

Summary
Background
Parkinson's disease (PD) is a common neurodegenerative disease, characterized by progressive loss of dopaminergic (DA) neurons in the substantia nigra. Recent investigations have shown that mitochondrial fragmentation, an early event during apoptosis, is implicated in the degeneration of DA neurons in PD, and more importantly, preventing mitochondrial fragmentation could rescue cell death in several PD models. Therefore, mitochondrial dynamics may be a therapeutic target for early intervention in PD. However, much remains unknown about the mechanism underlying mitochondrial fragmentation in PD.
Methods
The alterations in mitochondrial morphology, cell apoptosis, and mitochondrial shaping protein levels were detected after SH‐SY5Y cells were treated with various doses of MPP+ or rotenone.
Results
Mitochondrial fragmentation is an early event during apoptosis caused by MPP+ but not rotenone, and Trichostatin A (TSA), a commonly used histone deacetylase (HDAC) inhibitor, selectively rescues mitochondrial fragmentation and cell death induced by lower doses of MPP+. Mitochondrial fragmentation triggered by lower doses of MPP+ may be a result of Mfn2 down‐regulation, which could be completely reversed by TSA. Further investigation suggests that TSA prevents MPP+‐induced Mfn2 down‐regulation via inhibiting histone deacetylation over Mfn2 promoter and alleviating its transcriptional dysfunction.
Conclusions
Histone deacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection in PD cell model induced by MPP+. Hence, HDAC inhibitors may be a potential early treatment for PD.
Keywords: Early neuroprotection, Histone deacetylase inhibitors, Mitochondrial fragmentation, Parkinson's disease
Introduction
Mitochondria in living cells are organized into tubular networks that undergo frequent fission and fusion. The fission/fusion balance is necessary for maintaining mitochondrial shape and physiological functions, whereas perturbation of the balance is associated with many neurodegenerative diseases 1, 2. Mitochondrial fission and fusion are membrane‐remodeling processes regulated by mitochondrial shaping proteins, including optic atrophy 1 (Opa1), mitofusins (Mfn1 and Mfn2), dynamin‐like protein 1 (Drp1) and Fis1. Most of the proteins are localized to mitochondria, except that Drp1 is mainly located in the cytoplasm 3, 4. Mitochondrial fission requires Fis1 and mitochondrial translocation of Drp1, while mitochondrial fusion is mediated by Opa1 and mitofusins 3, 4.
Parkinson's disease (PD) is an age‐related neurodegenerative disorder, characterized by progressive loss of dopaminergic (DA) neurons in the substantia nigra. The precise mechanisms underlying the destruction of nigrostriatal DA pathway remain elusive, and so far, there are no available treatments to stop or even reverse the progression of this disease. Even so, a great deal of progress has been made in recent years, and especially, accumulating evidence suggests that mitochondrial fragmentation, the result of excessive fission or deficient fusion, may be an early and causal event in the degeneration of DA neurons in PD 5, 6, 7. Mitochondrial fragmentation has been reported in both the MPP+ and the rotenone models, and preventing mitochondrial fragmentation by manipulating mitochondrial fission and fusion rescues neuronal cells in several PD models 8, 9, 10. This implicates that mitochondrial dynamics may be a potential therapeutic target for early intervention in PD; however, the mechanism underlying mitochondrial fragmentation in PD is still unclear.
We report here that mitochondrial fragmentation is an early cell death event in the MPP+ model, whereas it may not be required for cell apoptosis in the rotenone model. Mitochondrial fragmentation triggered by MPP+ of lower doses might result from Mfn2 down‐regulation, which could be reversed by Mfn2 overexpression. More intriguingly, Trichostatin A (TSA), a commonly used broad‐spectrum histone deacetylase (HDAC) inhibitor, selectively rescues mitochondrial fragmentation and cell death initiated by lower doses of MPP+. TSA prevents MPP+‐induced Mfn2 down‐regulation by inhibiting histone hypoacetylation over Mfn2 promoter and reversing its transcriptional dysfunction. These indicate that mitochondrial fragmentation in PD may be associated with Mfn2 deficiency due to histone hypoacetylation and transcriptional dysfunction, and HDAC inhibitors could reverse this process, rescue mitochondrial fragmentation, and elicit early neuroprotection.
Materials and methods
SH‐SY5Y Cell Culture
SH‐SY5Y cells were obtained from American Tissue Culture Collection and used within 20 passages of the original vial. SH‐SY5Y cells were grown in Dulbecco's modified eagle's medium (DMEM)/F12 medium supplemented with 15% fetal bovine serum (FBS), 100 IU/mL penicillin, 0.1 mg/mL streptomycin, and 3.7 g/L NaHCO3. Cell cultures were all kept at 37°C in a saturated humidity air atmosphere containing 5% CO2. At 80% confluence, growth medium was changed to 1% FBS containing medium for 2 h before addition of test agents. MPP+, Rotenone, TSA, suberoylanilide hydroxamic acid (SAHA), and Bax inhibiting peptide V5 were all purchased from Sigma.
Mitochondrial Morphology Assessment
SH‐SY5Y cells were plated in glass‐bottom culture dishes for mitochondrial observation on live cell imaging system. Mitochondria were visualized by transfection with a mitochondria‐targeted RFP kit (Molecular Probes, Eugene, OR, USA) according to the manufacturer's instructions. Images of 20 cells from random view fields were taken from each group on UltraView VoX 3D live cell imaging system (PerkinElmer, Waltham, MA, USA) using a 100× objective, Z‐stacks were taken at 0.5 μm intervals, and the experiment was repeated four times. Cell images were analyzed for lengths of major and minor axes of the ellipse equivalent to the mitochondrion using Volocity 5.0 (Improvision, Lexington, MA, USA) software. From these values, we calculated mitochondrial length (or aspect ratio, the ratio between the major and minor axes of the ellipse equivalent to the mitochondrion) as described before 11.
Apoptosis Analysis
Detection of apoptosis by flow cytometry (FACScan; BD Company, Franklin Lakes, NJ, USA) was performed using the Annexin V‐FITC apoptosis detection kit (BD Company) following the manufacturer's instructions. Briefly, SH‐SY5Y cells were collected after drug treatments, and re‐suspended at a concentration of 1 × 106 cells/mL. Then cells were incubated with FITC Annexin V and PI for 15 min before analyzed by flow cytometry. A minimum of 10,000 cells were analyzed in each treatment.
Isolation of Mitochondria
Mitochondria were isolated from SH‐SY5Y cells as described before 11. Mitochondria were isolated by centrifuging SH‐SY5Y cell homogenates at 1500 g for 5 min at 4°C. We adjusted the supernatant to 14% Percoll and recentrifuged at 12,000 g for 10 min, and resuspended the loose mitochondrial pellet and recentrifuged at 8000 g for 10 min. Highly purified mitochondria were used for immunoblotting assay.
Western Blotting Analyses
SH‐SY5Y cells were collected after drug treatments or transfection. Cell lysates or mitochondrial lysates were prepared and ran on 10–12% SDS‐PAGE gels. Antibodies used were as follows: rabbit polyclonal antibax (1:1,000, Cell signaling, Beverly, MA, USA; #2772), rabbit antisera against Fis1 (1:500, BioVision, Milpitas, CA, USA; #3491‐100), Drp1 (1:500, ab54038), Mfn2 (1:1,000, ab50838) and Opa1 (1:500, ab42364; all from Abcam, Cambridge, MA, USA), chicken polyclonal anti‐Mfn1 (1:1,000, Novus Biologicals, Littleton, CO, USA; NB110‐58853), mouse antibody against β‐actin (1:20,000, Sigma, St. Louis, MO, USA; A3854).
RNA Extraction, RT‐PCR, and Real‐time PCR
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA (2 μg) was reverse‐transcribed using PrimeScript RT reagent Kit (Takara, Otsu, Japan), and the single stranded cDNA was amplified by quantitative real‐time PCR using SYBR Premix Ex Taq Kit (Takara) on Mastercycler ep realplex real‐time PCR system (Eppendorf) and data calculated using the DCt method (2−DDCt). The following primers were used: Mfn 2, 5′‐CTCTCGATGCAACTCTATCGTC‐3′ and 5′‐CTTGGCAGTGACAAAGTGCTT‐3′; Mfn1, 5′‐GGACAAGCAGTGGGAAGA‐3′ and 5′‐TTATCTCCATCAGTTCCTTC‐3′; GAPDH, 5′‐CCACTCCTCCACCTTTGAC‐3′ and 5′‐ACCCTGTTGCTGTAGCCA‐3′. Data are presented as means ± SEM of four independent experiments in three real‐time PCR replicates.
Chromatin Immunoprecipitation (ChIP) Assay
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP assay kit (Millipore, Billerica, MA, USA) according to the manufacturer's protocol. Briefly, after drug treatment, SH‐SY5Y cells were cross‐linked with formaldehyde, lysed, sonicated, and subjected to chromatin shearing. Immunoprecipitation was performed using antibodies to acetyl‐H3 and acetyl‐H4. As a negative control, samples were immunoprecipitated with nonimmune rabbit IgG. Immunoprecipitated DNA was subjected to both semi‐quantitative and quantitative real‐time PCR analysis using primers specific for Mfn2 promoter (5′‐AAATACAGCGGTGGATGTTAGAGA‐3′ and 5′‐CCAGGCCTAGGGTGAAGTGA‐3′), Mfn1 promoter (5′‐TCAGCCACCACCATCATT‐3′ and 5′‐GTTTGTTGTGGGCAGAGA‐3′) and GAPDH promoter (5′‐AGGCTGGATGGAATGAAAGG‐3′ and 5′‐GCGGAGGACAGGATGGCT‐3′). After around 30 cycles of amplification, 20 mL of the PCR product was analyzed on a 2% agarose gel. Real‐time PCR was performed as described above.
Transient Transfection
SH‐SY5Y cells were transfected using FuGENE HD Transfection Reagent (Roche) following the instruction of manufacturer. Full length human Mfn1 or Mfn2 cDNA was subcloned into pCMV6‐AC‐IRES‐GFP expression vector (Origene, Rockville, MD, USA). Plasmid DNA was first diluted in OPTI‐MEM I (Gibco‐BRL, Rockville, MD, USA) and then mixed with FuGENE HD Transfection Reagent. Following a 20‐min incubation period at room temperature, the DNA‐FuGENE HD Transfection Reagent mixture was added to the antibiotic‐free transfection medium (DMEM containing glutamine). The cells were maintained in a 37°C incubator with a saturated humidity air atmosphere containing 5% CO2. The medium was replaced with normal culture medium 6 h later. Cells were used for various experiments 48 h after transfection.
Statistical Analyses
The data are expressed as means ± SE and were subjected to statistical analysis via one‐way or two‐way ANOVA, followed by Bonferroni post hoc analysis, using GraphPad Prism software (GraphPad software, San Diego, CA, USA). The level of statistical significance was set at P < 0.05.
Results
TSA Rescues Mitochondrial Fragmentation and Cell Apoptosis Induced by MPP+ but not Rotenone
Firstly, after transfected with a mitochondria‐targeted RFP for 24 h, SH‐SY5Y cells were treated with different doses of MPP+ (1 mM, M1; 2 mM, M2; 3 mM, M3) or rotenone (1 μM, R1; 2 μM, R2; 3 μM, R3). Mitochondrial morphology was observed on live cell imaging system at 4 and 8 h of drug treatments; meanwhile, cell apoptosis was determined by flow cytometry. Mitochondrial length was calculated as we described before 11; it has a minimal value of 1 as a perfect circle and the value increases when mitochondria become elongated. As shown in Figure 1A,C, significant mitochondrial fragmentation was detected at 4 h of exposure to different doses of MPP+, and it became more striking at 8 h. However, appreciable cell apoptosis was only observed at 8 h of MPP+ treatments (Figure 1D), suggesting that mitochondrial fragmentation was triggered prior to cell apoptosis in the MPP+ model. On the contrary, cell death was induced before mitochondrial fragmentation in R2 group (Figure 1A,C,D). More intriguingly, mitochondria were not shortened but slightly elongated in R1 group (Figure 1A,C), even when significant cell apoptosis was provoked at 8 h (Figure 1D). The data above indicate that mitochondrial fragmentation plays different roles in apoptosis in those two PD models: it may be an early event during apoptosis in the MPP+ model, yet it might not be required for rotenone‐initiated cell apoptosis.
Figure 1.
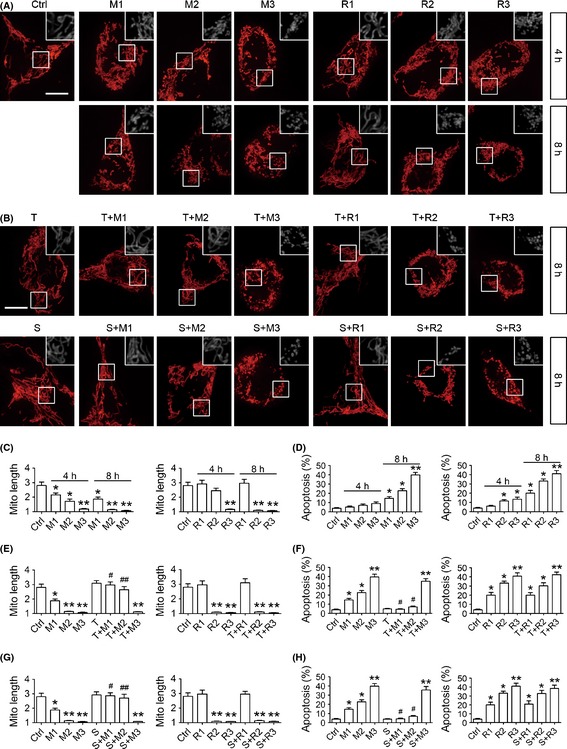
The effects of histone deacetylase inhibitors on mitochondrial fragmentation and cell apoptosis in MPP+ or rotenone cell model. After transfected with a mitochondria‐targeted RFP for 24 h, SH‐SY5Y cells were treated with different doses of MPP+ (1 mM, M1; 2 mM, M2; 3 mM, M3) or rotenone (1 μM, R1; 2 μM, R2; 3 μM, R3). Representative images taken by live cell imaging system (A) and quantitative analysis of mitochondrial length (C) show that MPP+ and rotenone both induce mitochondrial fragmentation in SH‐SY5Y cells in a dose‐ and time‐dependent fashion, except that mitochondria are not shortened but even slightly elongated in R1 group. Flow cytometry results (D) demonstrate that mitochondrial fragmentation is triggered prior to cell apoptosis in the MPP+ but not yet in the rotenone model. Co‐treatment with 0.1 nM Trichostatin A (T) or 0.1 μM suberoylanilide hydroxamic acid (SAHA, S) for 8 h selectively rescues mitochondrial fragmentation (B, E, G) and cell apoptosis (F, H) in the cells exposed to 1 or 2 mM MPP+, but does not have protective effect in other groups. n = 4; *P < 0.05, **P < 0.01 versus control; # P < 0.05 T + M/S + M group versus M group of the same dose. Scale bars for 10 μm.
Next, TSA (0.1 nM) was administered to SH‐SY5Y cells for 8 h with different doses of MPP+ or rotenone to test its influence on mitochondrial fragmentation and cell apoptosis. As seen from Figure 1B,E,F, TSA completely rescued mitochondrial fragmentation and further prevented cell apoptosis in M1 and M2 groups, yet it had no protective effect in M3 group or rotenone groups. To test whether TSA elicits this protection through HDAC inhibition or its nonspecific effects, we applied another broad‐spectrum HDAC inhibitor SAHA (0.1 μM) 12, and observed similar results (Figure 1B,G,H). In addition, neither 0.1 nM TSA nor 0.1 μM SAHA affected mitochondrial morphology or cell survival in normal cells (Figure 1E–H). This indicates that TSA rescues mitochondrial fragmentation and exerts early neuroprotection in the MPP+ model via HDAC inhibition.
TSA Prevents Mfn1 and Mfn2 Down‐regulation in the MPP+ Model
We then tested the effects of TSA on mitochondrial shaping protein levels and Drp1 cellular localization in MPP+ or rotenone model. SH‐SY5Y cells were collected after drug treatments for 8 h, and whole cell lysates and mitochondrial lysates were prepared for immunoblot assay. As shown in Figure 2B, mitochondrial Drp1 levels were remarkably increased in R2 and R3 groups, while no change was seen in the levels of total Drp1 or other mitochondrial shaping proteins, implying that mitochondrial fragmentation in the rotenone model may be caused by Drp1 mitochondrial recruitment. In the MPP+ cell model, Mfn1 and Mfn2 levels were both reduced in a time‐ and dose‐dependent fashion, and Mfn2 levels were decreased even earlier and more significantly than Mfn1; the levels of other mitochondrial shaping proteins were not affected, except that mitochondrial Drp1 levels were robustly elevated in M3 group (Figure 2A). It implies that mitochondrial fragmentation initiated by MPP+ might be a result of Mfn1 and Mfn2 down‐regulation, and it may also be associated with Drp1 mitochondrial translocation in M3 group. Co‐treatment with TSA entirely reversed Mfn1 and Mfn2 down‐regulation in MPP+ groups (Figure 2A), yet it had no influence on Drp1 mitochondrial translocation in M3 group (Figure 2A) or rotenone groups (Figure 2B). Even when TSA restored Mfn1 and Mfn2 levels in M3 group, it could not at all prevent mitochondrial fragmentation (Figure 1B,E), indicating that mitochondrial fragmentation in M3 group may be mainly caused by Drp1 mitochondrial translocation.
Figure 2.
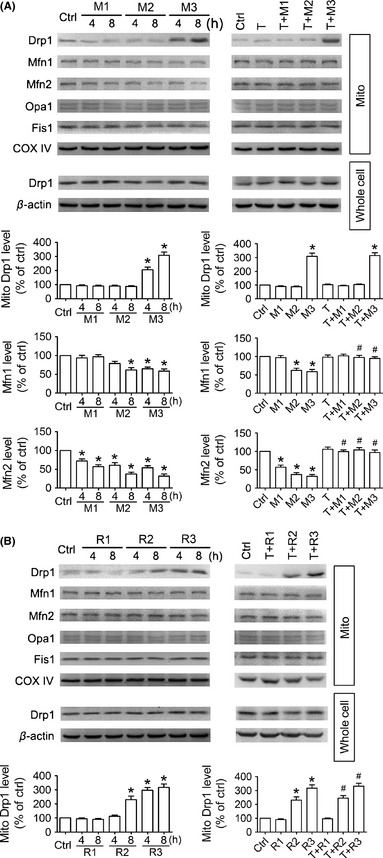
Trichostatin A (TSA) blocks the down‐regulation of Mfn1 and Mfn2 in MPP+ cell model. SH‐SY5Y cells were treated with different doses of MPP+ (1 mM, M1; 2 mM, M2; 3 mM, M3) or rotenone (1 μM, R1; 2 μM, R2; 3 μM, R3). Representative immunoblotting images and semi‐quantitative analysis of mitochondrial shaping protein levels (Drp1, Mfn1, Mfn2, Opa1, and Fis1) (A, B) show that Mfn1 and Mfn2 levels are both decreased in a dose‐ and time‐dependent fashion in MPP+‐exposed cells. Total Drp1 levels are not affected during drug exposure, while mitochondrial Drp1 levels are elevated in M3, R2, and R3 groups. Co‐treatment with 0.1 nM TSA (T) for 8 h entirely blocks the down‐regulation of Mfn1 and Mfn2 induced by MPP+, yet has no influence on Drp1 mitochondrial recruitment in the MPP+ or rotenone cell model. n = 4; *P < 0.05, **P < 0.01 versus control; # P < 0.05 T + M group versus M group of the same dose or T + R group versus R group of the same dose.
TSA Attenuates Transcriptional Dysfunction of Mfn1 and Mfn2 in the MPP+ Model
To explore the mechanism by which TSA prevents Mfn1 and Mfn2 down‐regulation in the MPP+ model, the mRNA levels of Mfn1 and Mfn2 were detected after SH‐SY5Y cells were exposed to 2 mM MPP+ for 8 h. As demonstrated in Figure 3A, Mfn1 and Mfn2 mRNA levels were remarkably decreased by MPP+, which were both totally reversed by co‐treatment with TSA. TSA alone barely had any effect on Mfn1 and Mfn2 mRNA levels in normal cells.
Figure 3.
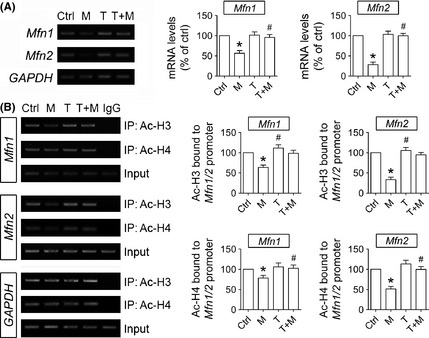
Trichostatin A (TSA) inhibits histone deacetylation over Mfn1 and Mfn2 promoters and alleviates their transcriptional dysfunction induced by MPP+. (A) Representative semi‐quantitative PCR analysis and quantitative real‐time PCR analysis show that Mfn1 and Mfn2 mRNA levels in SH‐SY5Y cells are both reduced after exposure to 2 mM MPP+ for 8 h, which are reversed by co‐treatment with 0.1 nM TSA. (B) Representative semi‐quantitative PCR analysis and quantitative real‐time PCR analysis of ChIP samples show that the acetylation levels of H3 and H4 over Mfn1 and Mfn2 promoter in SH‐SY5Y cells are both decreased after exposure to 2 mM MPP+ for 8 h, which is completely inhibited by TSA. n = 4; *P < 0.05 versus control; # P < 0.05 T + M group versus M group.
Next, ChIP was performed after cells were treated with 2 mM MPP+ for 8 h, to examine the acetylation status of core histones H3 and H4 physically associated with Mfn1 and Mfn2 promoters. As seen from Figure 3B, the acetylation of H3 and H4 over Mfn1 and Mfn2 promoters was significantly reduced by MPP+ treatment, yet it was completely blocked by TSA.
The results above implicate that TSA might prevent MPP+‐induced Mfn1 and Mfn2 down‐regulation by reversing histone hypoacetylation over Mfn1 and Mfn2 promoters and attenuating their transcriptional dysfunction.
The Overexpression of Mfn2 but not Mfn1 Rescues Mitochondrial Fragmentation and Cell Apoptosis Induced by a Lower Dose of MPP+
We further questioned whether mitochondrial fragmentation and cell apoptosis in the MPP+ model could be rescued by Mfn1 or Mfn2 overexpression. SH‐SY5Y cells were transfected with constructed plasmids containing human Mfn1 or Mfn2 cDNA followed by IRES‐GFP. This provides an important benefit that GFP is translated from the same transcript with Mfn1/2 yet not attached to them, thus GFP could serve as an expression marker without risking disturbing the structures or functions of Mfn1/2. Highly purified mitochondria were isolated after transfection for 2–4 days and used for immunoblot assay. Mfn1 protein levels in Mfn1‐overexpressed cells were more than 4‐fold over those in the cells transfected with an empty vector (Figure 4D), and so were Mfn2 levels in Mfn2‐overexpressed cells (Figure 4E).
Figure 4.
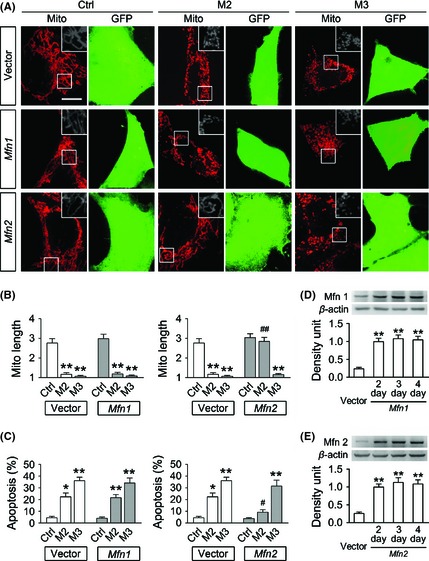
The effects of Mfn1/2 overexpression on mitochondrial fragmentation and cell apoptosis in the MPP+ cell model. SH‐SY5Y cells were first transfected with constructed plasmids containing human Mfn1 or Mfn2 cDNA followed by IRES‐GFP, and 2 days later transfected with a mitochondria‐targeted RFP. After 24 h, cells were treated with 2 or 3 mM MPP+ (M2, M3) for 8 h. Representative images taken by live cell imaging system (A) and quantitative analysis of mitochondrial length (B) show that MPP+ induces significant mitochondrial fragmentation in the cells transfected with an empty vector. Mfn2 overexpression selectively prevents mitochondrial fragmentation in M2 yet not M3 group, and Mfn1 overexpression has no effect on mitochondrial fragmentation induced by 2 or 3 mM MPP+. (C) Flow cytometric analysis shows that MPP+ induces severe cell apoptosis in the cells transfected with an empty vector. Mfn2 overexpression selectively attenuates cell apoptosis in M2 but not M3 group, while Mfn1 overexpression has no effect on cell damage caused by 2 or 3 mM MPP+. (D, E) Representative immunoblotting images and semi‐quantitative analysis show that Mfn1 or Mfn2 protein levels are robustly elevated after transfection for 2–4 days. n = 4; *P < 0.05, **P < 0.01 versus control; # P < 0.05, ## P < 0.01 Mfn2 group versus vector group of the same dose of MPP+. Scale bars for 10 μm.
Two days after the first transfection, those cells were then overexpressed with a mitochondria‐targeted RFP, and 24 h later, they were exposed to 2 or 3 mM MPP+ for 8 h. Mitochondrial morphology and cell apoptosis were assayed as described above. As demonstrated in Figure 4A–C, severe mitochondrial fragmentation and cell apoptosis were provoked in the cells transfected with an empty vector after treated with 2 or 3 mM MPP+ for 8 h. Mfn2 overexpression selectively prevents mitochondrial fragmentation and cell apoptosis triggered by 2 mM MPP+, whereas Mfn1 overexpression has no protective effect, suggesting that the deficiency of Mfn2 but not Mfn1 may play a central role in mitochondrial fragmentation and cell apoptosis in M2 group.
TSA Blocks Bax Translocation to Mitochondria Induced by a Lower Dose of MPP+
Previous studies have implicated Bax activation and mitochondrial translocation in MPP+‐induced cell apoptosis 13, 14. Recent investigations indicate that Bax might also be a regulator of mitochondrial morphogenesis, as Bax translocation to mitochondria leads rapidly to mitochondrial fragmentation 15, 16. To investigate the role of Bax mitochondrial translocation in MPP+‐induced mitochondrial fragmentation, firstly, mitochondrial Bax levels were detected after SH‐SY5Y cells were exposed to 2 or 3 mM MPP+ for 8 h. As seen from Figure 5A, mitochondrial Bax levels were modestly increased in M2 group, and they were elevated even higher in M3 group. Coadministration with TSA selectively suppressed the elevation of mitochondrial Bax levels in M2 group.
Figure 5.
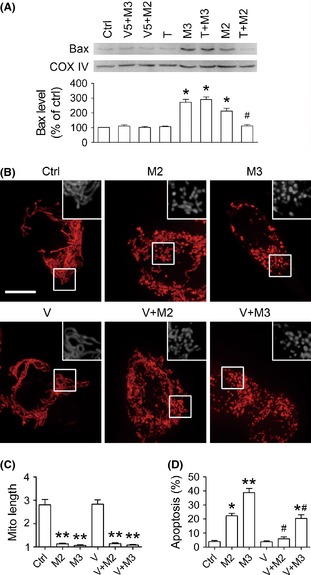
Blocking Bax translocation to mitochondria does not prevent mitochondrial fragmentation in the MPP+ cell model. (A) Representative immunoblotting image and semi‐quantitative analysis show that mitochondrial Bax levels are robustly increased after exposed to 2 or 3 mM MPP+ for 8 h, which is entirely blocked by Bax inhibiting peptide V5 (V5, 100 μM). Trichostatin A selectively reverses the elevation of mitochondrial Bax levels in the cells treated with 2 mM MPP+. (B) Representative images taken by live cell imaging system shows mitochondrial morphology in SH‐SY5Y cells. Co‐treatment with peptide V5 has no impact on mitochondrial fragmentation in the cells exposed to MPP+. (C) Quantitative analysis of changes in mitochondrial length per cell. Peptide V5 has no impact on the decrease of mitochondrial length in the cells exposed to MPP+. (D) Flow cytometry results show that Peptide V5 remarkably inhibits cell apoptosis in the cells exposed to MPP+. n = 4; *P < 0.05, **P < 0.01 versus control; # P < 0.05 T + M/V + M group versus M group of the same dose. Scale bars for 10 μm.
Next, Bax inhibiting peptide V5 (100 μM) was administered to SH‐SY5Y cells along with MPP+. Peptide V5 is commonly used to prevent Bax translocation on mitochondria and Bax‐mediated cell death 17. Mitochondrial morphology and cell apoptosis were observed at 8 h of drug exposure. Although peptide V5 totally blocked Bax translocation to mitochondria (Figure 5A) and prevented cell apoptosis in M2 and M3 groups (Figure 5D), it had little influence on mitochondrial fragmentation in either group (Figure 5B,C). This suggests that mitochondrial fragmentation in the MPP+ model may not result from Bax mitochondrial translocation. Hence, TSA preventing Bax translocation to mitochondria might not be involved in its protection against mitochondrial fragmentation in the MPP+ model.
Discussion
MPP+ and rotenone are widely used for PD models and both recognized as mitochondrial toxins that selectively inhibit complex I 18, 19, however, MPP+ is actually a much weaker inhibitor than rotenone is, and they induce cell apoptosis through distinct mechanisms 20. Although mitochondrial fragmentation has been reported in both MPP+ and rotenone cell models 7, 8, 9, our results suggest that it plays different roles in those two PD models: it may be an early event during apoptosis in the MPP+ model, but it might not be required for rotenone‐initiated cell apoptosis. Therefore, it is very likely that the MPP+ cell model is more suitable to study the mechanism underlying mitochondrial fragmentation in PD and screen drugs for early treatments for PD targeting at mitochondrial dynamics.
To investigate the mechanisms underlying mitochondrial fragmentation in the MPP+ model, we tested a serial of mitochondrial shaping proteins, and found that although Mfn1 and Mfn2 levels are both reduced by MPP+ exposure, the deficiency of Mfn2 but not Mfn1 may play a central role in mitochondrial fragmentation and cell apoptosis provoked by lower doses of MPP+. Mfn2 is a large GTPase localized to mitochondrial outer membrane and directs mitochondrial fusion 3, 21. It has been reported that silencing of Mfn2 causes mitochondrial fragmentation and enhances cell sensitivity to apoptotic stimuli 22, whereas Mfn2 overexpression, in addition to promoting mitochondrial fusion, results in delayed apoptotic death 4, 22, 23. Furthermore, a recent study has suggested that the loss of Mfn2 but not Mfn1 results in mitochondrial fragmentation and progressive degeneration of DA neurons in the nigrostriatal circuit 24. Those together imply that Mfn2 may be specifically required for the survival of DA neurons not only in physiological condition but more importantly, in the MPP+ model. Therefore, Mfn2 is very likely a potential target for early intervention in PD.
Wang et al. 7 showed that mitochondrial fragmentation in the MPP+ cell model may be mediated by Drp1 expression elevation and its translocation to mitochondria, but they did not explore how other mitochondrial shaping proteins are involved in the process. We found in this study that mitochondrial fragmentation in the MPP+ model may result from imbalance in mitochondrial shaping proteins: Mfn2 down‐regulation in M1 and M2 groups, and Drp1 mitochondrial recruitment in M3 group. Contradictory results have been reported about the role of Drp1‐dependent mitochondrial fragmentation in cell apoptosis. Although some earlier studies showed that inhibiting Drp1‐dependent mitochondrial fragmentation is able to prevent cell death 7, 25, others imply that Drp1 knockdown only inhibits mitochondrial fragmentation yet fails to rescue cell apoptosis 26, 27. Therefore, it is still debatable whether Drp1 is another potential target for intervention in PD. Since our present work was carried out on SH‐SY5Y cells, which are far less sensitive to MPP+ toxicity than DA neurons, further investigations are needed to confirm our results in an in vivo PD model.
A link between mitochondrial fission and Bax activation was firstly suggested by the finding that activated Bax forms discrete foci with Drp1 and Mfn2 at mitochondrial fission sites 15. Recent studies showed that Bax is required for mitochondrial fusion in healthy cells 28, yet Bax activation during apoptosis might promote mitochondrial fission 16. However, Yuan et al. 29 reported that mitochondrial fragmentation in response to nitric oxide is an upstream and required event for Bax accumulation on mitochondria in neurons. Similar results were found in our present study that inhibiting Bax translocation to mitochondria with peptide V5 prevents cell apoptosis, but fails to rescue mitochondrial fragmentation in MPP+ groups, implying that Bax activation may be a downstream event of mitochondrial fragmentation in the MPP+ model, as well.
Accumulating evidence suggests that HDAC inhibitors elicit significant protection against neurodegeneration 30, 31. Nevertheless, the mechanism of action in how histone acetylation is transferred to neuroprotection remains elusive, as it is an enormous task to identify every consequence of any specific treatment. So far, multiple genes have been identified under the regulation of HDAC inhibitors, but not all of them are involved in neuroprotection 32. We found here that TSA prevents MPP+‐induced Mfn1 and Mfn2 down‐regulation via inhibiting histone deacetylation over Mfn1 and Mfn2 promoters and alleviating their transcriptional dysfunction; meanwhile, TSA has no effect on the levels of other mitochondrial shaping proteins. Although Mfn1 and Mfn2 are both under the regulation of TSA, Mfn2 is more likely involved in TSA protection against MPP+, for that mitochondrial fragmentation provoked by MPP+ of lower doses is mainly associated with Mfn2 deficiency and could be reversed by the overexpression of Mfn2 but not Mfn1. There may also be some other mechanism underlying TSA protection against mitochondrial fragmentation and cell death in the MPP+ model, and further studies are required.
As seen from our results above, TSA reverses mitochondrial fragmentation and cell apoptosis triggered by MPP+, yet it had no protection in the rotenone cell model. Previous studies also reported distinct responses to HDAC inhibitors treatment in different PD models 33, 34. A possible explanation would be that HDACs act on a wide variety of substrates, and they may orchestrate distinct responses to different neurotoxins in diverse cell types. In conclusion, we showed here for the first time that TSA exerts early neuroprotection against mitochondrial fragmentation and cell apoptosis in the MPP+ model. Our present work not only sheds light into the pathogenesis of PD, implicating that mitochondrial fragmentation, an early event during neuronal apoptosis in PD, may be a result of alteration in the acetylase/deacetylase balance, but also indicates that HDAC inhibitors might be a potential early treatment for PD. Although the use of broad‐spectrum small molecule HDAC inhibitors as a treatment for neurodegeneration has provided some promising results, the beneficial effect is usually compromised by unintended harmful side effects 30, 31, 32. As such, the development of effective and more selective HDAC inhibitors with fewer adverse effects is eagerly needed.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by grants from Shanghai Natural Science Foundation of China (10ZR1406100), Shanghai Scientific and Technical Supporting Program of China (12401901003), Shanghai Postdoctoral Foundation (13R21411600), China Postdoctoral Foundation (200902205), and Ph.D. Programs Foundation of Ministry of Education of China (200802461038).
References
- 1. Cho DH, Nakamura T, Lipton SA. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci 2010;67:3435–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol 2013;23:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 2005;14:R283–289. [DOI] [PubMed] [Google Scholar]
- 4. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev 2008;22:1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xie W, Chung KK. Alpha‐synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem 2012;122:404–414. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson's disease‐associated DJ‐1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem 2012;121:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X, Su B, Liu W, et al. DLP1‐dependent mitochondrial fragmentation mediates 1‐methyl‐4‐phenylpyridinium toxicity in neurons: implications for Parkinson's disease. Aging Cell 2011;10:807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barsoum MJ, Yuan H, Gerencser AA, et al. Nitric oxide‐induced mitochondrial fission is regulated by dynamin‐related GTPases in neurons. EMBO J 2006;25:3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meuer K, Suppanz IE, Lingor P, et al. Cyclin‐dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ 2007;14:651–661. [DOI] [PubMed] [Google Scholar]
- 10. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 2008;105:1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu M, Li W, Lu C. Role of alpha‐synuclein protein levels in mitochondrial morphology and cell survival in cell lines. PLoS ONE 2012;7:e36377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci 2011;31:3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1‐mediated inhibition of 1‐methyl‐4‐phenylpyridinium ion neurotoxicity in human SH‐SY5Y neuroblastoma cells. J Biol Chem 2005;280:2132–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaib‐Oukadour I, Gil C, Rodriguez‐Alvarez J, Ortega A, Aguilera J. Tetanus toxin H(C) fragment reduces neuronal MPP+ toxicity. Mol Cell Neurosci 2009;41:297–303. [DOI] [PubMed] [Google Scholar]
- 15. Karbowski M, Lee YJ, Gaume B, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 2002;159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 2004;164:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane‐permeable peptides designed from the Bax‐binding domain of Ku70. Nat Cell Biol 2003;5:352–357. [DOI] [PubMed] [Google Scholar]
- 18. Ramsay RR, Krueger MJ, Youngster SK, Gluck MR, Casida JE, Singer TP. Interaction of 1‐methyl‐4‐phenylpyridinium ion (MPP+) and its analogs with the rotenone/piericidin binding site of NADH dehydrogenase. J Neurochem 1991;56:1184–1190. [DOI] [PubMed] [Google Scholar]
- 19. Ramachandiran S, Hansen JM, Jones DP, Richardson JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase‐3 activation. Toxicol Sci 2007;95:163–171. [DOI] [PubMed] [Google Scholar]
- 20. Giordano S, Lee J, Darley‐Usmar VM, Zhang J. Distinct effects of rotenone, 1‐methyl‐4‐phenylpyridinium and 6‐hydroxydopamine on cellular bioenergetics and cell death. PLoS ONE 2012;7:e44610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 2003;160:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 2004;279:52726–52734. [DOI] [PubMed] [Google Scholar]
- 23. Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 2005;280:25060–25070. [DOI] [PubMed] [Google Scholar]
- 24. Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet 2012;21:4817–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009;119:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parone PA, James DI, Da Cruz S, et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak‐dependent apoptosis. Mol Cell Biol 2006;26:7397–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Estaquier J, Arnoult D. Inhibiting Drp1‐mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 2007;14:1086–1094. [DOI] [PubMed] [Google Scholar]
- 28. Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006;443:658–662. [DOI] [PubMed] [Google Scholar]
- 29. Yuan H, Gerencser AA, Liot G, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ 2007;14:462–471. [DOI] [PubMed] [Google Scholar]
- 30. Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 2008;8:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 2009;32:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dietz KC, Casaccia P. HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol Res 2010;62:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Wang X, Liu L. HDAC inhibitor trichostatin A‐inhibited survival of dopaminergic neuronal cells. Neurosci Lett 2009;467:212–216. [DOI] [PubMed] [Google Scholar]
- 34. Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre‐motor stage PD. Neuropharmacology 2012;62:2409–2412. [DOI] [PubMed] [Google Scholar]