

Summary
Introduction
Patients with temporal lobe epilepsy (TLE) often suffer from comorbid psychiatric diagnoses such as depression, anxiety, or impaired cognitive performance. Endocannabinoid (eCB) signaling is a key regulator of synaptic neurotransmission and has been implicated in the mechanisms of epilepsy as well as several mood disorders and cognitive impairments.
Aims
We employed a pilocarpine model of TLE in C57/BJ mice to investigate the role of eCB signaling in epileptogenesis and concomitant psychiatric comorbidities.
Methods and Results
We sought to alter the neuronal levels of a known eCB receptor ligand, 2‐arachidonylglycerol (2‐AG), through the use of RHC80267 or JZL184. Pilocarpine‐treated mice were treated with RHC80267 (1.3 μmol) or JZL184 (20 mg/kg) immediately after the termination of status epilepticus (SE), which was followed by daily treatment for the next 7 days. Our results indicated that RHC80267 treatment significantly reduced the percentage of mice suffering from spontaneous recurrent seizures (SRS) in addition to decreasing the duration of observed seizures when compared to vehicle treatment. Furthermore, RHC80267 attenuated depression and anxiety‐related behaviors, improved previously impaired spatial learning and memory, and inhibited seizure‐induced hippocampal neuronal loss during the chronic epileptic period. In contrast, JZL184 administration markedly increased the frequency and the duration of observed SRS, enhanced the previously impaired neuropsychological performance, and increased hippocampal damage following SE.
Conclusions
These findings suggest that RHC80267 treatment after the onset of SE could result in an amelioration of the effects found during the chronic epileptic period and yield an overall decrease in epileptic symptoms and comorbid conditions. Thus, alterations to endocannabinoid signaling may serve as a potential mechanism to prevent epileptogenesis and manipulation of this signaling pathway as a possible drug target.
Keywords: Endocannabinoid, Epilepsy, Epileptogenesis, Hippocampus, JZL184, Neuropsychological performance, RHC80267
Introduction
Epilepsy is a common neurological disorder that is frequently complicated by psychiatric comorbidities such as depression, anxiety, and impaired cognitive performance 1. Such comorbidities often decrease the quality of life in patients with epilepsy, oftentimes being more debilitating than the primary epilepsy diagnosis itself 2. Moreover, mounting evidence showed that psychiatric disorders themselves may act as risk factors for certain kinds of epilepsy and may even exacerbate the progression of this disease 3. Due to the strong link between psychiatric disorders and epilepsy, there is an increased need to develop novel disease‐modifying therapeutic strategies for the effective treatment of epileptic patients exhibiting these comorbid conditions.
Endocannabinoid signaling is a key regulator of synaptic neurotransmission throughout the brain, and its main function is to modulate neuronal excitability and protect against hyperexcitability 4, 5. To this end, cannabinoid compounds, as well as activation of CB1 receptors or increasing the level of endocannabinoid through other means, have been shown to have anticonvulsant effects in several in vitro and in vivo seizure models 6, 7, 8, 9, 10. Importantly, recent findings have suggested that the endocannabinoid signaling pathway is disrupted in chronic epilepsy 11, 12, 13 and that altered endocannabinoid tone may contribute to the pathophysiological mechanisms underlying epileptogenesis 14, 15, 16. In addition, both direct and indirect activation of CB1 receptors have been found to produce a broad range of effects on emotional 17, 18 and cognitive regulation 19, 20, 21.
Thus far, the most well‐characterized endocannabinoids include arachidonoylethanolamide (AEA) and 2‐arachidonoylglycerol (2‐AG). Unlike AEA, 2‐AG is present at relatively high levels in the central nervous system and acts as a full agonist at type 1 cannabinoid receptors (CB1R) 22, 23. It is also the most abundant molecular species of monoacylglycerol found in mouse and rat brain 24. However, the specific role of 2‐AG signaling in patients with epilepsy and psychiatric comorbid conditions remains unclear. Recently, many compounds that control 2‐AG formation and degradation are emerging, such as 1,6‐bis‐(cyclohexyloximinocarbonylamino)‐hexane (RHC80267) and 4‐nitrophenyl‐4‐(dibenzo [d] 1, 3 dioxol‐5‐yl [hydroxy] methyl) piperidine‐1‐carboxylate (JZL184), thereby providing an important means for the study of 2‐AG. RHC80267 is an inhibitor of diacylglycerol lipase (DGL‐α), an enzyme that generates 2‐AG from diacylglycerol, while JZL184 is an irreversible inhibitor for monoacylglycerol lipase (MAGL), which is the primary enzyme responsible for degrading 2‐AG 25. Previous studies have shown that administration of these compounds resulted in a dramatic alteration of brain 2‐AG levels, subsequently leading to several cannabinoid‐related behavioral effects 26, 27. Accordingly, in this study, using a pilocarpine model of temporal lobe epilepsy, we aimed to investigate the effects of RHC80267 and JZL184 on the epileptogenesis and neuropsychological and cognitive performances in epileptic mice.
Materials and Methods
Animals
Male C57/BJ mice at 8–10 weeks of age were obtained from the Experimental Animal Center of the Fourth Military Medical University (FMMU, Xi'an, China). Their weight ranged from 20 to 25 g at the time of acquisition, after which they were housed under closely monitored environmental conditions (temperature, 24–25°C; humidity, 50–60%) and on a 12‐h light/dark cycle (lights on at 8:00 A.M.). Food and water were provided ad libitum. All experimental animals were habituated to the laboratory environment for 7 days prior to the start of the study. All procedures used were in strict accordance with the guidelines established by the U.S. NIH and were approved by the Fourth Military Medical University Animal Care Committee and were conducted such as to minimize animal number and distress.
This study consists of two experiments with different specific aims. Experiment 1 aimed to examine the effects of JZL184 on the epileptogenesis and neuropsychological and cognitive performances. Two groups of mice were used. The first group was the status epilepticus (SE) + saline (i.p.) group, in which subjects received a saline injection instead of JZL184 immediately after the interruption of SE, followed by twice daily i.p. injections of saline. The second group was the SE + JZL184 (i.p.) group; these mice received injections of JZL184 immediately after the termination of SE, which was followed by twice daily injections (i.p.) of JZL184. Experiment 2 aimed to investigate the effects of RHC80267 on the epileptogenesis, neuropsychological, and cognitive performances. In this experiment, each mouse received implantation of a stainless steel guide cannula into the right lateral ventricle under anesthesia (chloral hydrate, 400 mg/kg, i.p.). A stainless steel guide cannula was then inserted into the right lateral ventricle at the following coordinates: 0.4 mm posterior to bregma, 1.0 mm lateral from midline, and a depth of 2.5 mm from the skull surface. The location of the cannula tip in the ventricle was verified by visual observation of the free outflow of the cerebrospinal fluid through the cannula. After affixing each cannula with dental cement, all animals were singly housed and allowed to recover for 7 days until they had regained their preoperative body weight and had no visible signs of infection. Then, animals were randomly divided into two groups. One group of mice received a saline injection (i.c.v.) immediately after the termination of SE, which was followed by saline i.c.v. once a day (SE + saline (i.c.v.) group). Another group received RHC80267 i.c.v. immediately after the interruption of SE, which was then followed by RHC80267 i.c.v. once a day (SE + RHC [i.c.v.] group). In addition, a group of age‐matched controls without SE were also included in this study. The flowchart of experimental procedure was shown in Figure 1.
Figure 1.
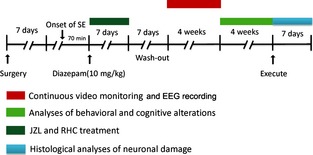
Schematic illustration of the experimental protocol used in this study. Surgery: mouse received implantation of stainless steel guide cannula and EEG electrode together 7 days before the induction of status epilepticus (SE).
Induction of Status Epilepticus
To verify the occurrence of both SE and reduction in mortality, a ramping‐up protocol was used according to previously published methods 28. Specifically, mice were given an i.p. dose of 100 mg/kg pilocarpine (0.9% saline vehicle; Sigma‐Aldrich, St. Louis, MO, USA) every 20 min until the onset of SE. We defined SE according to two acceptable categories: those that were continuous in nature (lasting for an hour to several hours if not terminated by diazepam) or intermittent seizures that did not have a full recovery between seizure episodes 29. The maximum number of repeated pilocarpine injections was restricted to fourteen injections. To reduce the peripheral effects of pilocarpine, mice were pretreated with methylscolpolamine (1 mg/kg, i.p.; Sigma‐Aldrich) 20 min prior to pilocarpine injection. After 70 min of SE, seizures were terminated with the administration of diazepam (10 mg/kg, i.p.).
Animals were randomly assigned to either the control or experimental group. Age‐matched controls (n = 5) received all pre‐ and postpharmacological treatments (methylscopolamine, diazepam), but were administered a vehicle injection of physiological saline rather than pilocarpine. All subjects were closely observed during periods of SE and given supportive care (e.g., food soaked in a 5% glucose solution for 3 days) until normal eating and drinking behavior resumed.
Drug Treatment
We determined the proper drug administration, dosage, and dosing interval from previously published work 19. JZL184 was obtained from Caymen Chemical (Ann Arbor, MI, USA) and dissolved in a vehicle consisting of: 15% DMSO, 4.25% polyethylene glycol 400, 4.25% Tween‐80, and 76.5% saline for chronic intraperitoneally (i.p.) administration (20 mg/kg, 7 days, twice daily). RHC80267 (Biomol, Plymouth Meeting, PA, USA) was dissolved in 100% N, N‐dimethylformamide (DMF) and delivered via a 5‐μL Hamilton syringe that had been threaded into the cannula of each restrained subject. Each injection was administered intracerebroventricularly (i.c.v., 1.3 μmol/animal) for 7 days at a frequency of once per day. Other reagents in this study were purchased from Sigma‐Aldrich and dissolved in a 75% saline vehicle prior to use. In this study, the volume of solutions to be administrated either i.p. or i.c.v. was 10 mL/kg and 1 μL, respectively.
Monitoring of Spontaneous Recurrent Seizures (SRS)
The mice were observed for the development of SRS from the third to the sixth week after pilocarpine‐induced SE. Epileptic mice were video‐monitored (HVR‐S270C; Sony, Tokyo, Japan) for 12 h per day. The frequency, severity, and duration of daily seizures were analyzed according to the observed behavioral seizures by video recordings. Recordings were scored by two independent and blind observers who did not know the results of group allocation. Behavioral seizures were assessed according to modified Racine's scale (1972). In addition to video monitoring, any spontaneous seizures that occurred during handling or other experimental manipulations were also recorded.
EEG data were used to confirm behavioral seizures in this study. For EEG recordings, mice were stereotaxically implanted with cortical EEG electrodes as described previously 30, after which animals were allowed to recover for 1 week. A monopolar depth electrode was implanted into the right hippocampal CA1 region (coordinates vs. bregma: −1.94 mm anteroposterior, −1.0 mm lateral, −1.25 mm depth). Three monopolar cortical electrodes, frontal left (+1.0 mm anteroposterior, −2.0 mm lateral) and occipital left/right (−4.0 mm anteroposterior, ±4 mm lateral), were also positioned on the dura mater. A ground electrode was placed in the left prefrontal bone. EEG recordings were conducted using Nihon Kohden 9200 Studio software (QP‐219BK; Nihon Kohden, Tokyo, Japan). Signals that were <1 Hz or >45 Hz were filtered out by the software. Within EEG recordings, nonictal discharges were distinguished from ictal discharges on the basis of waveform morphology, frequency, and the associated behavioral alterations as previously described 31. Finally, behavioral patterns that occurred during the EEG recordings were coded from time‐locked video recordings. Frequency and power analyses of EEG data were performed by uploading the data to automated program for EEG analysis (MATLAB R2013B; The Mathworks, Inc., Natick, MA, USA and AcqKnowledge 4.2; BIOPAC Systems, Inc., Worcester, UK, AD Instruments Ltd., Dunedin, New Zealand).
Behavioral Evaluations
Behavioral tests were started 6 weeks after SE and conducted over a time period of 4 weeks. Our control group consisted of an age‐matched, nonepileptic cohort (n = 5). These mice received all pre(methylscopolamine) and post(diazepam) pharmacological treatments for 7 days, except for pilocarpine.
Before each behavioral test, mice were habituated to the experimental room for 30 min. All tests were performed in a dim back room between 9:00 A.M. and 4:00 P.M. by the same two experimenters. Behavioral tests were conducted only when no SRS had been observed for at least 1 h prior to the start of testing. If an SRS happened at any point during the test, the subject was placed back into its cage and the test repeated 1 h later. Our behavioral battery included the open field test, elevated plus maze, tail suspension test, and Morris water maze, the sum of which was used to evaluate the level of spontaneous locomotor and exploratory activities, anxiety‐ and depressive‐like behaviors, as well as visual–spatial memory in all subjects.
Open Field Test
Mice were placed in the center of a square open field (50 × 50 × 40 cm) which was divided into 25 equal squares (5 × 5 cm) that all contained peripheral and center areas. Subjects were monitored for 5 min by a suspended camera that was connected to a computer. This system was used to record the path of each subject as well as count total distance covered, time spent in each zone (peripheral or center), and the number of times rearing occurred. Before each test, 20% alcohol was used to clean the open field to remove any odors from prior subjects.
Elevated Plus Maze
The elevated plus maze was consisted of two pairs of opposing arms (open arms dimensions of 30 × 5×0.5 cm and enclosed arms dimensions of 30 × 5×15 cm) that overlapped by a common central platform (5 × 5 cm) and was raised 40 cm above the floor. Each trial was begun by placing the subject in the center area and allowing them to face one of the open arms. All test sessions lasted for 5 min and were recorded by a camera fixed above the apparatus. We then analyzed the total distance moved and the time the subject spent in the open arms. Before each test, the maze was cleaned with 20% alcohol to remove any odors from prior subjects.
Tail Suspension Test
Each mouse was suspended 30 cm above the floor by a piece of adhesive tape which was affixed approximately 1 cm from the tail tip. Each test lasted for 6 min. We defined immobility as the amount of time each subject remained motionless. Each trial was manually scored by a blind observer and stopped when either the time elapsed or once the mouse climbed their tail or dropped from the attachment. All trials were conducted in a dimly lit and soundproofed room.
Morris Water Maze
A round swimming pool (120 cm diameter, 50 cm height) was used as the water maze apparatus, which was filled with water to a depth of 22 cm and maintained at a constant temperature of 22 ± 1°C. Milk power was dissolved in the water to make it opaque, thereby hiding a transparent escape platform (diameter of 10 cm). The escape platform was placed in the middle of one of the four quadrants. Digital tracking system and image analyzer were connected to a camera hanging vertically down to the center of the pool, which enabled them monitoring of subject swim patterns and escape latency.
The total time for this experiment was 7 days and included a familiarization phase, acquisition phase, and final probe test. In the familiarization phase, the platform was situated 1.0 cm below the water surface in the same quadrant each time. Mice were put into the maze facing the wall at one of the four starting positions and allowed 60 seconds to swim and find the platform. When they found the platform, subjects were allowed an additional 30 seconds to rest. Subjects that failed to find the platform in the allotted time were gently guided in the correct direction by the experimenter. In these instances, the escape latency was recorded as 60 seconds. Going in a clockwise manner, animals were placed into another two quadrants, different from the initial starting points as previously described. After 24 h from the end of the last acquisition session, the platform was removed from the maze for a single‐trial probe test, which is used to evaluate the spatial memory ability of subjects. The trial lasted for 60 seconds. At the start, mice were placed in the center of the maze, and the number of times they crossed over the four areas previously inhabited by the escape platform was recorded.
Histological Assessment of Neurodegeneration
Following behavioral testing, both pilocarpine‐treated mice and age‐matched controls were deeply anesthetized with chloral hydrate and transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Cresyl‐violet staining was used on 25‐μm‐thick slices derived from both sets of tissue. The number of hippocampal neurons was counted in every sixth section in three areas of the hippocampus: CA1 (four fields), CA3 (two fields), and the lateral area of the dentate gyrus (DG) in a mediolateral direction. Cells with a clear membrane and a visible nucleolus were counted in five sections from each animal. This measurement was then used to determine the relative change in the number of neurons between the experimental groups and controls. Any cells that had a somatic size of <3 μm were characterized as either glial or necrotic and were excluded from further analysis 32. Visual assessment was conducted by two blind observers, and all images were captured on an Olympus BX51 microscope equipped with a digital imaging system (Olympus DP70 digital camera and software, Optronics, Goleta, CA, USA). Images were analyzed using Image Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD, USA).
Statistics
Parametric or nonparametric statistical tests were used based on the normal or nonnormal distribution of the data. Fisher's exact test was used to calculate significant differences in the incidence of spontaneous seizures. Mann–Whitney U‐test was used to calculate significant differences in the seizure frequency, the duration, and the severity of observed seizures. EEG, behavioral, and histological data were analyzed using an ANOVA with subsequent post hoc testing for individual differences via the Bonferroni test. If data were not normally distributed, an ANOVA for nonparametric data (Kruskal–Wallis test), followed by post hoc testing for individual differences (Dunn's test), was used. Data management and statistical analyses were performed in SPSS v16.0 (SPSS, Chicago, IL, USA). All statistical significance was determined at P < 0.05.
Results
Induction of SE by Pilocarpine and Development of Spontaneous Seizures after SE
Among all 60 mice injected with pilocarpine, 44 developed SE and survived to the end of testing, 14 mice died during or shortly after SE, and two mice died during the video‐monitoring period. The final size of the pilocarpine‐treated groups used in this study was 11 (SE + saline [i.p.] group), 10 (SE + JZL [i.p.] group), 11 (SE + saline [i.c.v.] group), and 12 (SE + RHC [i.c.v.] group). SE was characterized by continuous seizure activity in the limbs, rearing and falling of the animal, straub tail, and repeated head twitches. Occasional running and jumping seizures as well as generalized clonic–tonic seizures were also observed during SE. Seizures were not observed during any time point during experimental testing in control mice (n = 5). Treatment with JZL184 or RHC80267 after SE had no effect on either the EEG recordings or the behavior that was assessed by video recordings (data not shown).
After the induction of SE and subsequent drug washout, we used video recordings to measure the total number, severity, and duration of observed SRS (Racine 4 or 5) for 4 weeks during the chronic epileptic phase. In a subset of mice (n = 4 in SE + saline [i.p.] group; n = 3 in SE + JZL [i.p.] group; n = 4 in SE + saline [i.c.v.] group; n = 4 in SE + RHC [i.c.v.] group), the EEG was recorded via cortical electrodes, illustrating that clinical seizures were associated with paroxysmal EEG alterations similar to those previously described 28. During the period of video recordings, observed SRSs were associated with paroxysmal high‐frequency spike activity with abrupt onset and termination in the EEG (Figure 2A). Furthermore, interictal EEG abnormalities (frequent spikes occurring either isolated or in short bursts) were also recorded (not illustrated). We did not observed distinct differences among SE groups in the power spectrum analysis of interictal EEG signals in chronic period (Figure 2 B,C).
Figure 2.
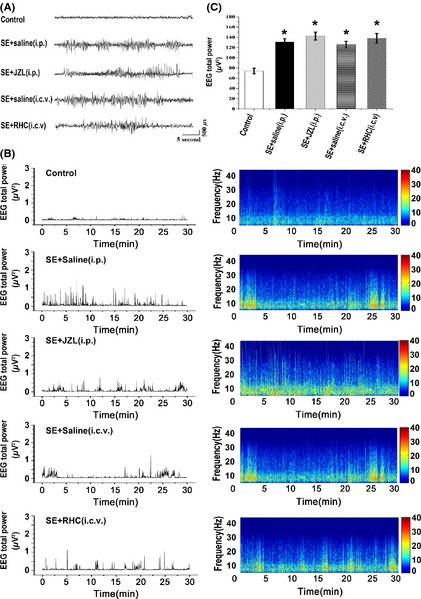
Cortical EEG recording from mice during chronic epileptic period. (A) normal EEG in a control mouse, and typical paroxysmal EEG alterations in SE + saline (i.p.) group, JZL‐treated SE group, SE + saline (i.c.v.) group, or RHC‐treated SE group. (B) graphs show total EEG power (left) and the frequency and amplitude parameters (right) during chronic epileptic period in each group. (C) statistical analysis of data by ANOVA indicates that total EEG power is higher in SE mice compared with control mice (P < 0.05). However, there is no significant difference among SE groups in the power spectrum analysis of interictal EEG signals (P > 0.05).
During the video‐monitoring period that occurred after SE, spontaneous seizures were found in 11 (100%) of the SE + saline (i.p.) group, 11 (100%) of the SE + JZL (i.p.) group, 10 (90.9%) of the SE + saline (i.c.v.) group, and 9 (75%) of the SE + RHC (i.c.v.) group (Figure 3A). Results indicate that RHC80267 treatment significantly reduced the percentage of mice that experienced SRS (P < 0.05, compared with SE + saline [i.c.v.] group). Within a similar median seizure frequency in vehicle‐treated (SE + saline [i.p.] group, 19 per week; SE + saline [i.c.v.] group, 20 per week) and RHC‐treated mice (18 per week), the JZL‐treated mice exhibited a markedly increased seizure frequency (26 per week; P < 0.05, compared with SE + saline [i.p.] group; Figure 3B). There was no statistical difference in the severity of SRS across all groups (Figure 3C; P > 0.05). The averaged duration of observed seizures was 28 ± 0.8 seconds in SE + saline (i.p.) group, 31 ± 1.0 seconds in SE + JZL (i.p.) group, and 29 ± 1.3 seconds in SE + saline (i.c.v.) group. When compared to vehicle‐treated SE mice, RHC‐treated mice showed significantly decreased seizure duration (16 ± 1.6 seconds; Figure 3D; P < 0.05). These results indicate that early treatment with RHC80267 after SE decreased both the occurrence and the duration of SRS.
Figure 3.
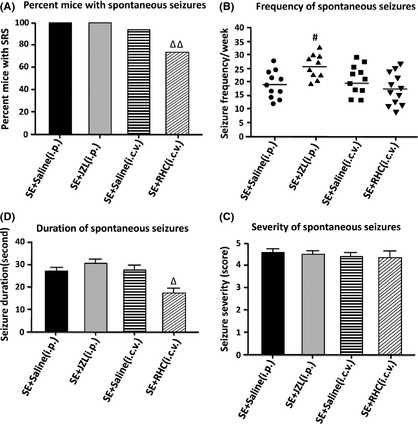
Effects of drug treatments on development of spontaneous recurrent seizures (SRS). SRS were recorded by video and EEG monitoring from 3 to 6 weeks following SE. (A) the percentage of mice per group that exhibited spontaneous seizures during video/EEG monitoring. (B) the frequency of spontaneous seizures. (C) seizure severity. (D) seizure duration. Data in C and D are shown as mean ± SEM, while individual values and median seizure frequency are shown in B. Analysis of data in A by Fisher's exact test indicated that RHC treatment significantly reduced the percentage of mice that had experienced SRS compared with the vehicle‐treated group (P < 0.05). Analysis of data in B and C indicated that JZL‐treated mice exhibited a markedly increased seizure frequency (P < 0.05), while the duration of observed seizures was significantly decreased in RHC‐treated mice (P < 0.05). Analysis of data in D indicated that there was no statistical difference in the severity of SRS across all groups. (# P < 0.05 compared with SE + saline [i.p.] group; Δ P < 0.05 compared with SE + saline [i.c.v.] group).
Behavioral Alterations during Epileptogenesis
The open field test and elevated plus maze (EPM) were both used to examine the effect of RHC80263 or JZL184 on anxiety‐like behaviors in epileptic mice. Results showed that the SE + JZL (i.p.) group spent significantly more time near the periphery of the open field and shorter amount of time near the center when compared to SE mice treated with saline, indicating an increase in anxiety‐like behaviors in the JZL‐treated group (Figure 4, P < 0.05). Additionally, rearing activity and the distance traveled by JZL‐treated mice were also significantly decreased when compared to vehicle‐injected SE mice (Figure 4, P < 0.05).
Figure 4.
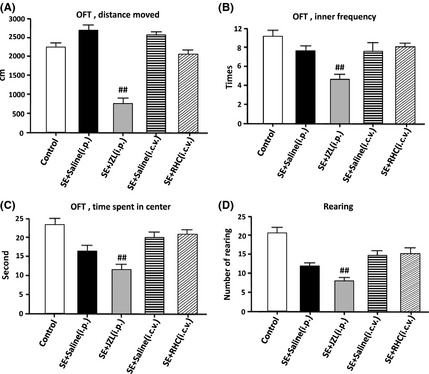
Behavioral alterations of drug‐treated epileptic mice in the open field test. SE mice without spontaneous recurrent seizures were excluded, and all remaining mice used for data analysis had developed epilepsy during the chronic stage. Behavioral testing in the open field was performed 7–10 weeks after SE. (A) the total distance that the mice moved during the open field test. (B): (C): the time that mice spent in the center of the open field. (C) the frequency that mice enter the center of the open field. (D) the number of rearings in the open field. Analysis of data by ANOVA indicated significant differences between JZL‐treated mice and SE mice treated with vehicle‐injected saline. JZL‐treated group spent a significantly smaller amount of time in the center of the open field (P < 0.01). Rearing activity and the distance traveled by JZL‐treated mice in the open field were also significantly reduced (P < 0.01). (## P < 0.01 compared with SE + saline [i.p.] group).
In the elevated plus maze, JZL‐treated mice tended to travel less and remain in the open arms for a shorter amount of time (Figure 5). Contrastingly, RHC‐treated mice spent a greater percentage of time in the open arms than vehicle‐injected controls. Altogether, these results demonstrate that administration of JZL184 increases anxiety‐like responses, whereas RHC80267 treatment slightly attenuates anxiety‐like behaviors.
Figure 5.
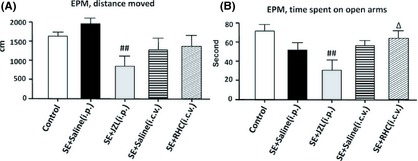
Behavior alterations of drug‐treated epileptic mice in the elevated plus maze. (A) the total distance that the mice moved during the test. (B) the time that mice spent in the open arms of the maze. JZL‐treated mice tended to travel and stay for a smaller amount of time in the open arm of the maze (P < 0.01). RHC‐treated mice spent a greater percentage of time on the open arms than vehicle‐injected controls (P < 0.05). (## P < 0.01 compared with SE + saline [i.p.] group; Δ P < 0.05 compared with SE + saline [i.c.v.] group).
The tail suspension test indicated that JZL‐treated mice were immobile for significantly longer periods of time when compared to vehicle‐injected controls (P < 0.01, compared with SE + saline (i.p.) group; Figure 6). Conversely, immobilility was remarkably reduced by RHC80267 administration (P < 0.05, compared with SE + saline (i.c.v.) group; Figure 6).
Figure 6.
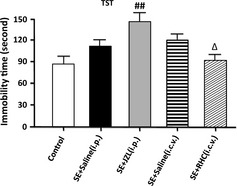
Behavior alterations of drug‐treated epileptic mice in the tail suspension test. Analysis of data by ANOVA indicated that immobility time was increased significantly in subjects administered JZL184 (P < 0.01) and reduced in subjects given RHC (P < 0.05). (## P < 0.01 compared with SE + saline [i.p.] group; Δ P < 0.05 compared with SE + saline [i.c.v.] group).
In the Morris water maze, epileptic mice exhibited significant learning and memory impairments when compared to controls without SE. This conclusion is based on an increased time to locate the platform (escape latency) over the whole training period when compared with control (Figure 7A). Results revealed that RHC‐treated SE animals learned significantly better than SE animals receiving vehicle injections (P < 0.05). However, JZL‐treated animals learned significantly more slowly than vehicle‐injected control animals (P < 0.05) and also tended to swim in circles away from the destination rather than in a more strategic route. These differences were not likely to be accounted for by differences in swim speed, as an analysis of mean quadrant crossing time revealed no significant differences between groups (P > 0.05, data not shown). During probe trial test (escape platform removed), the number of target area crossings was recorded. JZL‐treated mice showed an obscure preference for the target platforms than vehicle‐injected mice, whereas RHC‐treated mice performed significantly better than the vehicle‐treated mice in this same test (Figure 7B).
Figure 7.
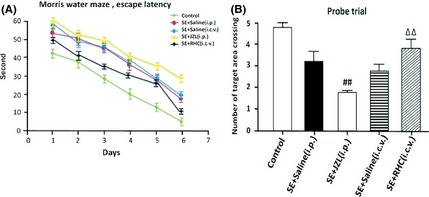
Learning of platform location by drug‐treated epileptic mice in the Morris water maze. (A) the mean time to reach the hidden platform (“escape latency”) during the 7 days of water maze testing (acquisition period). Comparison of escape latencies of day 1 versus day 7 indicated a significant learning effect for all groups. (B) data from the probe trial in which the platform was removed and the crossings of the former platform position during a single trial were recorded. Statistical analysis of data by ANOVA revealed that RHC‐treated SE animals learned significantly better than SE animals receiving only vehicle injection (P < 0.05). JZL‐treated animals learned significantly more slowly than vehicle‐injected control animals (P < 0.05). (## P < 0.01 compared with SE + saline [i.p.] group; ΔΔ P < 0.01 compared with SE + saline [i.c.v.] group).
Effects of Drug Treatment on Hippocampal Damage
Hippocampal damage was determined at 10 weeks after SE and was characterized by neurodegeneration in the CA1, CA3, and DG areas (Figure 8). The number of neurons in all three areas (CA1, CA3, and DG) was significantly decreased in all groups that had experienced epileptic seizures (P < 0.01, compared with control; Figure 8). Remarkably, JZL‐treated mice (n = 10) showed much greater neuronal loss in the CA1, CA3, and DG than vehicle‐treated SE mice (P < 0.05). We did not find a significant difference of hippocampal damage in either the CA3 or DG in mice that had been administered RHC80267 (n = 12, P > 0.05). However, the CA1 region revealed significantly more cell loss in vehicle‐injected control than in the RHC‐treated group (P < 0.05). These results indicate a protective effect exerted by RHC80267 administration.
Figure 8.
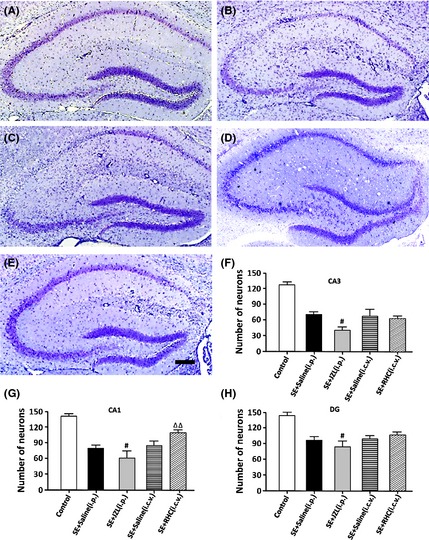
Representative thionin‐stained coronal sections of the hippocampal formation and severity of neuronal damage in the hippocampal formation of epileptic mice. Hippocampal neuronal loss was assessed in control (A), vehicle‐treated (i.p.) SE mice (B), JZL‐treated mice (C), vehicle‐treated (i.c.v.) SE mice (D), and RHC‐treated mice (E) at 10 weeks after SE. SE mice showed substantial neuronal loss in the CA3, CA1, and DG regions of the hippocampus. (F–H) Quantitative analysis demonstrated that JZL184 treatment led to much more neuronal loss in the CA3, CA1, and DG areas than vehicle‐treated control (P < 0.05), while RHC treatment significantly attenuated the neuronal loss in the CA1 area (P < 0.01). # P < 0.05 compared with SE + saline (i.p.) group; ΔΔ P < 0.01 compared with SE + saline [i.c.v.] group. Scale bars: 200 μm.
Discussion
In this study, we have shown that treatment with RHC80267, which is an inhibitor of diacylglycerol lipase, suppressed SRS during the chronic epileptic phase in a mouse model of temporal lobe epilepsy (TLE). Furthermore, RHC80267 had beneficial effects on the psychiatric and cognitive performances of animals with chronic seizures. Finally, administration of RHC80267 resulted in an inhibition of seizure‐induced hippocampal neuronal loss. In contrast, inhibition of the enzyme primarily responsible for degrading 2‐AG mediated by application of JZL184 resulted in an increase in both the frequency and the duration of observed SRS. Moreover, we found increases in subjects' neuropsychologic impairment as well as increased hippocampal damage following SE. Thus, our findings suggest that RHC80267 might have a beneficial effect on subjects after SE through decreasing the level of endogenous CB receptor ligands.
CB1Rs are located mainly on presynaptic nerve terminals and modulate neuronal excitability by suppressing the release of other neurotransmitters (e.g., glutamate, GABA, and dopamine) 33, 34. The concentrations of 2‐AG, which is one of the main endogenous CB receptor ligands, can be enhanced by administration of JZL184 or reduced by RHC80267 19. We found a significant decrease in the number and duration of SRS in the RHC80267‐treated group. This result suggests that endogenous CB receptor activation may contribute to the process of epileptogenesis after SE. Given that CB1 receptors play a critical role in inducing persistent changes in limbic networks during seizures 16, future experiments will be needed to determine the mechanism responsible for interaction between 2‐AG attenuation and epileptogenesis suppression.
In both animal models of TLE and human patients with TLE, CB1 receptor agonists have been found to be potent anticonvulsants 7, 35. Although the exact reason behind the protective actions of acute CB1 receptor activation is not fully known, the most likely mechanism is the activity‐dependent depression of glutamate release 35, 36. However, recent data have shown that the activity‐dependent rise in endocannabinoids and the rapid downstream activation of CB1 receptors may also be important in triggering long‐term hyperexcitability after an insult like SE 15. Prolonged exposure to cannabinoids results in the development of tolerance to the anticonvulsant effects of cannabinoids and an exacerbation of seizure activity in a hippocampal neuronal culture model 37. Further, CB1R blockade immediately following a brain insult prevents the emergence of chronic limbic hyperexcitability caused by febrile seizures in the developing rat brain 16 and abolishes the long‐term increase in seizure susceptibility caused by head injury in rats 15. Consistent with these studies, our present results support the idea that the acute downregulation of levels of endogenous CB receptor ligands during the epileptogenic latency phase following SE suppressed chronic spontaneous seizures in a mice model of TLE.
Consistent with previous findings 38, in this study, epileptic mice exhibited significant increases in depressive‐ and anxiety‐like behaviors, as well as severely impaired spatial learning and memory. The high level of expression of CB1 receptors in brain areas involved in the regulation of mood functions and cognition (e.g., amygdala, cortex, and hippocampus) implies that the endocannabinoid system is likely involved in emotional and cognitive processing 39. In line with this concept, there is the potential for pharmacological manipulation of the endocannabinoid system as a novel approach for emotional and cognitive regulation 39. A decrease in the functioning of endocannabinoid receptors is considered a predisposing factor for major depression 18. The link between endocannabinoids and anxiety is more complicated and often results in widely differing effects that depend on the drug, the dose, the species, and the model used 17. Nevertheless, the majority of preclinical studies have found that CB1 receptor agonists are anxiogenic at high doses 19, 40, 41 and ineffective at low doses 42, 43. Our results have shown that RHC80267 treatment during the latency period following induction of SE attenuates depressive‐ and anxiety‐like behaviors during the chronic epileptic period. These data point to a potential positive effect of modulation of endocannabinoid levels on mood regulation after SE. Although the neurobiological role of 2‐AG in psychiatric disorders is still poorly understood, the present data indicate that relatively prolonged exposure to an inhibitor of diacylglycerol lipase, with subsequent decrease in 2‐AG levels during the latency phase following pilocarpine‐induced SE, might be beneficial to some of the cognitive comorbidities found in chronic epilepsy.
It is well known that marijuana and other cannabinoid receptor agonists produce disturbances in animal models of learning and memory 19, 20, 21. 2‐AG has been proposed to modulate a number of signaling pathways critically implicated in the deleterious effect of cannabinoids on learning and memory 44. Recent evidence revealed that JZL184 increased 2‐AG levels in the hippocampus, prefrontal cortex, and cerebellum and resulted in disrupted short‐term spatial memory performance via a CB1R‐dependent mechanism of action 45. In parallel with these findings, our study demonstrated that a reduction in 2‐AG levels, and subsequent CB1R activation, may prevent spatial memory impairment after SE. However, the possibility that the antiepileptogenic effect of RHC80267 is working through other mechanisms to produce beneficial regulation on mood functions and cognition cannot be excluded at this time.
As a result of their ability to normalize glucose homeostasis, prevent calcium influx, and reduce oxidative injury through the activation of CB1Rs in neurons or astrocytes, cannabinoids have been proposed as promising neuroprotective molecules 36, 46, 47. In addition, cannabinoids have been found to decrease local inflammatory events by modulating peripheral blood lymphocytes, interfering with migration of lymphocytes across the blood‐brain barrier, and controlling microglial/macrophage activation 48. However, these data show that hippocampal damage was attenuated in mice that had received RHC80267, whereas JZL‐treated mice showed significantly greater neuronal loss. The discrepancy between this result and previous studies can be interpreted in two ways. First, CB1Rs are not only present on neurons, but are also found on astrocytes, albeit at lower concentrations. Activation of astrocytic CB1Rs induces release of calcium from intracellular stores and triggers glutamate release, which can result in an increase in the injury of neighboring neurons 49, 50, 51. Second, hippocampal damage was examined at a chronic stage in our experiment, whereas the neuroprotective effects of cannabinoids were observed in acute seizure models (hours to several days) in previous studies. Thus, antiepileptogenic effects of RHC80267 treatment during the latency phase in this model might partly account for its neuroprotective effects during chronic stage of TLE.
One limitation of our study is that we did not examine the levels of 2‐AG in our subjects. However, prior studies using our same protocol have shown that both JZL184 and RHC80267 induced changes in 2‐AG concentration 19, 27. In addition, both RHC80267 and JZL184 have multiple biological effects other than regulating 2‐AG level in the brain. RHC80267 inhibit cholinesterase and potentiates cholinergic activities 52. Although JZL184 increases brain 2‐AG level by inhibition of MAGL, it exhibits a lack of selectivity and 2‐AG is not the only substrate of MAGL 53. Therefore, 2‐AG regulation might not be the exclusive explanation for the effects of RHC80267 and JZL184 in the present epilepsy model. Moreover, the proconvulsant nature of CB1R blockade 7 is a drawback that must be addressed before it can be employed as a possible therapy. That being said, it is unlikely that RHC80267 treatment would aggravate pilocarpine‐induced seizures in the present study given that we did not record any increase in EEG‐recorded or behavioral seizures after the termination of SE.
In summary, the data reported here suggest that RHC80267 treatment after the onset of SE during the latency phase of epileptogenesis may produce significant beneficial effects over a chronic timeline. These results reiterate the importance of endocannabinoid signaling in SE and present it as a target for novel agents to prevent epileptogenesis and therefore improve patients' quality of life.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by Grants from the Natural Science Foundation of China (grant numbers 81071051, 81271432), from the Program for Changjiang Scholars and Innovative Research Team in University from Ministry of Education of China (IRT1053) and from the Science and Technology Research and Development Program of Shaanxi Province (2013K12‐18‐03).
The first two authors contributed equally to this work.
References
- 1. Tellez‐Zenteno JF, Patten SB, Jette N, Williams J, Wiebe S. Psychiatric comorbidity in epilepsy: A population‐based analysis. Epilepsia 2007;48:2336–2344. [DOI] [PubMed] [Google Scholar]
- 2. Jones R, Rickards H, Cavanna AE. The prevalence of psychiatric disorders in epilepsy: A critical review of the evidence. Funct Neurol 2010;25:191–194. [PubMed] [Google Scholar]
- 3. Lin JJ, Mula M, Hermann BP. Uncovering the neurobehavioural comorbidities of epilepsy over the lifespan. Lancet 2012;380:1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 2012;35:529–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wenner P. The effects of endocannabinoid signaling on network activity in developing and motor circuits. Ann N Y Acad Sci 2013;1279:135–142. [DOI] [PubMed] [Google Scholar]
- 6. Wallace MJ, Wiley JL, Martin BR, DeLorenzo RJ. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur J Pharmacol 2001;428:51–57. [DOI] [PubMed] [Google Scholar]
- 7. Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther 2003;307:129–137. [DOI] [PubMed] [Google Scholar]
- 8. Blair RE, Deshpande LS, Sombati S, Falenski KW, Martin BR, DeLorenzo RJ. Activation of the cannabinoid type‐1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. J Pharmacol Exp Ther 2006;317:1072–1078. [DOI] [PubMed] [Google Scholar]
- 9. Coomber B, O'Donoghue MF, Mason R. Inhibition of endocannabinoid metabolism attenuates enhanced hippocampal neuronal activity induced by kainic acid. Synapse 2008;62:746–755. [DOI] [PubMed] [Google Scholar]
- 10. Jones NA, Hill AJ, Smith I, et al. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo . J Pharmacol Exp Ther 2010;332:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ludanyi A, Eross L, Czirjak S, et al. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci 2008;28:2976–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falenski KW, Carter DS, Harrison AJ, Martin BR, Blair RE, DeLorenzo RJ. Temporal characterization of changes in hippocampal cannabinoid CB(1) receptor expression following pilocarpine‐induced status epilepticus. Brain Res 2009;1262:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falenski KW, Blair RE, Sim‐Selley LJ, Martin BR, DeLorenzo RJ. Status epilepticus causes a long‐lasting redistribution of hippocampal cannabinoid type 1 receptor expression and function in the rat pilocarpine model of acquired epilepsy. Neuroscience 2007;146:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goffin K, Van Paesschen W, Van Laere K. In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain 2011;134(Pt 4):1033–1040. [DOI] [PubMed] [Google Scholar]
- 15. Echegoyen J, Armstrong C, Morgan RJ, Soltesz I. Single application of a CB1 receptor antagonist rapidly following head injury prevents long‐term hyperexcitability in a rat model. Epilepsy Res 2009;85:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen K, Neu A, Howard AL, et al. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci 2007;27:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tambaro S, Bortolato M. Cannabinoid‐related agents in the treatment of anxiety disorders: Current knowledge and future perspectives. Recent Pat CNS Drug Discov 2012;7:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorzalka BB, Hill MN. Putative role of endocannabinoid signaling in the etiology of depression and actions of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry 2011;35:1575–1585. [DOI] [PubMed] [Google Scholar]
- 19. Busquets‐Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R, Ozaita A. Differential role of anandamide and 2‐arachidonoylglycerol in memory and anxiety‐like responses. Biol Psychiatry 2011;70:479–486. [DOI] [PubMed] [Google Scholar]
- 20. Niyuhire F, Varvel SA, Martin BR, Lichtman AH. Exposure to marijuana smoke impairs memory retrieval in mice. J Pharmacol Exp Ther 2007;322:1067–1075. [DOI] [PubMed] [Google Scholar]
- 21. Heyser CJ, Hampson RE, Deadwyler SA. Effects of delta‐9‐tetrahydrocannabinol on delayed match to sample performance in rats: Alterations in short‐term memory associated with changes in task specific firing of hippocampal cells. J Pharmacol Exp Ther 1993;264:294–307. [PubMed] [Google Scholar]
- 22. Savinainen JR, Jarvinen T, Laine K, Laitinen JT. Despite substantial degradation, 2‐arachidonoylglycerol is a potent full efficacy agonist mediating CB(1) receptor‐dependent G‐protein activation in rat cerebellar membranes. Br J Pharmacol 2001;134:664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugiura T, Kodaka T, Nakane S, et al. Evidence that the cannabinoid CB1 receptor is a 2‐arachidonoylglycerol receptor. Structure‐activity relationship of 2‐arachidonoylglycerol, ether‐linked analogues, and related compounds. J Biol Chem 1999;274:2794–2801. [DOI] [PubMed] [Google Scholar]
- 24. Kondo S, Kondo H, Nakane S, et al. 2‐Arachidonoylglycerol, an endogenous cannabinoid receptor agonist: Identification as one of the major species of monoacylglycerols in various rat tissues, and evidence for its generation through CA2+‐dependent and ‐independent mechanisms. FEBS Lett 1998;429:152–156. [DOI] [PubMed] [Google Scholar]
- 25. Sutherland CA, Amin D. Relative activities of rat and dog platelet phospholipase A2 and diglyceride lipase. Selective inhibition of diglyceride lipase by RHC 80267. J Biol Chem 1982;257:14006–14010. [PubMed] [Google Scholar]
- 26. Long JZ, Li W, Booker L, et al. Selective blockade of 2‐arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 2009;5:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gregg LC, Jung KM, Spradley JM, et al. Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase‐alpha initiates 2‐arachidonoylglycerol formation and endocannabinoid‐mediated analgesia. J Neurosci 2012;32:9457–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groticke I, Hoffmann K, Loscher W. Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp Neurol 2007;207:329–349. [DOI] [PubMed] [Google Scholar]
- 29. Lowenstein DH. Status epilepticus: An overview of the clinical problem. Epilepsia, 1999;40(Suppl 1):S3–S8; discussion S21–S22. [DOI] [PubMed] [Google Scholar]
- 30. Mazzuferi M, Kumar G, Rospo C, Kaminski RM. Rapid epileptogenesis in the mouse pilocarpine model: Video‐EEG, pharmacokinetic and histopathological characterization. Exp Neurol 2012;238:156–167. [DOI] [PubMed] [Google Scholar]
- 31. Kharlamov EA, Jukkola PI, Schmitt KL, Kelly KM. Electrobehavioral characteristics of epileptic rats following photothrombotic brain infarction. Epilepsy Res 2003;56:185–203. [DOI] [PubMed] [Google Scholar]
- 32. Jung KH, Chu K, Lee ST, et al. Cyclooxygenase‐2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine‐induced status epilepticus. Neurobiol Dis 2006;23:237–246. [DOI] [PubMed] [Google Scholar]
- 33. Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron 2012;76:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science 2002;296:678–682. [DOI] [PubMed] [Google Scholar]
- 35. Monory K, Massa F, Egertova M, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 2006;51:455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marsicano G, Goodenough S, Monory K, et al. CB1 cannabinoid receptors and on‐demand defense against excitotoxicity. Science 2003;302:84–88. [DOI] [PubMed] [Google Scholar]
- 37. Blair RE, Deshpande LS, Sombati S, Elphick MR, Martin BR, DeLorenzo RJ. Prolonged exposure to WIN55,212‐2 causes downregulation of the CB1 receptor and the development of tolerance to its anticonvulsant effects in the hippocampal neuronal culture model of acquired epilepsy. Neuropharmacology 2009;57:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muller CJ, Groticke I, Bankstahl M, Loscher W. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine‐induced status epilepticus in C57BL/6 mice. Exp Neurol 2009;219:284–297. [DOI] [PubMed] [Google Scholar]
- 39. Parolaro D, Realini N, Vigano D, Guidali C, Rubino T. The endocannabinoid system and psychiatric disorders. Exp Neurol 2010;224:3–14. [DOI] [PubMed] [Google Scholar]
- 40. McGregor IS, Issakidis CN, Prior G. Aversive effects of the synthetic cannabinoid CP 55,940 in rats. Pharmacol Biochem Behav 1996;53:657–664. [DOI] [PubMed] [Google Scholar]
- 41. Arevalo C, de Miguel R, Hernandez‐Tristan R. Cannabinoid effects on anxiety‐related behaviours and hypothalamic neurotransmitters. Pharmacol Biochem Behav 2001;70:123–131. [DOI] [PubMed] [Google Scholar]
- 42. Patel S, Cravatt BF, Hillard CJ. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology 2005;30:497–507. [DOI] [PubMed] [Google Scholar]
- 43. Bortolato M, Campolongo P, Mangieri RA, et al. Anxiolytic‐like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology 2006;31:2652–2659. [DOI] [PubMed] [Google Scholar]
- 44. Puighermanal E, Busquets‐Garcia A, Maldonado R, Ozaita A. Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos Trans R Soc Lond B Biol Sci 2012;367:3254–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wise LE, Long KA, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH. Dual fatty acid amide hydrolase and monoacylglycerol lipase blockade produces THC‐like Morris water maze deficits in mice. ACS Chem Neurosci 2012;3:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Naidoo V, Nikas SP, Karanian DA, et al. A new generation fatty acid amide hydrolase inhibitor protects against kainate‐induced excitotoxicity. J Mol Neurosci 2011;43:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fernandez‐Ruiz J, Garcia C, Sagredo O, Gomez‐Ruiz M, de Lago E. The endocannabinoid system as a target for the treatment of neuronal damage. Expert Opin Ther Targets 2010;14:387–404. [DOI] [PubMed] [Google Scholar]
- 48. Sanchez AJ, Garcia‐Merino A. Neuroprotective agents: Cannabinoids. Clin Immunol 2012;142:57–67. [DOI] [PubMed] [Google Scholar]
- 49. Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2012;68:113–126. [DOI] [PubMed] [Google Scholar]
- 50. Coiret G, Ster J, Grewe B, et al. Neuron to astrocyte communication via cannabinoid receptors is necessary for sustained epileptiform activity in rat hippocampus. PLoS One 2012;7:e37320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Navarrete M, Araque A. Endocannabinoids mediate neuron‐astrocyte communication. Neuron 2008;57:883–893. [DOI] [PubMed] [Google Scholar]
- 52. Ghisdal P, Vandenberg G, Hamaide MC, Wibo M, Morel N. The diacylglycerol lipase inhibitor RHC‐80267 potentiates the relaxation to acetylcholine in rat mesenteric artery by anti‐cholinesterase action. Eur J Pharmacol 2005;517:97–102. [DOI] [PubMed] [Google Scholar]
- 53. Vandevoorde S. Overview of the chemical families of fatty acid amide hydrolase and monoacylglycerol lipase inhibitors. Curr Top Med Chem 2008;8:247–267. [DOI] [PubMed] [Google Scholar]