

Summary
Background and purpose
Our previous studies have demonstrated adenosine triphosphate‐sensitive potassium channel (KATP channel) openers could protect against inflammatory response in brain disease, but little is known about the mechanisms involved in KATP channel openers inhibiting neuroinflammation.
Methods and results
In the present study, we found that oxygen–glucose deprivation (OGD) resulted in BV‐2 cells activation, significantly increased tumor necrosis factor‐alpha and interleukin‐1beta (IL‐1β) levels, accompanied by downregulating Kir6.1 subunit. Pretreatment with nicorandil, a KATP channel opener, could attenuate OGD‐induced BV‐2 cells activation and inhibit pro‐inflammatory factors release. Further study demonstrated that OGD activated Toll‐like receptor‐4 (TLR4) signaling pathway and NOD‐like receptor pyrin domain containing three inflammasome, thereby increased IL‐1β production. Pretreatment with nicorandil could reverse the two pathways involved in IL‐1β production.
Conclusions
Our findings reveal that KATP channel openers could protect against OGD‐induced neuroinflammation via inhibiting inflammasome activation and TLR4 signal transduction.
Keywords: ATP‐sensitive potassium channel, Inflammasome, Neuroinflammation, Nicorandil, Oxygen–glucose deprivation
Introduction
Stroke is the third leading cause of death, which is only next to the heart disease and cancer. At present, the only globally approved treatment for stroke is tissue plasminogen activator (tPA). However, this treatment only suits for <5% patients, owing to its narrow time‐dependent therapy and complications 1. It is urgent to reveal the pathophysiological mechanisms of stroke, which is essential for drug development. Increasing evidence suggests that post‐ischemic inflammation is involved in the progress of ischemic cascade, from acute brain damage to tissue repair 2. At the early stage of ischemic stroke onset, microglia is activated rapidly and produces lots of pro‐inflammatory mediators, which trigger parenchymal inflammation and recruit infiltrating immune cells from peripheral system 3. Microglia is the resident immune cells in the brain, which is also the major resource of inflammatory cytokines at the initial stage of brain ischemia 4. Despite the fact that there are intensive studies of post‐ischemic inflammation, the molecular mechanisms that activate resident microglia and drive the production of inflammatory cytokines remain unclear. Interleukin‐1beta (IL‐1β) is one of the most potent mediators in inflammation, which is produced from an inactive precursor (pro‐IL‐1β) by immune cells such as macrophages or microglia during diseases or after injury 5, 6. Caspase‐1 is the key protease required for cleaving pro‐IL‐1β into IL‐1β, its activation is controlled by its recruitment to multimolecular scaffolds named inflammasome 7. NOD‐like receptor pyrin domain containing three (NLRP3) is implicated as a sensor of sterile injury 8. Whether production of IL‐1β in response to oxygen–glucose deprivation (OGD) is due to inflammasome activation remains unclear.
Adenosine triphosphate (ATP)‐sensitive potassium channels (KATP channels) couple cellular energetic metabolism with electric activity 9. Accumulating evidence indicates Kir6.1‐contained KATP channels mainly express in glia 10, 11, 12. Our previous study has demonstrated that Kir6.1‐contained KATP channels are involved in brain ischemic injury, Kir6.1 knockdown aggravates ischemic injury via augmenting reactive glia and inflammatory responses 13. In addition, other studies also elucidate that Kir6.1‐contained KATP channels are pivotal targets for regulating inflammatory responses, which is implicated in disease process 14, 15. But little is known about whether and how KATP channels are concerned with post‐ischemic inflammation and microglial responses in stroke.
The present study was to determine the impacts of KATP channel opener nicorandil on OGD‐induced inflammatory responses in BV‐2 cell lines and clarify the involved mechanisms. Our work showed that OGD could induce production of IL‐1β, which was involved with upregulation of Toll‐like receptor 4 (TLR4), NLRP3, and cleaved caspase‐1. However, pretreatment with nicorandil could reverse the above phenomena and decrease production of IL‐1β. These results suggest that KATP channels are involved in regulating the production of IL‐1β in microglia after OGD injury.
Materials and Methods
Cell Cultures and Treatment
BV‐2 cells were generated by immortalizing primary murine microglial cells with a v‐raf/v‐myc oncogene carrying retrovirus, as seen in previous studies 14. BV‐2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing fetal bovine serum (10%) (GIBCO, Carlsbad, CA, USA), penicillin (100 U/mL), and streptomycin (100 mg/L). The cells were gently suspended in DMEM and plated in six‐well plates at a density of 1 × 105 cells/mL. In all experiments, cells were pretreated with nicorandil (a KATP channel opener) (TOCRIS, Bristol, UK) and PNU37883 (a selective Kir6.1 blocker) (TOCRIS) for 1 h before OGD. Control samples contained the same volume of vehicle. The experimental groups were divided as follows: (1) control, (2) PNU37883 (20 μM), (3) nicorandil (10 μM), (4) OGD, (5) OGD + PNU37883 (20 μM), and (6) OGD + nicorandil (10 μM).
Oxygen–Glucose Deprivation
After washing twice, BV‐2 cells were immersed in 1 mL deoxygenated custom DMEM medium without glucose (GIBCO), oxygen removed, and the six‐well plates were placed inside an incubator (Thermo scientific, Waltham, MA, USA) for 1 h with a pre‐mixed gas (1% O2, 94% N2, 5% CO2). Control cultures were treated similarly but with normoxic DMEM supplemented with 3 mM D‐glucose (GIBCO) in a normoxic incubator.
Cell Viability Analysis
The 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay was used to detect cell viability as Zhou et al. 11 described before. BV‐2 cells were cultured in 96‐well plates at a density of 5 × 104 cells per well. After different time‐course (0.5 h/1 h/1.5 h) of OGD and reoxygenation 24 h, the MTT was dissolved in DMEM medium as a final concentration of 0.5 mg/mL and incubated at 37°C for 4 h. Then, we replaced the medium with 150 μL dimethyl sulfoxide (DMSO) to dissolve the formazan product. The absorbance of each well was obtained using a Dynatech MR5000 plate counter (Dynatech Corp., Burlington, MA, USA) at a test wavelength of 570 nm with a reference wavelength of 630 nm. All data are expressed as mean ± SEM of three independent experiments.
Lactate Dehydrogenase Release
After different time‐course (0.5 h/1 h/1.5 h) of OGD and reoxygenation 24 h, lactate dehydrogenase (LDH) release in culture medium was measured by using the LDH diagnostic kit (Jiancheng Bioengineering, Nanjing, China), according to manufacturer's instructions. LDH activity was calculated by measuring absorbance at 490 nm. All the detail experimental procedures were as described before by Xie et al. 16.
Western Blotting
The cytosolic protein in samples was extracted according to the KEYGEN protein extraction kit (KeyGen Biotech. Co. Ltd., Nanjing, China). Protein concentrations were determined using the Micro BCA Kit (Pierce Biotechnology, Rockford, IL, USA). The supernatants (50 μg protein) were separated by Tris–glycine SDS‐PAGE, transferred to PVDF membranes (Millipore, Billerica, MA, USA) with the electrophoretic transfer system (Trans‐blot Semi‐dry Transfer Cell; Bio‐Rad, Hercules, CA, USA) and blocked with 10% nonfat dry milk in Tris–HCl buffer saline (TBS, pH 7.4) containing 0.1% Tween 20 (TBS‐T) for 1 h at room temperature. Then, the PVDF membranes were incubated with primary antibody against Kir6.1 (1:200; Santa Cruz, Dallas, TX, USA), SUR1 (1:100; Santa Cruz), SUR2 (1:100; Santa Cruz), Caspase‐1 (1:400; Santa Cruz), TLR4 (1:400; Santa Cruz), pIKKa/β (1:800; CST, Boston, MA, USA), NLRP3 (1:200; Santa Cruz), and IL‐1β (R&D Systems, Minneapolis, MN, USA) overnight at 4°C. After being washed in TBS‐T, the membranes were incubated with corresponding secondary antibody for 1 h at room temperature. Finally, visualization of the signal was performed by enhanced chemiluminescence (Ultra‐Lum, Claremont, CA, USA). Quantification of bands was made by scanned densitometric analysis and Image J analysis system.
Enzyme‐Linked Immunosorbent Assay
After 24‐h reoxygenation, we collected the medium from BV‐2 cells. Measurement of key inflammatory cytokines (TNF‐a, IL‐1β, and IL‐10) released into the culture supernatant was performed using specific enzyme‐linked immunosorbent assays (ELISAs) (TNF‐a and IL‐10 were obtained from R&D Systems; IL‐1β was obtained from Biogot technology co., Ltd., Nanjing, China) according to manufacturers guidelines.
Statistical Analysis
Data are shown as mean ± SEM. Unless stated otherwise, we carried out all statistical quantitative assessments in a blinded manner. For two groups paired t‐test (two‐tailed), for three or more groups one‐way or two‐way analysis of variance (ANOVA) followed by Student–Newman–Keuls tests. Differences were considered significant for P < 0.05.
Results
OGD Downregulates Protein Expression of Kir6.1 Subunit in BV‐2 Cell Lines
Microglia is the resident immune cells in the brain. Once stressed, they can be activated and produce some cytokines. However, the functions are determined by the activated state of microglia. As in the present study, BV‐2 cells released lots of LDH to the medium since OGD 1 h and reoxygenation 24 h, but there was no significant change in cell viability (Figure 1A,B). After OGD 1.5 h and reoxygenation 24 h, there was a significant change in LDH release and cell viability (Figure 1A,B). These data suggested that OGD 1.5 h and reoxygenation 24 h could induce BV‐2 cells death.
Figure 1.
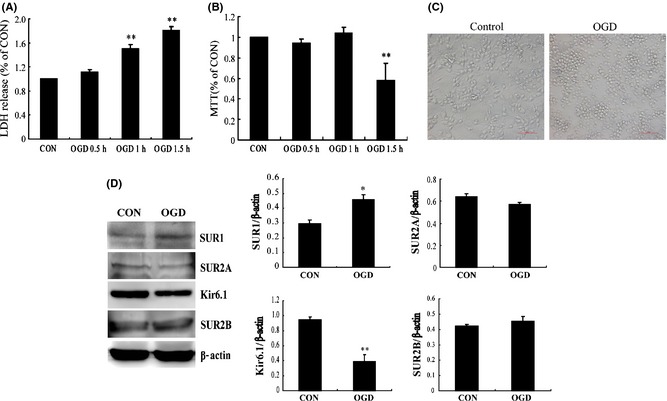
Oxygen–glucose deprivation (OGD) induces downregulation of Kir6.1 subunit in activated BV‐2 cells. The timecourse of lactate dehydrogenase (LDH) release (A) and MTT activity (B) of BV‐2 cells after OGD/reoxygenation. (C) The representative morphological alteration of BV‐2 cells after OGD 1 h and reoxygenation 24 h. Scale bar: 40 μM. (D) Western blotting analyses and represented immunoblots of KATP channels subunits in BV‐2 cells. *P < 0.05, **P < 0.01 versus control group. Results are shown as mean ± SEM of every three individual experiments.
Under normoxia condition, BV‐2 cells showed long fusiform in shape with slender processes. Exposure to OGD for 1 h and reoxygenation 24 h, the morphology of BV‐2 cells became round with shorter processes, as shown in Figure 1C. Moreover, OGD‐induced changes in protein expressions of KATP channel subunits (Figure 1D). The protein expressions of Kir6.1 subunit were downregulated by 50% after OGD 1 h and reoxygenation 24 h (P < 0.05), whereas SUR1 subunit was upregulated about 53% (P < 0.01). OGD did not alter the protein levels of SUR2A and SUR2B. Thus, these results indicated Kir6.1/KATP channels expressed in BV‐2 cells might be involved in OGD‐induced microglial activation.
Nicorandil Inhibits OGD‐Induced Releases of Pro‐Inflammatory Cytokines from BV‐2 Cells
To identify the effects of KATP channels expressed in microglia on OGD‐induced inflammation, we respectively treated BV‐2 cells with KATP channel opener (Nicorandil, 10 μM) and blocker (PNU37883, 20 μM) 1 h prior to OGD. As shown in Figure 2A, nicorandil treatment prevented OGD‐induced variance of cellular morphology and BV‐2 cells remained ramified instead of being activated. However, PNU37883 failed to prevent BV‐2 cells being activated. Furthermore, we detected inflammatory cytokines in the medium. After OGD 1 h and reoxygenation 24 h, pro‐inflammatory factors tumor necrosis factor‐alpha (TNF‐α) (619.62 ± 85.16 pg/mL) (Figure 2B) and IL‐1β (119.51 ± 5.05 pg/mL) (Figure 2C) produced from BV‐2 cells were greatly enhanced (P < 0.01), compared with control group (TNF‐α: 315.71 ± 27.41 pg/mL, IL‐1β: 60.93 ± 4.42 pg/mL). Moreover, it could also induce significant downregulation of IL‐10 (P < 0.05) (Figure 2D). However, pretreatment with nicorandil (10 μM) significantly reduced TNF‐α (reduction by 34.53%) and IL‐1β (reduction by 17.23%) production from BV‐2 cells after OGD (P < 0.05). While pretreatment with PNU37883 (20 μM) failed to decrease inflammatory factors release after OGD. Both nicorandil and PNU37883 did not affect OGD‐induced downregulation of IL‐10. All these results suggested that opening KATP channels inhibited pro‐inflammatory cytokines release from microglia induced by OGD.
Figure 2.
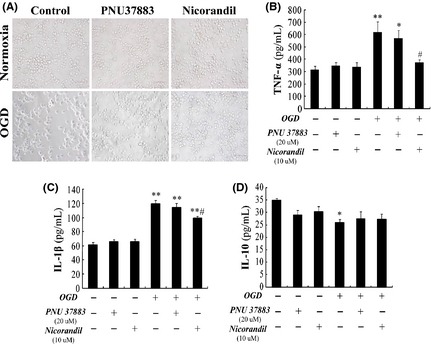
Opening KATP channels decreases production of pro‐inflammatory cytokines from activated BV‐2 cells. (A) The represented appearance of BV‐2 cells under microscope. Scale bar: 40 μM. ELISA assays of tumor necrosis factor‐alpha (TNF‐α) (B), Interleukin‐1beta (IL‐1β) (C), and IL‐10 (D) in the medium of BV‐2 cells. *P < 0.05, **P < 0.01 versus lane 1; #P < 0.05 versus lane 4. Results are shown as mean ± SEM of every seven individual experiments.
Nicorandil Downregulates TLR4 and Inhibits Phosphorylation of IKK
Because activation of microglial cells in response to OGD is characterized by upregulation of TLRs, which drives downstream signaling pathway to promote the expression of inflammatory genes and cytokines. We next investigated how this process initiated after OGD and how did KATP channels work. Our results showed that the expression of TLR4 (Figure 3A,B) and phosphorylation of IkappaB kinase (IKK) (Figure 3A,C) increased notably after OGD 1 h and reoxygenation 24 h (P < 0.01). However, treatment with nicorandil (10 μM) remarkably decreased the expression of TLR4 (reduction by 29.39%) and phosphorylated IKK (reduction by 26.91%) (P < 0.01), but closing Kir6.1/KATP channels by PNU37883 (20 μM) could not produce this potency. These obtained results indicated that opening KATP channels inhibited TLR4/IKK signaling pathway and thereby prevented neuroinflammation induced by OGD. High mobility group box 1 (HMGB1) is a DNA‐binding nuclear protein, which can be released into the medium during the state of stress. Released HMGB1 is a biomarker of necrosis. As shown in Figure 3D, expression of HMGB1 did not change after OGD 1 h and reoxygenation 24 h, and pretreatment with nicorandil or PNU37883. These results suggested that BV‐2 cells were only activated when there was no cell death.
Figure 3.
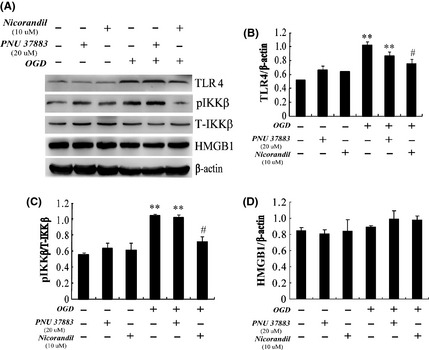
Opening KATP channels downregulates Toll‐like receptor‐4 (TLR4) and phosphorylated IkappaB kinase (IKK) complex in activated BV‐2 cells. (A) Represented immunoblots of TLR4, IKKβ, and high mobility group box 1 (HMGB1). Western blotting analyses of TLR4 (B), pIKK (C), and HMGB1 (D) in BV‐2 cells. **P < 0.01 versus lane 1; #P < 0.05 versus lane 4. Results are shown as mean ± SEM of every three individual experiments.
Nicorandil Inhibits the Activation of NLRP3 Inflammasome
As we know IL‐1β is one of the most important pro‐inflammatory mediators. Previous findings have shown that NLRP3 inflammasome is most significantly associated with maturation of IL‐1β and involved in sterile inflammation. According to these findings, we further wanted to confirm whether activation of NLRP3 inflammasome was involved in OGD‐induced production of IL‐1β. We found that OGD could lead to upregulation of mature IL‐1β (increase about 3‐fold). Pretreatment with nicorandil not PNU37883 could reverse this phenomenon (P < 0.05) (Figure 4A,D). Furthermore, we examined expressions of NLRP3 and cleaved Caspase‐1 by Western blotting (Figure 4A). Consistently, the expressions of NLRP3 (Figure 4B) and cleaved caspase‐1 (Figure 4C) were both significantly increased after OGD (P < 0.05), but pretreatment with nicorandil (10 μM) suppressed the upregulation of NLRP3 and cleaved caspase‐1 in response to OGD. However, the blocker PNU37883 was ineffective. These results suggested that opening the Kir6.1/KATP channels could inhibit inflammasome activation and ameliorated OGD‐induced production of IL‐1β in BV‐2 cells, although its molecular mechanisms should need to be further investigated in future study.
Figure 4.
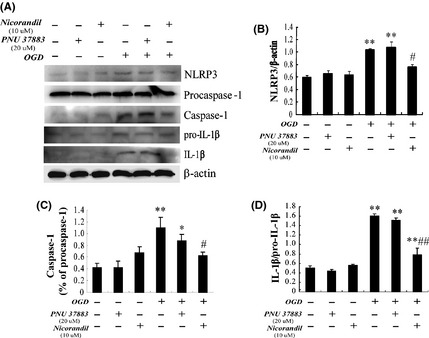
Opening KATP channels suppresses inflammasome activation and production of interleukin‐1beta (IL‐1β) induced by oxygen–glucose deprivation (OGD). (A) Represented immunoblots of NLRP3, cleaved Caspase‐1, and IL‐1β. Western blotting analyses of NLRP3 (B), cleaved Caspase‐1 (C), and maturation of IL‐1β (D). *P < 0.05, **P < 0.01 versus lane 1; #P < 0.05, ##P < 0.01 versus lane 4. Results are shown as mean ± SEM in every four individual experiments.
Discussion
Post‐ischemic inflammation is widely believed to exacerbate neuron death in cerebral ischemia and reoxygenation 17. Microglia is the main resource of pro‐inflammatory cytokines at the early stage of cerebral ischemia 18. So, the regulation of microglial activation and microglia‐mediating neuroinflammation is considered to be an urgent therapeutic strategy for stroke. Kir6.1‐contained KATP channels are the main subtypes expressed in microglia 11, 12. In the present study, we found that OGD downregulated Kir6.1 subunit expression in BV‐2 cell lines. Downregulation of Kir6.1 might be concerned with prompt energy exhaust or glucose deprivation induced KATP channels trafficking 13, 19. Moreover, it was reported that Kir6.1 gene knockout mice were susceptible to LPS stimulus, which lead to endotoxemia and death 15. Our previous study has also clarified Kir6.1 knockdown aggravates microglal activation and inflammatory responses after cerebral ischemia injury 13. In the present study, we found that microglia was activated after OGD 1 h and reoxygenation 24 h and released massive pro‐inflammatory factors, such as TNF‐a and IL‐1β. Moreover, we found that OGD downregulated Kir6.1 subunit expression in BV‐2 cells. Then, we pretreated BV‐2 cells with the KATP channel opener (nicorandil) and blocker (PNU37883), respectively. The data showed that pretreatment with nicorandil could attenuate BV‐2 cells activation and prevented the productions of TNF‐a and IL‐1β, but PNU37883 failed to inhibit inflammatory responses induced by OGD in BV‐2 cells. These findings were consistent with other reports that opening KATP channels was favorable for sterile inflammation 11, 12. However, the molecular mechanisms remain unclear.
Recent evidence implicates the importance of TLR signaling in response to inflammatory injury 20, 21. TLR4 is an important member of transmembrane receptor family, which is consistently expressed in microglia and mediates innate immunity. The present study showed OGD 1 h and reoxygenation 24 h induced upregulation of TLR4 and phosphorylation of the IKK complex in BV‐2 cells. It is reported that expression of TLR4 upregulated in microglia in response to hypoxia or ischemia; meanwhile, expression of phosphorylated IKK complex increased and promoted nuclear factor kappa B (NF‐κB) translocation to the nucleus, all these lead to production of pro‐inflammatory cytokines 20, 22. Consistently, TLR4 knockout mice showed significant smaller infarct size, improved behavioral outcomes and suppressed IKK complex phosphorylation, NF‐кB activity and production of pro‐inflammatory cytokines including TNF‐a and IL‐1β 23, 24. These suggested that OGD/reoxygenation (OGD/R)‐induced production of pro‐inflammatory cytokines associated with TLR4/IKK signaling pathway. Additionally, we found that OGD/reoxygenation could also induce upregulation of NLRP3 and cleaved caspase‐1. NLRP3 expression depends on priming stimulus. After acute injury such as cerebral ischemia, there are many endogenous molecules, which can be served as priming stimulus to initiate NLRP3 expression. But its mechanisms remain poorly characterized. Recently, it has been confirmed that NLRP3 inflammasome can be activated by diverse molecules including extracellular ATP, Aβ and elevated plasma glucose 25, 26, 27. The latest study reported that acute‐phase protein serum amyloid A (SAA) acted as priming stimulus on glial cells resulting in inflammasome activation 28. Additionally, extracellular PH or rodex also exerted great contribution to activating NLRP3 inflammasome 29, 30. However, which is the main contributor to the upregulation of NLRP3 and caspase‐1 activation under OGD condition needs to be further studied. At least, our data indicated OGD/R‐induced inflammatory responses via regulating TLR4/IKK signaling pathway and NLRP3 inflammasome activation. To explore the molecular mechanisms of inflammasome activation under OGD/R condition, its intervention deserves to be focused.
The accumulating evidence indicates that KATP channels correlate with glial inflammation. Besides, opening KATP channels could inhibit inflammatory cytokines transcription. Our present study showed that pretreatment with nicorandil significantly suppressed the expressions of TLR4, NLRP3, phosphorylation of IKK complex and cleaved caspase‐1 induced by OGD, and thereby reduced overproduction of inflammatory factors, such as TNF‐a and IL‐1β. Nicorandil is a potassium channel opener and nitric oxide (NO) donor, which is clinically utilized as a nitrovasodilator and reduce smooth muscle contraction. There is increasing evidence suggests that nicorandil is involved in modulation of inflammation, but its molecular mechanisms remain unclear 31, 32. These data revealed that nicorandil could inhibit TLR4/IKK signaling pathway and inflammasome activation, which protected BV‐2 cells against inflammatory responses after OGD/R injury. However, PNU37883 (Kir6.1 blocker) failed to do it. These suggest that Kir6.1‐contained KATP channels might play an essential part in glial neuroinflammation.
In summary, our data revealed that opening Kir6.1‐contained KATP channels expressed in microglia could inhibit inflammasome activation and TLR4 signal transduction and consequently prevented OGD/R‐induced neuroinflammation. These findings suggest that opening KATP channels is favorable for post‐ischemic inflammation, contributing to limiting microglial activation and overproduction of pro‐inflammatory cytokines. However, the detailed molecular mechanisms involved in Kir6.1‐contained KATP channels regulating inflammasome activation need to be clarified in future study.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No.81273495), the Major Project of Jiangsu Provincial Department of Education (No.12KJA310002), and the National Key Basic Research Program of China (No.2009CB521906).
The first two authors contributed equally to this work.
References
- 1. Zhang L, Zhang ZG, Chopp M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol Sci 2012;33:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Macrez R, Ali C, Toutirais O, et al. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol 2011;10:471–480. [DOI] [PubMed] [Google Scholar]
- 3. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol 2010;87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brough D, Tyrrell PJ, Allan SM. Regulation of interleukin‐1 in acute brain injury. Trends Pharmacol Sci 2011;32:617–622. [DOI] [PubMed] [Google Scholar]
- 6. Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab 2008;28:1707–1721. [DOI] [PubMed] [Google Scholar]
- 7. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–832. [DOI] [PubMed] [Google Scholar]
- 8. Cassel SL, Sutterwala FS. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol 2010;40:607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 2006;440:470–476. [DOI] [PubMed] [Google Scholar]
- 10. Thomzig A, Wenzel M, Karschin C, et al. Kir6.1 is the principal pore‐forming subunit of astrocyte but not neuronal plasma membrane KATP channels. Mol Cell Neurosci 2001;18:671–690. [DOI] [PubMed] [Google Scholar]
- 11. Zhou F, Yao HH, Wu JY, Ding JH, Sun T, Hu G. Opening of microglial K(ATP) channels inhibits rotenone‐induced neuroinflammation. J Cell Mol Med 2008;12:1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X, Wu JY, Zhou F, et al. The regulation of rotenone‐induced inflammatory factor production by ATP‐sensitive potassium channel expressed in BV‐2 cells. Neurosci Lett 2006;394:131–135. [DOI] [PubMed] [Google Scholar]
- 13. Dong YF, Wang LX, Huang X, et al. Kir6.1 knockdown aggravates cerebral ischemia/reperfusion‐induced neural injury in mice. CNS Neurosci Ther 2013;19:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gade AR, Kang M, Akbarali HI. Hydrogen sulfide as an allosteric modulator of ATP‐sensitive potassium channels in colonic inflammation. Mol Pharmacol 2013;83:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kane GC, Lam CF, O'Cochlain F, et al. Gene knockout of the KCNJ8‐encoded Kir6.1 K(ATP) channel imparts fatal susceptibility to endotoxemia. FASEB J 2006;20:2271–2280. [DOI] [PubMed] [Google Scholar]
- 16. Xie J, Duan L, Qian X, Huang X, Ding J, Hu G. K(ATP) channel openers protect mesencephalic neurons against MPP+‐induced cytotoxicity via inhibition of ROS production. J Neurosci Res 2010;88:428–437. [DOI] [PubMed] [Google Scholar]
- 17. Lo EH. T time in the brain. Nat Med 2009;15:844–846. [DOI] [PubMed] [Google Scholar]
- 18. Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2005;196:290–297. [DOI] [PubMed] [Google Scholar]
- 19. Lim A, Park SH, Sohn JW, et al. Glucose deprivation regulates KATP channel trafficking via AMP‐activated protein kinase in pancreatic beta‐cells. Diabetes 2009;58:2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll‐like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 2007;104:13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll‐like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 2007;115:1599–1608. [DOI] [PubMed] [Google Scholar]
- 22. Ock J, Jeong J, Choi WS, et al. Regulation of Toll‐like receptor 4 expression and its signaling by hypoxia in cultured microglia. J Neurosci Res 2007;85:1989–1995. [DOI] [PubMed] [Google Scholar]
- 23. Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia‐reperfusion injury in Toll‐like receptor 4 deficient mice. Biochem Biophys Res Commun 2007;353:509–514. [DOI] [PubMed] [Google Scholar]
- 24. Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll‐like receptor 4 is involved in subacute stress‐induced neuroinflammation and in the worsening of experimental stroke. Stroke 2008;39:1314–1320. [DOI] [PubMed] [Google Scholar]
- 25. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228–232. [DOI] [PubMed] [Google Scholar]
- 26. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 2008;9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010;11:136–140. [DOI] [PubMed] [Google Scholar]
- 28. Savage CD, Lopez‐Castejon G, Denes A, Brough D. NLRP3‐inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front Immunol 2012;3:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajamaki K, Nordstrom T, Nurmi K, et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem 2013;288:13410–13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukoc Biol 2012;92:951–958. [DOI] [PubMed] [Google Scholar]
- 31. Heywood GJ, Thomas PS. Nicorandil inhibits degranulation and TNF‐alpha release from RBL‐2H3 cells. Inflamm Res 2002;51:176–181. [DOI] [PubMed] [Google Scholar]
- 32. Wei XM, Heywood GJ, Di Girolamo N, Thomas PS. Nicorandil inhibits the release of TNFalpha from a lymphocyte cell line and peripheral blood lymphocytes. Int Immunopharmacol 2003;3:1581–1588. [DOI] [PubMed] [Google Scholar]