ. Dietary vitamin D3 restriction differentially alters quadriceps contractile proteins in both sexes in the transgenic G93A mouse model of amyotrophic lateral sclerosis: a pilot study. FASEB J 2012;26:255.8 (abst.). [Google Scholar]
- 126. Garcia LA, King KK, Ferrini MG, Norris KC, Artaza JN. 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011;152:2976–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. de Boland AR, Boland R. In vitro cellular muscle calcium metabolism. Characterization of effects of 1,25‐dihydroxy‐vitamin D3 and 25‐hydroxy‐vitamin D3. Z Naturforsch C 1985;40:102–108. [DOI] [PubMed] [Google Scholar]
- 128. Walters MR, Ilenchuk TT, Claycomb WC. 1,25‐Dihydroxyvitamin D3 stimulates 45Ca2+ uptake by cultured adult rat ventricular cardiac muscle cells. J Biol Chem 1987;262:2536–2541. [PubMed] [Google Scholar]
- 129. Giuliani DL, Boland RL. Effects of vitamin D3 metabolites on calcium fluxes in intact chicken skeletal muscle and myoblasts cultured in vitro. Calcif Tissue Int 1984;36:200–205. [DOI] [PubMed] [Google Scholar]
- 130. Birge SJ, Haddad JG. 25‐hydroxycholecalciferol stimulation of muscle metabolism. J Clin Invest 1975;56:1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. de Boland AR, Albornoz LE, Boland R. The effect of cholecalciferol in vivo on proteins and lipids of skeletal muscle from rachitic chicks. Calcif Tissue Int 1983;35:798–805. [DOI] [PubMed] [Google Scholar]
- 132. Burne TH, McGrath JJ, Eyles DW, Mackay‐Sim A. Behavioural characterization of vitamin D receptor knockout mice. Behav Brain Res 2005;157:299–308. [DOI] [PubMed] [Google Scholar]
- 133. Wergeland S, Torkildsen O, Myhr KM, Aksnes L, Mork SJ, Bo L. Dietary vitamin D3 supplements reduce demyelination in the cuprizone model. PLoS ONE 2011;6:e26262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chabas JF, Stephan D, Marqueste T, et al. Cholecalciferol (vitamin D(3)) improves myelination and recovery after nerve injury. PLoS ONE 2013;8:e65034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Verity AN, Wyatt TL, Lee W, et al. Differential regulation of glial cell line‐derived neurotrophic factor (GDNF) expression in human neuroblastoma and glioblastoma cell lines. J Neurosci Res 1999;55:187–197. [DOI] [PubMed] [Google Scholar]
- 136. Neveu I, Naveilhan P, Jehan F, et al. 1,25‐dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res Mol Brain Res 1994;24:70–76. [DOI] [PubMed] [Google Scholar]
- 137. Cornet A, Baudet C, Neveu I, Baron‐Van Evercooren A, Brachet P, Naveilhan P. 1,25‐Dihydroxyvitamin D3 regulates the expression of VDR and NGF gene in Schwann cells in vitro. J Neurosci Res 1998;53:742–746. [DOI] [PubMed] [Google Scholar]
- 138. Saporito MS, Brown ER, Hartpence KC, Wilcox HM, Vaught JL, Carswell S. Chronic 1,25‐dihydroxyvitamin D3‐mediated induction of nerve growth factor mRNA and protein in L929 fibroblasts and in adult rat brain. Brain Res 1994;633:189–196. [DOI] [PubMed] [Google Scholar]
- 139. Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1,25‐dihydroxyvitamin D3 regulates NT‐3, NT‐4 but not BDNF mRNA in astrocytes. NeuroReport 1994;6:124–126. [DOI] [PubMed] [Google Scholar]
- 140. Kihira T, Yoshida S, Yoshimasu F, Wakayama I, Yase Y. Involvement of Onuf's nucleus in amyotrophic lateral sclerosis. J Neurol Sci 1997;147:81–88. [DOI] [PubMed] [Google Scholar]
- 141. Mannen T, Iwata M, Toyokura Y, Nagashima K. Preservation of a certain motoneurone group of the sacral cord in amyotrophic lateral sclerosis: its clinical significance. J Neurol Neurosurg Psychiatry 1977;40:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium‐binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol 1994;36:846–858. [DOI] [PubMed] [Google Scholar]
- 143. Ince P, Stout N, Shaw P, et al. Parvalbumin and calbindin D‐28k in the human motor system and in motor neuron disease. Neuropathol Appl Neurobiol 1993;19:291–299. [DOI] [PubMed] [Google Scholar]
- 144. Elliott JL, Snider WD. Parvalbumin is a marker of ALS‐resistant motor neurons. NeuroReport 1995;6:449–452. [DOI] [PubMed] [Google Scholar]
- 145. Morrison BM, Gordon JW, Ripps ME, Morrison JH. Quantitative immunocytochemical analysis of the spinal cord in G86R superoxide dismutase transgenic mice: neurochemical correlates of selective vulnerability. J Comp Neurol 1996;373:619–631. [DOI] [PubMed] [Google Scholar]
- 146. Sasaki S, Warita H, Komori T, Murakami T, Abe K, Iwata M. Parvalbumin and calbindin D‐28k immunoreactivity in transgenic mice with a G93A mutant SOD1 gene. Brain Res 2006;1083:196–203. [DOI] [PubMed] [Google Scholar]
- 147. Beers DR, Ho BK, Siklos L, et al. Parvalbumin overexpression alters immune‐mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem 2001;79:499–509. [DOI] [PubMed] [Google Scholar]
- 148. Alexianu ME, Robbins E, Carswell S, Appel SH. 1Alpha, 25 dihydroxyvitamin D3‐dependent up‐regulation of calcium‐binding proteins in motoneuron cells. J Neurosci Res 1998;51:58–66. [DOI] [PubMed] [Google Scholar]
- 149. Patel BP, Hamadeh MJ. Nutritional and exercise‐based interventions in the treatment of amyotrophic lateral sclerosis. Clin Nutr 2009;28:604–617. [DOI] [PubMed] [Google Scholar]
- 150. Bruijn LI, Becher MW, Lee MK, et al. ALS‐linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1‐containing inclusions. Neuron 1997;18:327–338. [DOI] [PubMed] [Google Scholar]
- 151. Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine‐nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001;29:160–165. [DOI] [PubMed] [Google Scholar]
- 152. Hand CK, Khoris J, Salachas F, Gros‐Louis F, Lopes AA, Mayeux‐Portas V, et al. A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q. Am J Hum Genet 2002;70:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sapp PC, Hosler BA, McKenna‐Yasek D, Chin W, Gann A, Genise H, et al. Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am J Hum Genet 2003;73:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004;74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nishimura AL, Mitne‐Neto M, Silva HC, et al. A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004;75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Marques VD, Barreira AA, Davis MB, et al. Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve 2006;34:731–739. [DOI] [PubMed] [Google Scholar]
- 157. Conforti FL, Sprovieri T, Mazzei R, et al. A novel Angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul Disord 2008;18:68–70. [DOI] [PubMed] [Google Scholar]
- 158. Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 2006;38:411–413. [DOI] [PubMed] [Google Scholar]
- 159. Sreedharan J, Blair IP, Tripathi VB, et al. TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 161. Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet 2003;33:455–456. [DOI] [PubMed] [Google Scholar]
- 163. Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010;133(Pt 2):591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 2011;477:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Al‐Saif A, Al‐Mohanna F, Bohlega S. A mutation in sigma‐1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 2011;70:913–919. [DOI] [PubMed] [Google Scholar]
- 166. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gros‐Louis F, Lariviere R, Gowing G, et al. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem 2004;279:45951–45956. [DOI] [PubMed] [Google Scholar]
- 168. Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Couthouis J, Hart MP, Erion R, et al. Evaluating the role of the FUS/TLS‐related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010;465:223–226. [DOI] [PubMed] [Google Scholar]
- 171. Elden AC, Kim HJ, Hart MP, et al. Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet 1994;3:1757–1761. [DOI] [PubMed] [Google Scholar]
- 173. Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006;67:1074–1077. [DOI] [PubMed] [Google Scholar]
- 174. Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG 4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 2009;84:85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mitchell J, Paul P, Chen HJ, et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D‐amino acid oxidase. Proc Natl Acad Sci U S A 2010;107:7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wu CH, Fallini C, Ticozzi N, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012;488:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011;68:1440–1446. [DOI] [PubMed] [Google Scholar]
- 178. Couthouis J, Hart MP, Shorter J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A 2011;108:20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]