

Summary
Background and purpose
As a molecular chaperone, acetylcholinesterase (AChE; EC 3.1.1.7) plays a critical role in the pathogenesis of Alzheimer's disease (AD). The peripheral anionic site (PAS) of AChE has been indicated as the amyloid‐β (Aβ) binding domain. The goal of this study was to determine other motifs in AChE involved in Aβ aggregation and deposition.
Methods and results
The β‐hairpin in monomeric Aβ is the key motif of nucleation‐dependent Aβ self‐aggregation. As AChE could induce Aβ aggregation and deposition, we searched AChE for β‐hairpin structures. In A11‐specific dot blot assay, AChE was detected by an oligomer‐specific antibody A11, implying the existence of β‐hairpin structures in AChE as β‐hairpin was the core motif of oligomers. A molecular superimposing approach further revealed that the N‐terminal region, from Glu7 to Ile20, in AChE (AChE 7–20) was similar to the β‐hairpin domain in Aβ. The results of further dot blot assays, thioflavin T fluorescence assays, and electron microscopy imaging experiments, indicated that the N‐terminal synthetic peptide AChE 7–20 had nearly the same ability as AChE with regard to triggering Aβ aggregation and deposition.
Conclusions
AChE 7–20, a β‐hairpin region in AChE, might be a new motif in AChE capable of triggering Aβ aggregation and deposition. This finding will be helpful to design new and more effective Aβ aggregation inhibitors for AD treatment.
Keywords: Acetylcholinesterase, Alzheimer's disease, Amyloid‐β, Oligomer
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease that poses a serious threat to the life expectancy of elderly people 1. Except for familial AD caused by mutations in certain known genes, most cases of AD occur sporadically with unknown etiology 2. The neuropathological hallmarks of AD are the presence of senile plaques (SPs) and neurofibrillary tangles in the brain 3. SPs are mainly composed of amyloid‐β (Aβ), a 40‐ to 42‐amino acid fragment of the Amyloid precursor protein (APP), surrounded by dystrophic dendrites, reactive astrocytes, and activated microglia 4. Although there are intracellular and extracellular SPs in most AD brains, substantial numbers of SPs are occasionally observed in the brains of nondemented individuals 5. This suggests that Aβ deposition is not the sole cause of dementia and that there must exist some molecular factors underlying the amyloidogenic process that make the SPs pathogenic 6. Recently, a group of proteins have been identified and termed “Aβ pathological chaperones,” due to the ability to promote the conformational transformation of Aβ and to stabilize its abnormal structure. Acetylcholinesterase (AChE; EC 3.1.1.7) is one of the so‐called pathological chaperones and has received much attention in recent years.
AChE plays a critical role in the regulation of cholinergic neurotransmission by rapidly hydrolyzing acetylcholine (ACh) 7. AChE inhibitors have been used for AD therapy throughout the last decade 8. In addition to its catalytic activity, AChE has recently been regarded as an important molecular chaperone for Aβ aggregation. AChE is colocalized with Aβ in preamyloid diffuse deposits, mature senile plaques, and cerebral blood vessels in the brain of patients with AD 9. Studies have also found that the total amount and activity of AChE in AD brains are reduced, while those in the amyloid plaques are increased 10. Furthermore, in vitro studies have revealed that AChE is able to induce Aβ aggregation and form stable AChE–Aβ complexes that are more toxic than aggregated Aβ 11, 12, 13. The SPs in the brains of AChE–APP double transgenic mice emerged earlier and larger than those emerged in the brains of the single APP transgenic models. Furthermore, the memory impairment was more severe in the double transgenic models 14. Therefore, it is believed that the local increment of AChE in specific areas of the brain is the culprit to trigger Aβ aggregation and deposition and eventually causes neuropathological and behavioral events in patients with AD. However, the mechanism of the AChE–Aβ interaction has not been clearly defined. The peripheral anionic site (PAS) in AChE has been determined to be an Aβ binding site. AChE is thought to induce Aβ aggregation through PAS by mediating electrostatic interactions with the cationic area of Aβ 15. Recently, the bivalent ligand strategy has been used to design AChE inhibitors with dual binding effects, targeting the catalytic and peripheral sites. Such inhibitors could ameliorate the cognitive deficit by elevating ACh levels and delaying the formation of SPs in AD models 16. As Aβ aggregation is a highly complicated kinetic process, establishing the existence of other interaction motifs aside from PAS is of great importance for candidate drug and strategy discovery.
AD is considered to be a protein conformational disease, because it is characterized by the kinetic aggregation of Aβ, which is a typical nucleation‐dependent polymerization process 17. The β‐hairpin structure in monomeric Aβ is considered to be the key motif for the nucleation‐dependent Aβ self‐aggregation 18. As AChE can trigger and promote Aβ to aggregate, there is a high possibility that there are some β‐hairpin structures in AChE that may mediate a conformational binding with Aβ. In this study, the results of a dot blot assay showed that AChE could be recognized by A11, an oligomer‐specific antibody, indicating some oligomer‐like structure may exist in AChE. By superimposing the structures of AChE and Aβ, we discovered that the N‐terminal region of AChE had high structural similarity with the β‐hairpin domain of Aβ. Furthermore, the synthetic peptide–AChE7–20, corresponding to the 14 amino acid residues from Glu7 to Ile20 in AChE, could also be immunoblotted by oligomer‐specific antibody A11. Using electron microscopy, we observed that AChE7–20 could induce Aβ aggregation, similar to AChE. Taken together, the results suggest that in addition to PAS, AChE 7–20 may be another Aβ binding motif in AChE that triggers Aβ aggregation and deposition. Our findings may also aid development of new Aβ aggregation inhibitors.
Materials and methods
Materials and Reagents
Thioflavin T (ThT, Basic Yellow 1), AChE lyophilized powder of human source, hexafluoroisopropanol (HFIP), and absolute DMSO poured over molecular sieves were acquired from Sigma‐Aldrich (St. Louis, MO, USA). Aβ1–40 supplied as trifluoroacetate salt and rabbit polyclonal antioligomer antibody A11 were purchased from Invitrogen (Carlsbad, CA, USA). The synthetic peptide AChE7–20 was synthesized by Chinese Peptide (Hangzhou, China). Mouse monoclonal antibody 6E10 reactive to amino acid residues 1–16 of Aβ was purchased from Covance (Emeryville, CA, USA). Horseradish peroxidase–conjugated anti‐rabbit or anti‐mouse IgG were acquired from Promega Biosciences (San Luis Obispo, CA, USA). Water was deionized and double distilled (ddH2O). Buffers and other chemicals were of analytical grade.
Peptide Preparation
Lyophilized Aβ1–40 powder was initially dissolved to 500 μM in HFIP at room temperature with intermittent vibration for 2 h. Samples were centrifuged for 15 min at 14,000 × g. The supernatant was transferred to a new tube and subjected to a gentle stream of N2 for 5–10 min to evaporate HFIP. DMSO was then added so that the final concentration of Aβ1–40 was 2.3 mM and stored at −20°C. Lyophilized AChE and the synthetic peptide AChE7–20 were dissolved to 2.875 μM in ddH2O and stored at −20°C.
ThT‐Based Fluorometric Assay
The ThT‐based fluorometric assay was performed according to a published method with some modifications 19. For the self‐aggregation experiments, Aβ1–40 was incubated for 24 h at room temperature in 0.215 M sodium phosphate buffer (pH 8.0) in a final volume of 20 μL (final concentration was 125 μM). For induced aggregation experiments, AChE or AChE7–20 (final concentration was 1.25 μM) was added to 125 μM Aβ1–40 solutions and then coincubated for 24 h at room temperature. After incubation, solutions were diluted with 50 mM glycine–NaOH buffer (pH 8.5), containing 10 μM ThT, to achieve a final volume of 100 μL. Fluorescence was monitored using a PE LS45 spectrophotometer (Perkin Elmer, Waltham, MA, USA), with excitation at 446 nm and emission at 490 nm.
Molecular Superimposing
The nuclear magnetic resonance (NMR) structure of Aβ was retrieved from the RCSB Protein Data Bank (RCSB PDB, access code: 2OTK) 18. The crystallographic structure of AChE was obtained from RCSB PDB (access code: 2X8B) 20. All β‐hairpin motifs of AChE were aligned with Aβ to calculate the sequence similarity and then superimposed to estimate the structural root mean standard deviation (RMSD). All computations were carried out in Molecular Operating Environment (CCG, Canada).
Dot Blot Assay
Aβ (230 μM) was incubated with or without AChE/AChE7–20 (2.3 μM) in 0.215 M phosphate buffer (PB; pH 8.0) at 37°C for appropriate time. After the incubation period, 5 μL of each sample was applied to a nitrocellulose membrane (Millipore, Bedford, MA, USA) and blocked with 3% BSA in Tris‐buffered saline (TBS), overnight at 4°C. The membrane was incubated for 1 h at room temperature with the oligomer‐specific antibody A11 (1 μg/mL in 2% BSA in TBS with 0.01% Tween‐20 [TBS‐T]), or anti‐Aβ antibody 6E10 (1 μg/mL in 2% BSA in TBS‐T). After washed three times, the membrane was incubated with horseradish peroxidase–conjugated anti‐rabbit or anti‐mouse IgG (1:10,000) and incubated for 1 h at room temperature. The blots were developed with chemiluminescence detection kit (Pierce Chemical, Rockford, IL, USA). The remaining samples were used for transmission electron microscopy imaging 21.
Circular Dichroism Assay
Far‐UV circular dichroism (CD) assay spectra were obtained using a Jasco J‐510 spectropolarimeter (Jasco, Japan) 22. The synthetic peptide solution (100 μM) in 0.215 M PB (pH 8.0) was incubated for 2 h, 4 h, 16 h, 24 h, and 48 h at 37°C. The spectra were recorded immediately after the incubation, with background spectra recorded in the absence of peptide subtracted from the sample spectra.
Electron Microscopy
Aβ (230 μM) was incubated with or without AChE/AChE7–20 (2.3 μM) in 0.215 M PB (pH 8.0), at 37°C for appropriate time. A volume of 5 μL of each solution was applied to 150‐mesh copper grids coated with Formvar/carbon film (EM Science, Fort Washington, PA, USA) for 10 min. Excess solution was absorbed with filter paper. The grids were then stained with 2% filtered uranyl acetate for 30 seconds, absorbed with filter paper, washed twice with ddH2O, dried at room temperature, and then examined with transmission electron microscope (TEM, JEOL JEM‐1230, Japan) at 80 kV 23. Alternatively, the grids were stained with 3.5% filtered phosphotungstic acid for 2 min, the excess solution was absorbed, and the grids were dried at room temperature and scanned immediately with a transmission electron microscope (TEM, Hitachi H7650, Japan) at 80 kV 24.
Statistical Analysis
Data were presented as mean ± standard error of the mean (SEM) and were analyzed by analysis of variances (anova) followed by Dunnett's test using GraphPad Prism 5.0. Significant difference was set at P < 0.05.
Results
AChE‐Induced Aβ Aggregation and Deposition
Fluorescence intensity of ThT indicated the aggregated proteins or peptides in solution. As shown in Figure 1A, after incubation for 24 h, the ThT fluorescence intensity of Aβ was increased from 1.574 ± 0.210 to 7.354 ± 0.355 (P < 0.001), indicating a self‐aggregation of Aβ. When compared with Aβ alone, coincubation of Aβ with AChE significantly increased the ThT fluorescence to 11.130 ± 0.244 (P < 0.001). However, incubation of AChE alone only showed weak fluorescence intensity of 0.859 ± 0.149, suggesting that AChE could augment Aβ aggregation, but could not aggregate by itself.
Figure 1.
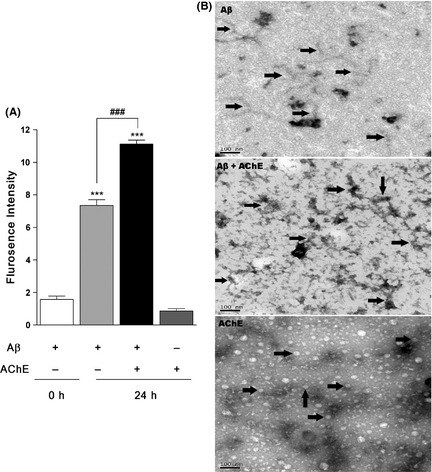
AChE induces Aβ aggregation and deposition. (A) ThT assay of AChE inducing Aβ aggregation. Solution of 125 μM Aβ in the presence and absence of 1.25 μM AChE were incubated in 0.215 M PB (PH 8.0) at 37°C for 24 h. Solutions were diluted with 10 μM ThT and the fluorescence was monitored on Varioskan Flash at an excitation of 446 nm and emission of 490 nm. Data are the mean values ± SEM (n = 4). ***P < 0.001, # # # P < 0.001. (B) Effects of AChE on inducing Aβ deposition by TEM experiments. Aβ was incubated alone or co‐incubated with AChE for 7 days at 37°C; AChE was incubated alone as a control. Samples were dropped into the grids, stained with 2% filtered uranyl acetate. Arrows indicate Aβ fibrils, senile plaques and G4 AChE, respectively. The morphologies were imaged by JOEL JEM‐1230 TEM at 80 kV. The scale bars represent 100 nm.
For AChE‐induced Aβ deposition, the morphologies were observed in TEM experiments. As shown in Figure 1B, incubation of 230 μM Aβ alone for 7 days yielded multiple fibrils. When Aβ was coincubated with 2.3 μM AChE, a mesh of numerous mature fibrils were intertwined and deposited throughout the visualized field and that was a reminiscent of the SPs in the AD brain 25. AChE is prone to form amphiphilic globular tetramers (G4 AChE), both in vivo and after incubated in vitro 26. Therefore, AChE‐alone group only showed some oligomer‐like structures in the study.
Identification of the Homologous β‐Hairpin Structure in AChE
β‐Hairpin is a crucial intermediate conformation for monomeric Aβ aggregating into fibrils 18, so searching AChE for β‐hairpin structures that can mediate a conformational binding with Aβ is very meaningful. Dot blot assays and molecular superimposing experiments were used to determine the β‐hairpin structures in AChE in this study. A11 is a conformation‐specific polyclonal antibody usually used for recognizing Aβ oligomers 27. Aβ oligomers, the most toxic Aβ aggregates, are composed mainly of β‐hairpin structures; therefore, in dot blot assays, we used A11 to detect the similar conformation. As shown in Figure 2A, Aβ incubated alone could only be identified by the A11 antibody at 48 h, but not at other shorter or longer incubation periods (e.g., 1 h, 72 h, and 120 h). This suggests that the optimal incubation time for Aβ oligomers is 48 h, as A11 could not recognize monomers or mature fibers of Aβ. After being coincubated with AChE, blots were darker than Aβ alone and could always be seen at all the time points (Figure 2A). Interestingly, AChE incubated alone for 48 h could be detected by A11, but not by 6E10, which is a sequence‐specific antibody for Aβ (Figure 2B). AChE itself, rather than Aβ, seemed to have a stronger affinity for A11, although AChE was not involved in the catalog of oligomeric proteins recognized by A11. As β‐hairpin is the core structure of oligomers, the above results strongly suggest that some β‐hairpin structures may exist in AChE.
Figure 2.
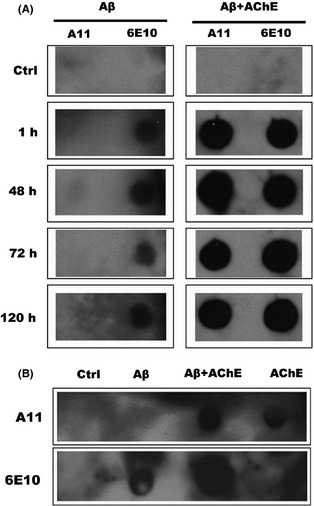
Identification of the oligomeric structure in AChE by dot‐blot assay. 230 μM Aβ was incubated alone or with 2.3 μM AChE in 0.215 M PB (pH 8.0) for (A) 1 h, 48 h, 72 h and 120 h at 37°C. (B) AChE was incubated in the presence or absence of 230 μM Aβ for 48 h at 37°C. Samples were applied to a nitrocellulose membrane and probed with either oligomer‐specific antibody A11 or sequence‐specific antibody 6E10. Buffer was used as control.
In molecular superimposing experiments, the β‐hairpin structure of Aβ, mainly Aβ16–40, was compared with all the β‐hairpin‐related structures in AChE. There were eight such structures aligned with AChE; the locations of them are listed in Table 1. The sequence similarities, which were calculated by sequence alignment, ranged from 4.5% to 36.4%, and only one of them was more than 30%, indicating little homology among them. In addition, the structural differences calculated as RMSD are shown in Table 1. The results of structural superposition are depicted in Figure 3, and it provided direct comparisons between Aβ and β‐hairpin‐related structures of AChE. It was observed that β‐hairpin A and β‐hairpin H in AChE were most similar to β‐hairpin structures in Aβ, with a structural RMSD of 2.081 Å and 2.019 Å, respectively. As β‐hairpin H located near the C‐terminal domain of AChE was hidden in the nonamphiphilic soluble tetrameric G4 form, β‐hairpin A was considered to be the most possible interaction motif to induce Aβ amyloidogenesis in studies above.
Table 1.
Sequence similarity and structural RMSD between Aβ 16–40 and all the β‐hairpin‐related structures of AChE
| β‐Hairpin structure | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| Location | 5–29 | 98–120 | 131–152 | 170–202 | 203–230 | 311–332 | 396–431 | 506–527 |
| Sequence similarity (%) | 20.0 | 8.7 | 4.5 | 12.1 | 10.7 | 36.4 | 13.9 | 13.6 |
| Structural RMSDa (Å) | 2.081 | 4.335 | 4.723 | 4.958 | 4.380 | 5.144 | 5.860 | 2.019 |
Root mean standard deviation.
Figure 3.

Structural superposition of Aβ 16–40 on all the β‐hairpin related structures in AChE. Aβ 16–40 was rendered as pink, while the β‐hairpin structures of AChE were rendered as orange. The structural differences were calculated as RMSD, as listed in Table 1. (A) β‐hairpin A (5–29) of AChE, (B) β‐hairpin B (98–120) of AChE, (C) β‐hairpin C (131–152) of AChE, (D) β‐hairpin D (170–202) of AChE, (E) β‐hairpin E (203–230) of AChE, (F) β‐hairpin F (311–332) of AChE, (G) β‐hairpin G (396–431) of AChE and (H) β‐hairpin H (506–527) of AChE.
Synthesis of the β‐Hairpin Structure in AChE
To reveal the effects of the β‐hairpin structure in AChE on Aβ amyloidogenesis, the core structure of β‐hairpin A (AChE 7–20) was synthesized, and the synthetic peptide AChE7–20 was incubated in water for 2, 4, 16, 24 and 48 h to detect its second structure by CD assay (Figure 4A). Except for the curve of water that has no absorption peak, all curves of AChE7–20 were well overlapped, with maximum absorption at 215 nm and minimum absorption at 198 nm. The results, which were calculated automatically by the spectropolarimeter, indicated that the proportion of random coil, β‐turn, and β‐sheet structures in solution was 59.9%, 25.7%, and 14.4%, respectively. Moreover, the assays were repeated under different incubatory pH or salt concentrations and showed the same results (data not shown), suggesting that AChE7–20 could keep the basic β‐hairpin‐like structure well through incubation, which was coincidental with the molecular simulation results. Furthermore, as shown in Figure 4B, synthesized peptide AChE7–20 also inherited the ability of AChE, which could be immunoblotted by A11.
Figure 4.
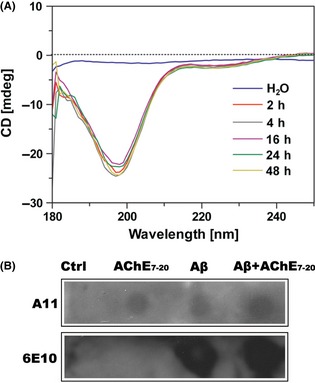
Structural identification of the synthetic peptide AChE 7–20. (A) CD spectra of AChE 7–20 secondary conformation. 100 μM AChE 7–20 was incubated in water for 2 h, 4 h, 16 h, 24 h and 48 h at 37°C. The top blue line was water used as control. (B) Identification of AChE 7–20 oligomeric structure by dot‐blot assay. AChE 7–20 or Aβ were incubated alone in 0.215 M PB (pH8.0) for 48 h, at 37°C. Buffer was used as control. Samples were applied to a nitrocellulose membrane and probed with either oligomer‐specific antibody A11 or sequence‐specific antibody 6E10. One of five repetitive experiments is shown.
Effects of the β‐Hairpin Structure in AChE
AChE7–20 inherited the capability of AChE to induce Aβ aggregation (Figure 5A). The ThT fluorescence intensity of the AChE–Aβ coincubation group was significantly increased from 6.237 ± 0.409 to 9.698 ± 1.019 (P < 0.001) in comparison with the group of Aβ that was incubated alone; this indicates that AChE7–20 could trigger Aβ aggregation in a similar manner to AChE. Little fluorescence was observed in the group of AChE7–20 alone (0.008 ± 0.002), which indicates that self‐aggregation did not occur.
Figure 5.
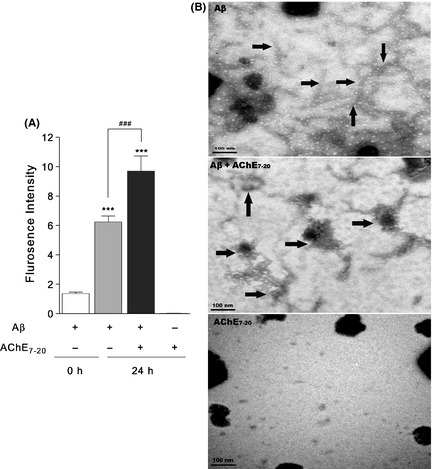
AChE 7–20 induced Aβ aggregation and deposition. (A) ThT assay of AChE 7–20 induced Aβ aggregation. Solution of 125 μM Aβ in the presence and absence of 1.25 μM AChE 7–20 were incubated 0.215 M PB (pH 8.0) at 37°C for 24 h. Solutions were diluted with 10 μM ThT, and the fluorescence was monitored on Varioskan Flash at an excitation of 446 nm and emission of 490 nm. Data are the mean values ± SEM (n = 4). ***P < 0.001, # # # P < 0.001. (B) Demonstration of the effects of AChE 7–20 induced Aβ deposition by TEM. 230 μM Aβ or 2.3 μM AChE 7–20 was incubated alone, or co‐incubated for 7 days at 37°C. Samples were dropped into grids and immediately stained with 3.5% filtered phosphotungstic acid. Arrows indicate Aβ fibrils and senile plaques respectively. The morphologies were imaged by Hitachi H7650 TEM at 80 kV. The scale bars represent 100 nm.
The morphologies of AChE7–20‐induced Aβ deposition were observed in TEM experiments. As shown in Figure 5B, coincubation of Aβ with AChE7–20 for 7 days yielded more mature fibrils than the Aβ‐alone group. AChE7–20 could also induce fibrils to intertwine and deposit similar to AChE. No visible structures were observed when incubating AChE7–20 alone.
Discussion
AChE is one of the molecules that could promote Aβ amyloidogenesis, and it is a so‐called molecular chaperone 25, 27. The results of our ThT assay and TEM experiments showed that Aβ could aggregate by itself after incubation in PB buffer, but the aggregation process was aggravated, and eventually more senile plaque‐like structures were developed in the presence of AChE (Figure 1).
A related conformational change exists when Aβ begins to aggregate. The random coil or α‐helix structure is the main structure of Aβ in its native condition. When the environment is changed or chemicals are added, native Aβ will convert into β‐sheet structure 28, 29. β‐hairpin is a conformation between α‐helix and β‐sheet, and it is a crucial intermediate conformation for monomeric Aβ to aggregate into fibrils 18. PAS of AChE, with its high negative charge density, can trigger the conformational change of Aβ from α‐helix to β‐hairpin through electrostatic effects. Therefore, PAS is regarded as the interaction motif for Aβ.
The crystal structure of AChE has been reported widely, and there is a particularly narrow gorge in the space structure of AChE, called the active gorge 20, 30, 31, 32. At the bottom of the active gorge, the active site of AChE is located where the hydrolysis of ACh occurs. PAS exists near the entrance of the gorge, where the substrates enter. Therefore, if AChE binds to Aβ in the PAS region, Aβ may block the substrates from going through the gorge, which in turn may decrease the catalytic activity of AChE, especially when Aβ forms extended fibrils. However, our in vitro AChE activity test produced inconsistent results. Compared with the control (103 ± 6%), coincubating AChE with Aβ slightly decreased the AChE activity to 92 ± 9% of control, while coincubating AChE with the PAS inhibitor propidium iodide (PI) could significantly decrease AChE activity to 80 ± 6% of control, P < 0.05 (data not shown). The results suggest that other Aβ binding sites, besides PAS, may exist, which could induce Aβ amyloidogenesis, without interfering with the catalytic effects of AChE. Indeed, because Aβ aggregation is a cascade process, PAS may play a role in helping the conformational change from α‐helix to β‐sheet, as the prolonging or depositing of aggregations seemed to be triggered by other motifs in AChE.
Alzheimer's disease is a protein conformational disease, which shows kinetic nucleation‐dependent aggregation, based on a common morphological β‐hairpin amyloid. It is thought that proteins may form an amyloid structure with Aβ through an allotypic interaction if they have generic β‐hairpin structures 17. Ten years ago, it was reported that there was a β‐hairpin structure near the C‐terminal domain of AChE, which has a similar sequence to Aβ and could aggregate in vitro 33, 34. It was suggested that the β‐hairpin region at the C‐terminal of AChE might be relevant to its interaction with Aβ. However, the C‐terminal domain of AChE is hidden inside the nonamphiphilic soluble AChE G4 tetramer, and G4 AChE is the dominant form that accounts for 80–90% of the total AChE in the brain 35. Therefore, it is impossible for AChE to bind Aβ in this area. Although this assumption seems to lack an overall understanding of the special AChE structure, it still indicated that we should pursue β‐hairpin structures for new Aβ binding motifs in AChE.
As β‐hairpin structure is the core of Aβ oligomers, we began by screening for the oligomeric structure. A11 antibody (Invitrogen) is a conformation‐specific antibody for identifying oligomeric structures. After being coincubated with AChE, Aβ was stained by A11 for the entirety of the incubation time. When Aβ was incubated alone, it was dark only at the time point of 48 h, and the color was lighter than those of the AChE‐coincubating groups. The aggregation process cannot remain at the stage of oligomer, as more fibrils will form as incubation time continues. As A11 cannot recognize fibrils, the oligomeric structure of AChE itself contributes to the dark blots of Aβ and AChE‐coincubated samples, and it is then confirmed by hybridizing AChE with A11. Furthermore, in vivo studies have revealed that SPs could be identified by A11 antibody 36. As more AChE–Aβ cosediments are observed in senile plaques, the recognition of SP by A11 should be attributed mainly to AChE, other than Aβ oligomers. Therefore, although AChE is not involved in the catalog of A11, some β‐hairpin‐like structures seemed to exist in it.
An ideal way to find out whether the oligomeric region exists would be to dock A11 into AChE. However, the sequence and structure of A11 are protected as commercial secrets. As Aβ aggregation can be induced by itself, which is mainly attributed to its β‐hairpin conformation, it is hypothesized that the exposed β‐hairpin motif may be an important factor in triggering aggregation. As a result, an alternate method to find the potential interaction motif by superimposing Aβ and AChE together is utilized. It has been demonstrated that the N‐terminal residues (7–20) of AChE (AChE 7–20) have a relatively small structural RMSD with Aβ, and this motif is exposed to the solvent (special basis for interaction). Additionally, the location of the N‐terminal 7–20 region of AChE is far from the active gorge and the C‐terminal hinge chain. Consequently, it was supposed that the N‐terminal 7–20 region of AChE could be a new motif involved in Aβ aggregation.
To prove this hypothesis, the synthesized N‐terminal peptide was produced and subjected to the described molecular biological assays. As the AChE‐induced Aβ aggregation relied upon a β‐hairpin core, not the whole N‐terminal 1–29 region, the β‐hairpin core AChE7–20 was synthesized. The AChE7–20 was characterized for its structural conformation and its ability to induce aggregation. CD study data showed the secondary structure of the synthesized peptide was mostly random coil and β‐turn. It almost maintained the same structure in solutions of different pH levels or different salt concentrations and for different incubation times. The β‐turn conformation of the synthetic peptide AChE7–20 basically inherited the secondary structure of the β‐hairpin region of AChE 7–20, and this formed the structural basis for the following molecular biological effects. Subsequent results of AChE7–20 in dot blot assays were coincident with the supposition that AChE 7–20 was an oligomeric region. AChE7–20 also possessed the capability of AChE to induce Aβ aggregation. The intensity of the fluorescence of ThT increased when Aβ was coincubated with AChE7–20, and the increased amount was almost equal to that of AChE. Moreover, in spite of slightly less mature fibrils, Aβ–AChE7–20‐coincubated samples showed similar SPs‐like morphology as the Aβ–AChE‐coincubated group. Self‐aggregation did not occur in AChE7–20, because it was just the core of a β‐hairpin region that was too short to self‐aggregate easily. Thus, the N‐terminal domain of AChE was exactly a core structure, which was independent of PAS, and could be another motif in Aβ amyloidogenesis.
In conclusion, our results have demonstrated that there is an oligomeric domain near the N‐terminal of AChE, and the synthetic peptide of this region shows similar immunological properties to AChE, and it is able to promote Aβ aggregation and deposition. Thus, in addition to PAS, the N‐terminal region of AChE (AChE 7–20) may be a new motif involved in Aβ pathogenesis. Furthermore, considering the relationship between Aβ, AChE and SPs in the AD brain, this finding paves a new way to design novel and more active Aβ aggregation inhibitors for AD treatment.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgment
This work was supported by the National Basic Research Program of China (Grant No. 2010CB529806), the National Natural Science Foundation of China to Hong‐Zhuan Chen (Grant Nos. 30973509 and 81173084), the International Science & Technology Cooperation Program of China (2011DFA33180), and the National Innovative Drug Development Project (Grant Nos. 2009ZX09103‐077 and 2012ZX09303‐003).
The first two authors contributed equally to this work.
References
- 1. Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 2001;81:741–766. [DOI] [PubMed] [Google Scholar]
- 3. Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006;314:777–781. [DOI] [PubMed] [Google Scholar]
- 4. Mattson MP. Pathways towards and away from Alzheimer's disease. Nature 2004;430:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muñoz FJ, Inestrosa NC. Neurotoxicity of acetylcholinesterase amyloid beta‐peptide aggregates is dependent on the type of Abeta peptide and the AChE concentration present in the complexes. FEBS Lett 1999;450:205–209. [DOI] [PubMed] [Google Scholar]
- 6. Cleary JP, Walsh DM, Hofmeister JJ, et al. Natural oligomers of the amyloid‐beta protein specifically disrupt cognitive function. Nat Neurosci 2005;8:79–84. [DOI] [PubMed] [Google Scholar]
- 7. Martorana A, Esposito Z, Koch G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease? CNS Neurosci Ther 2010;16:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mancuso C, Siciliano R, Barone E, Butterfield DA, Preziosi P. Pharmacologists and Alzheimer disease therapy: to boldly go where no scientist has gone before. Expert Opin Investig Drugs 2011;20:1243–1261. [DOI] [PubMed] [Google Scholar]
- 9. Morán MA, Mufson EJ, Gómez‐Ramos P. Colocalization of cholinesterases with beta amyloid protein in aged and Alzheimer's brains. Acta Neuropathol 1993;85:362–369. [DOI] [PubMed] [Google Scholar]
- 10. Ulrich J, Meier‐Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol 1990;80:624–628. [DOI] [PubMed] [Google Scholar]
- 11. Luo W, Li YP, He Y, et al. Synthesis and evaluation of heterobivalent tacrine derivatives as potential multi‐functional anti‐Alzheimer agents. Eur J Med Chem 2011;46:2609–2616. [DOI] [PubMed] [Google Scholar]
- 12. Reyes AE, Chacón MA, Dinamarca MC, Cerpa W, Morgan C, Inestrosa NC. Acetylcholinesterase‐Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: effect on rat beta‐amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am J Pathol 2004;164:2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura S, Kawashima S, Nakano S, Tsuji T, Araki W. Subcellular distribution of acetylcholinesterase in Alzheimer's disease: abnormal localization and solubilization. J Neural Transm Suppl 1990;30:13–23. [DOI] [PubMed] [Google Scholar]
- 14. Rees TM, Berson A, Sklan EH, et al. Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice. Curr Alzheimer Res 2005;2:291–300. [DOI] [PubMed] [Google Scholar]
- 15. Reyes AE, Perez DR, Alvarez A, et al. A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem Biophys Res Commun 1997;232:652–655. [DOI] [PubMed] [Google Scholar]
- 16. Xie Q, Wang H, Xia Z, et al. Bis‐(‐)‐nor‐meptazinols as novel nanomolar cholinesterase inhibitors with high inhibitory potency on amyloid‐beta aggregation. J Med Chem 2008;51:2027–2036. [DOI] [PubMed] [Google Scholar]
- 17. Kisilevsky R, Fraser PE. A beta amyloidogenesis: unique, or variation on a systemic theme? Crit Rev Biochem Mol Biol 1997;32:361–404. [DOI] [PubMed] [Google Scholar]
- 18. Hoyer W, Grönwall C, Jonsson A, et al. Stabilization of a beta‐hairpin in monomeric Alzheimer's amyloid‐beta peptide inhibits amyloid formation. Proc Natl Acad Sci USA 2008;105:5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc 2006;1:2406–2415. [DOI] [PubMed] [Google Scholar]
- 20. Carletti E, Colletier JP, Dupeux F, Trovaslet M, Masson P, Nachon F. Structural evidence that human acetylcholinesterase inhibited by tabun ages through O‐dealkylation. J Med Chem 2010;53:4002–4008. [DOI] [PubMed] [Google Scholar]
- 21. Yoshiike Y, Minai R, Matsuo Y, Chen YR, Kimura T, Takashima A. Amyloid oligomer conformation in a group of natively folded proteins. PLoS ONE 2008;3:e3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M, Jiji RD. Spectroscopic detection of β ‐sheet structure in nascent Aβ oligomers. J Biophotonics 2011;4:637–644. [DOI] [PubMed] [Google Scholar]
- 23. Fezoui Y, Teplow DB. Kinetic studies of amyloid beta‐protein fibril assembly. Differential effects of alpha‐helix stabilization. J Biol Chem 2002;277:36948–36954. [DOI] [PubMed] [Google Scholar]
- 24. Doi Y, Mizuno T, Maki Y, et al. Microglia Activated with the Toll‐Like Receptor 9 Ligand CpG Attenuate Oligomeric Amyloid β Neurotoxicity in in Vitro and in Vivo Models of Alzheimer's Disease. Am J Pathol 2009;175:2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inestrosa NC, Alvarez A, Pérez CA, et al. Acetylcholinesterase accelerates assembly of amyloid‐beta peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron 1996;16:881–891. [DOI] [PubMed] [Google Scholar]
- 26. Fernandez HL, Moreno RD, Inestrosa NC. Tetrameric (G4) acetylcholinesterase: structure, localization, and physiological regulation. J Neurochem 1996;66:1335–1346. [DOI] [PubMed] [Google Scholar]
- 27. Fernàndez‐Busquets X, Ponce J, Bravo R, et al. Modulation of amyloid beta peptide(1–42) cytotoxicity and aggregation in vitro by glucose and chondroitin sulfate. Curr Alzheimer Res 2010;7:428–438. [DOI] [PubMed] [Google Scholar]
- 28. Ma B, Nussinov R. Stabilities and conformations of Alzheimer's beta –amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): sequence effects. Proc Natl Acad Sci USA 2002;99:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carrell RW, Lomas DA. Conformational disease. Lancet 1997;350:134–138. [DOI] [PubMed] [Google Scholar]
- 30. Harel M, Schalk I, Ehret‐Sabatier L, et al. Quaternary ligand binding to aromatic residues in the active‐site gorge of acetylcholinesterase. Proc Natl Acad Sci USA 1993;90:9031–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanson B, Colletier JP, Xu Y, et al. Backdoor opening mechanism in acetylcholinesterase based on X‐ray crystallography and molecular dynamics simulations. Protein Sci 2011;20:1114–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheung J, Rudolph MJ, Burshteyn F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 2012;55:10282–10286. [DOI] [PubMed] [Google Scholar]
- 33. Cottingham MG, Hollinshead MS, Vaux DJ. Amyloid fibril formation by a synthetic peptide from a region of human acetylcholinesterase that is homologous to the Alzheimer's amyloid‐beta peptide. Biochemistry 2002;41:13539–13547. [DOI] [PubMed] [Google Scholar]
- 34. Cottingham MG, Voskuil JL, Vaux DJ. The intact human acetylcholinesterase C‐terminal oligomerization domain is alpha‐helical in situ and in isolation, but a shorter fragment forms beta‐sheet‐rich amyloid fibrils and protofibrillar oligomers. Biochemistry 2003;42:10863–10873. [DOI] [PubMed] [Google Scholar]
- 35. Collins FS, Drumm ML, Cole JL, Lockwood WK, Vande Woude GF, Iannuzzi MC. Construction of a general human chromosome jumping library, with application to cystic fibrosis. Science 1987;235:1046–1049. [DOI] [PubMed] [Google Scholar]
- 36. Rijal Upadhaya A, Capetillo‐Zarate E, Kosterin I, et al. Dispersible amyloid β‐protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol Aging 2012;33:2641–2660. [DOI] [PubMed] [Google Scholar]