

Summary
Aims
We recently described multifunctional tools (2a–c) as potent inhibitors of human Cholinesterases (ChEs) also able to modulate events correlated with Aβ aggregation. We herein propose a thorough biological and computational analysis aiming at understanding their mechanism of action at the molecular level.
Methods
We determined the inhibitory potency of 2a–c on Aβ 1–42 self‐aggregation, the interference of 2a with the toxic Aβ oligomeric species and with the postaggregation states by capillary electrophoresis analysis and transmission electron microscopy. The modulation of Aβ toxicity was assessed for 2a and 2b on human neuroblastoma cells. The key interactions of 2a with Aβ and with the Aβ‐preformed fibrils were computationally analyzed. 2a–c toxicity profile was also assessed (human hepatocytes and mouse fibroblasts).
Results
Our prototypical pluripotent analogue 2a interferes with Aβ oligomerization process thus reducing Aβ oligomers‐mediated toxicity in human neuroblastoma cells. 2a also disrupts preformed fibrils. Computational studies highlighted the bases governing the diversified activities of 2a.
Conclusion
Converging analytical, biological, and in silico data explained the mechanism of action of 2a on Aβ 1–42 oligomers formation and against Aβ‐preformed fibrils. This evidence, combined with toxicity data, will orient the future design of safer analogues.
Keywords: Alzheimer's disease, Amyloid beta oligomers, Amyloid beta peptides, Cholinesterase inhibitors, Molecular dynamics, Multifunctional ligands
Introduction
Alzheimer's disease (AD) is a multifactorial and fatal neurodegenerative disorder characterized by the neuropathological extracellular accumulation of Aβ plaques and the intracellular accumulation of hyperphosphorylated tau protein in the form of neurofibrillary tangles. Currently available therapies for AD only treat disease symptoms and do not address the underlying disease processes 1, thus making AD as the biggest unmet medical need in neurology. As age is the major risk factor, AD has become an urgent public health problem being projected to lead to epidemic levels unless a disease‐modifying anti‐Alzheimer's drug (DMAAD) can be found 2. Although the molecular mechanisms of AD pathogenesis have not been clearly understood, due to its complexity, the use of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitors represents the only therapeutic approach to the disease. Cholinesterase (ChE) inhibitors of catalysis apparently improve cognitive functions and do not have profound disease‐modifying effects 3, 4, 5, although AChE also accelerates the assembly of Aβ to amyloid fibrils 6.
The formation of Aβ deposits in the brain is a seminal step in the development of AD 7, and inhibiting Aβ oligomerization can provide a novel approach for treating the underlying cause of AD. Recent advances have indeed demonstrated the pathological assembly of Aβ as a causal factor in AD, and disease progression has been shown to closely correlate with the level of soluble Aβ oligomers. Prefibrillar, soluble oligomers of Aβ have been indicated as the early and key intermediates in AD‐related synaptic dysfunction 7.
The multifactorial nature of AD supports the current innovative therapeutic approach of multitarget directed ligands (MTDLs) 8. Based on preliminary data on bis‐tacrine ChE inhibitors (1a 9, 1b 10, Table 1), and to facilitate the identification of effective DMAADs, we recently described appropriately functionalized bis‐tacrine compounds as new pharmacological tools (2a–c, Table 1) 11 able to interfere with both spontaneous and induced Aβ aggregation while retaining potent antienzymatic (catalytic) properties (Table 1 second and third column).
Table 1.
Inhibition of human cholinesterases activities, Aβ 1–42 spontaneous and human AChE‐induced Aβ 1–40 aggregation by compounds 1a,b and 2a–c
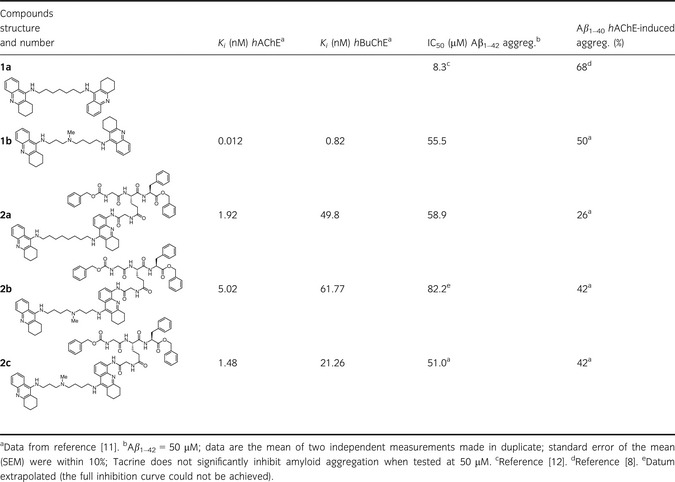
We herein propose a thorough investigation of the additional effect displayed by our prototypical multipotent compound 2a. Inhibition of spontaneous Aβ oligomerization process and disruption of the preformed fibrils induced by 2a and its analogues have been more in depth analyzed by combining in vitro aggregation experiments with cellular studies on human neuroblastoma cells (Table 1 and Figures 1 and 2) and with computational approaches (Figures 3 and 4). In particular, we hypothesized the binding mode of 2a with Aβ 1–42 and its interactions with the fibrils.
Figure 1.
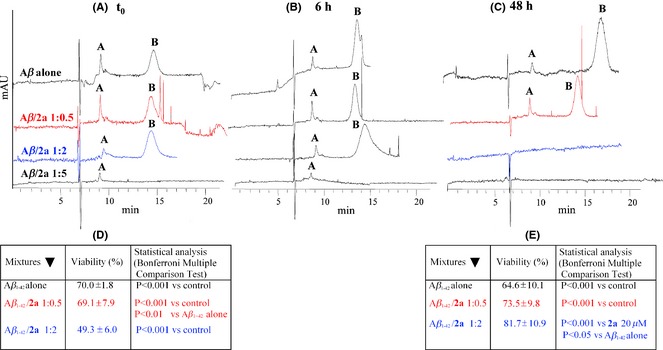
(A–C) Monitoring of Aβ 1–42 (100 μM) aggregation by capillary electrophoresis, in absence and presence of increasing concentrations of compound 2a (from top to bottom) and at increasing elapsed time from co‐incubation (from panel A to panel C); (D) cell viability on SH‐SY5Y neuroblastoma cells expressed as % of untreated cells for 10 μM Aβ 1–42 alone and 10 μM Aβ 1–42 co‐incubated with increasing concentrations of 2a, at t0 (immediately after solubilization), and (E) at t 48 h (48 h after solubilization). In C–D panels, data are normalized as % control (1.5% ethanol in phosphate buffer).
Figure 2.

Transmission electron microscopy images: (A) 100 μM Aβ 1–42; (B) 100 μM Aβ 1–42 co‐incubated with 2a (50 μM); (C) preformed Aβ 1–42 fibrils after incubation with 2a (50 μM). Images are taken at t = 5 day and are representative of those obtained for each of at least two replicates. Scale bar: 200 nm, magnification 60,000x.
Figure 3.
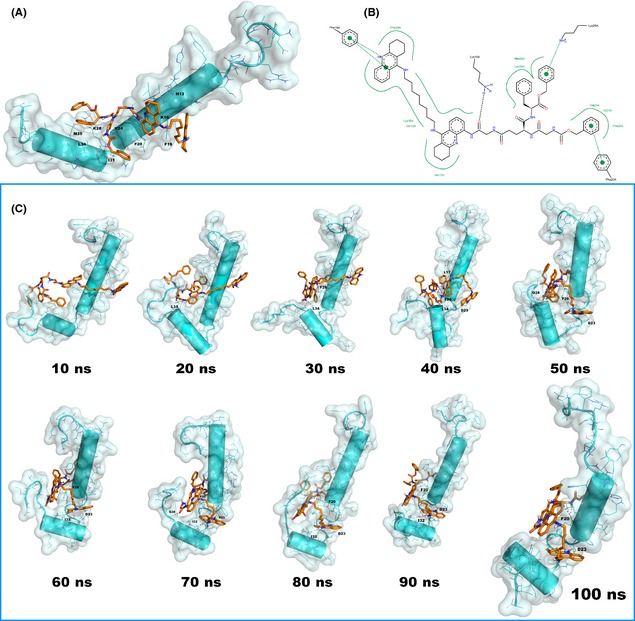
(A) Best docked pose of 2a (orange stick) in complex with Aβ 1–42 (cyan cartoon). H‐bonds are reported by gray dotted lines, the picture was generated by means of PyMOL 13, the nonpolar hydrogen atoms are omitted for the sake of clarity; (B) schematic representation of the interactions based on docking calculation. The H‐bonds are reported as black dotted lines while the π‐π or cation‐π stacking are reported as green dotted lines. The picture was generated by means of PoseView 14; (C) 2a (orange stick) and Aβ 1–42 peptide (cyan cartoon) complex: progression of MD simulation. The picture was generated by means of PyMOL 13, the nonpolar hydrogen atoms are omitted for the sake of clarity.
Figure 4.
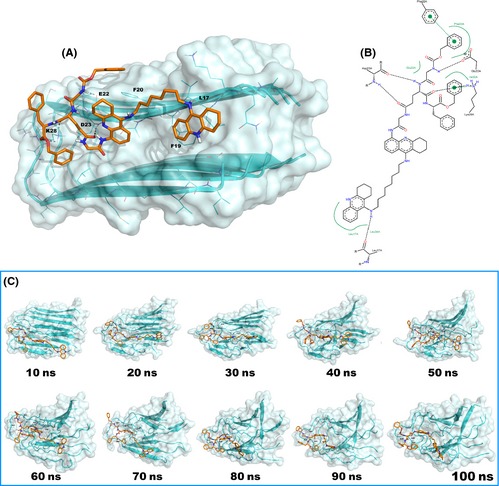
(A) Best docked pose of 2a (orange stick) in complex with Aβ 1–42 fibrils (cyan cartoon). H‐bonds are reported by gray dotted lines, the picture was generated by means of PyMOL 13, the nonpolar hydrogen atoms are omitted for the sake of clarity; (B) schematic representation of the interactions based on docking calculation. The H‐bonds are reported as black dotted lines while the π‐π or cation‐π stacking are reported as green dotted lines. The picture was generated by means of PoseView PyMOL 14; (C) 2a (orange stick) and fibrils form Aβ 1–42 (cyan cartoon) complex: progression of MD simulation. The picture was generated by means of PyMOL 13, the nonpolar hydrogen atoms are omitted for the sake of clarity.
Furthermore, we have analyzed, in comparison with tacrine (a drug recently discontinued in US due to its documented hepatotoxicity) 15, the toxicity profile of 2a–c in human fibroblast and hepatocytes (Table 2).
Table 2.
Cytotoxic effect on the NIH3T3 and human liver embryo WRL‐68 cell lines. Cell viability after 24 h of incubation was measured by the Neutral Red Uptake (NRU) test and data normalized as % control (Polystirene)a
| Tested doses ► Cmpds ▼ | 1 μM | 10 μM | 30 μM | 100 μM | 300 μM |
|---|---|---|---|---|---|
| NIH3T3 cells | |||||
| Tacrine | 98 ± 3 | 94 ± 6 | 83 ± 2* | 52 ± 4* | 26 ± 5* |
| 2a | 93 ± 4 | 30 ± 6* | 18 ± 4* | 0 | 0 |
| 2b | 103 ± 6 | 95 ± 5* | 91 ± 4* | 72 ± 7* | 54 ± 6* |
| 2c | 97 ± 4 | 91 ± 3* | 89 ± 4* | 71 ± 6* | 0 |
| WRL‐68 cells | |||||
| Tacrine | 56 ± 4* | 41 ± 2* | 39 ± 2* | 37 ± 2* | 17 ± 1* |
| 2a | 33 ± 3* | 33 ± 2* | 32 ± 4* | 20 ± 6* | 14 ± 2* |
| 2b | 56 ± 3* | 48 ± 2* | 42 ± 2* | 40 ± 1* | 38 ± 5* |
| 2c | 54 ± 5* | 38 ± 2* | 34 ± 2* | 23 ± 6* | 13 ± 2* |
aData are expressed as mean ± SD of three experiments repeated in six replicates. All compounds were tested at increasing concentrations ranging from 1 to 300 μM. *Values are statistically different versus control, P ≤ 0.05.
All these data will be crucial for driving the rational design of analogues characterized by improved druggability.
Methods
Determination of the Inhibitory Potency on Aβ 1–42 Self‐Aggregation
Procedure concerning the determination of the inhibitory potency of the presented compounds (Table 1) on Aβ 1–42 self‐aggregation is described in the Supporting Information.
Capillary Electrophoresis (CE) and Transmission Electron Microscopy (TEM)
Experimental procedures were performed as already reported 11. Details are provided in the Supporting Information.
Evaluation of Aβ 1–42 Toxicity Modulation by 2a and 2b in Human Neuroblastoma Cells
The assays were performed by means of a 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT) colorimetric assay on a human neuroblastoma SH‐SY5Y cells. Experimental details are provided in the Supporting Information.
Computational Procedure
Computational procedures based on molecular docking studies coupled to molecular dynamics (MD) simulation were carried out as reported 16, 17, 18. Molecular modeling details are provided in the Supporting Information.
Cellular Toxicity Evaluation
The cytotoxicity assays were performed on the NIH3T3 and WRL‐68 cells as described 17, 18, 19, 20. Further details are provided in the Supporting Information.
Results
With the aim of elucidating the mechanism of action of our identified tools, we have investigated compounds 2a–c for better studying their interference with spontaneous Aβ 1–42 self‐oligomerization process and with Aβ 1–42 pre‐ and postaggregation states.
Evaluation of the Inhibitory Potency on Aβ 1–42 Self‐Aggregation
To complete the panel of information relative to the inhibitory potency of compounds 2a–c on Aβ1–42 spontaneous aggregation, and as the IC50 value was previously determined only for 2b, a reinvestigation was performed by determining the IC50 for the other analogues (using a Thioflavin T‐based fluorometric assay). The full profile of the compounds is given in Table 1, where old data relative to the inhibition of hChEs (Ki values) and the hAChE‐induced Aβ1–40 aggregation are listed together with the new data. Although less potent than the bis‐tacrine analogue 1a, compounds 2a–c behave as two‐digit μM inhibitors of Aβ self‐aggregation and are characterized by similar potency.
Effect of Compound 2a on Pre‐Aggregation State of Aβ 1–42: CE Studies
The CE method enables a qualitative analysis over time of the oligomers formed along the fibrillogenesis process of Aβ 1–42 by the separation of the multimeric fraction responsible for Aβ 1–42 toxicity (peak B) from smaller oligomers (peak A) 11, 21, 22. This analysis can be performed also in the presence of small molecules, starting from their co‐solubilization with the peptide sample (t0).
In Figure 1 (panels A–C), Aβ 1–42 alone shows over time an increase of peak B at the expenses of peak A (not‐toxic oligomers). A quantitative evaluation of the oligomer peak areas is intrinsically inaccessible, as in the absence of standards a proper calibration curve cannot be built up. However, a semi‐quantitative analysis of the area percentages is possible (Figure S1). Ultrafiltration experiments and CE analysis of the filtered and the retained solutions have previously suggested that a conversion from smaller (trimers ≤ peak A ≤ undecamers) to larger (peak B >22mers) oligomers occurs 11. Following co‐incubation, compound 2a exhibits a concentration‐dependent effect on the formation of Aβ 1–42 toxic multimers. In detail, at lower concentrations of compound (50 μM, Aβ 1–42/2a 1:0.5) the growth of toxic oligomers is stabilized, while at higher concentrations a slow (200 μM, Aβ 1–42/2a 1:2, 48 h) or fast (500 μM, Aβ 1–42/2a 1:5 t0) oligomer depletion is induced. Notably, oligomer depletion nicely correlates with lower toxicity on SH‐SY5Y cells, as explained below.
Effect of Compounds 2a and 2b on Preaggregation State of Aβ 1–42: Viability Studies on SH‐SY5Y Neuroblastoma Cells
In line with the CE data and to investigate the toxicity of the Aβ aggregates formed after co‐incubation with tested compounds, we have performed cell viability studies on SH‐SY5Y neuroblastoma cells. Preliminary cell viability tests, at different concentration of 2a,b, demonstrated a comparable toxicity profile for these analogues (see Table S1A, 2a 5 μM = 61.7 vs. 2b 5 μM = 78.4, and 2a 20 μM = 40.5 vs. 2b 20 μM = 49.9, as viability %). In compliance with CE experiments, we have tested SH‐SY5Y neuroblastoma cells viability in the presence of Aβ 1–42 alone and of Aβ 1–42 mixed with the compounds in 1:0.5 and 1:2 ratios. In line with CE data, where at t0 peak B (toxic oligomers) is still detectable (Figure 1A red and blue traces), a comparable cell toxicity was found for Aβ 1–42 alone and for Aβ 1–42 mixed with 2a in 1:0.5 ratio (Figure 1D, red row). The same effect was also observed with 2b (Aβ 1–42/2b 1:0.5; 75% viability, Table S1B). Increasing the compound doses, in line with the intrinsic toxicity profile of the same, a decrease in viability was observed (Figure 1D blue row for 2a and Table S1B for 2b: Aβ 1–42/2b 1:2; 54.7% viability). Further, for tracing out the CE outcome where peak B was still present after 48 h of co‐incubation with Aβ 1–42/2a 1:0.5 (Figure 1C, red trace) and absent with Aβ 1–42/2a 1:2 (Figure 1C, blue trace), 2a was co‐incubated with Aβ 1–42 for 48 h and the cells were then treated with the resulting amorphous aggregates (Figure 1E, blue and red rows). Cell viability at 5 μM 2a (Figure 1E, red row) is unvaried and underwent a significant rise at 20 μM 2a (Figure 1E, blue row), thus indicating an overall toxicity lowering. These data are consistent with CE results which clearly evidence the capability of 2a, at 200 μM and in 48 h, in lowering toxic oligomers content, thus leading to the formation of amorphous aggregates, as evidenced from TEM results 11. Notably, the preincubated mixture is less toxic than Aβ 1–42 alone or 2a alone and also of their mixture at t0. Collectively, the data obtained with our lead 2a represent the proof‐of‐concept for the original hypothesis, which inspired the development of this class of multifunctional compounds.
Effect of Compound 2a on Postaggregation State of Aβ 1–42: TEM Data
Aβ 1–42 Fibrillogenesis Inhibition
As shown in Figure 2A, the control experiment, with Aβ peptide alone, exhibits typical nonbranching fibrils. The incubation of higher concentrations of 2a (i.e., 200 and 500 μM) has shown the absence of fibrils at TEM inspection 11. The amorphous aggregates of Figure 2B further show that this is the case also when a lower concentration (50 μM) of 2a was added. Taken altogether, the CE and TEM data make us reasonably conclude that either the stabilization (induced by the addition of 50 μM 2a) or the disaggregation (induced by the addition of 200 or 500 μM 2a) of toxic peak B (Figure 1C) correspond to a clear antifibrillogenic activity.
Aβ 1–42 Preformed Fibrils Disaggregation
Five days after solubilization in an aqueous medium is an adequate time window for Aβ 1–42 to grow classical mature fibrils, analogous to those shown in Figure 2A. Interestingly, the in vitro antiamyloid activity of compound 2a also applies to preformed fibrils, at all concentrations tested. The amorphous aggregates evident in Figure 2C are representative of the disaggregation effect of 2a at a concentration as low as 50 μM.
In Silico Studies of the Effect of Multifunctional AChE Modulator on Pre‐ and Postaggregation States of Aβ 1–42
In silico studies (molecular docking coupled to a MD simulation protocol) have been performed on 2a as the better characterized of our ligands both in vitro (fibrillogenesis, CE and TEM) and in cellular studies. The aim of the computational analysis was to understand at the molecular level the experimentally determined effects of the multifunctional compound 2a: (i) the prevention of Aβ 1–42 self‐aggregation and (ii) the disaggregation effect on the preformed fibrils.
Effect of Compound 2a on Preaggregation State of Aβ 1–42: Computational Studies
To rationalize the experimental data related to the potency of 2a in the inhibition of Aβ 1–42 peptide aggregation (Table 1) and in prevention of the misfolding event of amyloid leading to oligomerization (CE studies), intensive modeling studies were performed by using the crystal structure of Aβ 1–42 (PDB ID: 1IYT 23) and applying molecular docking and MD techniques as reported in literature 24, 25, 26. Our docking protocol employed GOLD software 27 using the scoring function GoldScore 28. The output (Figure 3) is referred to the most populated cluster found by docking. 2a strongly interacts by its bis‐tacrine system with H13 (hydrophobic contacts) and P19 by a π‐π stacking, while the peptide portion of the molecule forms a H‐bond with K16, a cation‐π stacking with K28 by the benzyl ester function and a π‐π stacking with F20 by the benzyl carbamate (Figure 3B). Our findings are in agreement with data recently obtained by others 24. The high GoldScore (83.59) and the favorable free‐binding energy (Prime MM/GBSA 29, ΔGbind = −115.65 kcal/mol) indicate a high affinity of 2a for the monomeric peptide.
Starting from the docking pose (Figure 3A), we performed MD simulation (Desmond) 30, 31 in water. Essentially, the trajectory of the MD simulation confirms the docking results. In fact, as observed in Figure 3C, the position of 2a is maintained between the central hydrophobic region and C‐terminal region of Aβ 1–42. The protonated terminal tacrine moiety initially lacks the contact with F19, forming a H‐bond with D23 and I32. These latter polar contacts maintain the protonated tacrine moiety in the same position for all the simulation thus allowing it to re‐establish a contact with F19. On the other hand, the peptide‐bound tacrine replaces the hydrophobic contact with H13 with a more favorable π‐π stacking with F20, which will persist for all the simulation. The peptide moiety preserves, although occasionally, the cation‐π stacking with K28, while it is involved in a strong H‐bond network with the backbones of F20, G38, and L34. Notably, the analysis of the MD trajectory (PoseView 14) revealed hydrophobic contacts for the benzyl carbamate moiety with M35, L17, and A21, residues which are constantly targeted during the simulation. The benzyl ester group is mainly involved in hydrophobic contacts with V24, V39, V40, and I41. These findings support a high affinity of 2a for Aβ 1–42 monomer and help in explaining its mechanism of action. In fact, 2a could prevent the misfolding of the C‐terminal region to β‐sheet, as during the 100 ns no change of secondary structure is observed (Figure 3) and the helix is constantly conserved. Concerning the amino‐acids of Aβ 1–42, their fluctuation plot (Figure S2) confirms the overall stability of the structure. In fact, as expected, only a restricted number of residues at the N‐ and at the C‐terminal ends show a relevant difference in root mean square fluctuation (RMSF), whereas for all the other residues no evident fluctuation is observed, in line with the evidence that the secondary structure appears stabilized. It is notable that the misfolding event at the C‐terminal end of Aβ 1–42 alone has been observed 32 already after 20 ns of simulation in water. Furthermore, based on a recently published model 33, the misfolded C‐terminal region (β‐hairpin) folds up toward the central hydrophobic region, thus triggering the step that drives to oligomerization. As observed in our calculations, 2a could physically prevent this type of arrangement, as it lies between the above‐mentioned key regions and does not allow the misfolding of Aβ 1–42.
In conclusion, our molecular docking and MD simulations demonstrated that 2a strongly binds to Aβ 1–42 and intensely interacts with the key regions of the α‐helical conformation of the peptide, thus leading to its stabilization. This event may reduce the potential interaction with other Aβ 1–42 monomers by preventing the folding of the C‐terminal region from helix to β‐sheet that has been identified as key step in the oligomerization process 34, 35.
The mechanism of action proposed for 2a is in line with CE and cellular studies (Figure 1). It is plausible that a similar mechanism could govern the activity of its structurally related analogues 2b,c, which displayed an antifibrillogenic activity comparable to that of 2a (Table 1).
Effect of Compound 2a on Postaggregation State of Aβ 1–42: Computational Studies
The observation by TEM analysis that 2a is able to disaggregate preformed fibrils of Aβ 1–42 after incubation of these latter with 50 (Figure 2C), 200 or 500 μM 11, prompted us to explain this event by applying to the fibrils the same protocol used for the Aβ 1–42 peptide. Molecular docking and/or MD protocols have been widely applied for observing and rationalizing the mechanism of action of different classes of compounds in fibrils disruption 36, 37, 38, 39, 40, 41, 42. The structure of the Aβ 1–42 fibrils was found in the PDB (PDB ID: 2BEG 43) and was managed as described 42, 44. In each peptide chain, the residues Q15‐K16 were added to the model in an extended conformation to ensure the β‐sheet complementarity of the U‐shaped pentamer 42, 44. The resulting structure was then used for our computational studies.
Docking calculation (Figure 4A) performed by means of GOLD software 27 revealed that the best cluster of docked solutions establishes a relevant number of polar contacts with key residues of the fibril complex. In fact, the protonated tacrine moiety is able to interact with L17 of chain A, while the tacrine bringing the peptide lies in proximity of the F19 of the same chain (Figure 4A). The peptide moiety interacts with chain A residues E22 and D23 by polar contacts and in particular with K28 by a cation‐π stacking with the benzyl ester (Figure 4B), while the benzyl carbamate establishes a π‐π stacking with F20 (chain A). It is noticeable that the targeted residues are widely accepted as pivotal for stabilizing and aggregating the fibrils. In fact, the hydrophobic central region L17‐E22 is involved in fibrils assembly 42, 44, 45, while the residues D23 and K28 are involved in a salt bridge that is necessary for the fibrils stability 42, 44, 45. Interestingly, the formation of H‐bonds between 2a and K28 and/or D23 may not be favorable to the formation of the mentioned salt bridge. The docking score value of 67.71 and the favorable estimated free‐binding energy (Prime MM/GBSA 29, ΔGbind = −162.03 kcal/mol) confirm the significant affinity of 2a for the Aβ fibrils. Relevantly, our MD simulation study performed using Desmond software 30, 31 is in agreement with the outcome obtained from the docking calculation. Accordingly, during 100 ns of MD simulation, 2a maintains the previously described contacts and strongly interacts with the fibrils (PoseView 14). Initially, F20 stacks with the benzyl carbamate, but a more favorable π‐π stacking with the peptide‐bound tacrine was observed already after 10 ns (Figure 4C). In fact, during the simulation, the benzyl carbamate moiety is principally involved in H‐bonds with K28 and/or D23 and not seldom with E22, V24 (hydrophobic contacts), G25, and S26. The benzyl ester is frequently involved in H‐bonds with N27, A30, I32 from chain A and L34 (chain B). Interestingly, the protonated tacrine forms H‐bond with I41 and the carbon linker of 2a establishes a series of hydrophobic contacts with L34 (chains A, B, C), V36 (chain B), V39, and V40 (chains A, B, C). The central tacrine is constantly involved in H‐bond with F19 backbone and establishes hydrophobic contacts with L17, F19 (frequently by π‐π stacking), F20, V40 of chain A. Notably, after about 40 ns of MD, the Aβ fibrils lack the ordered organization and disruption starts (Figure 4C).
At the end of the 100 ns of the MD simulation, the disordered state of the Aβ fibrils complex is largely evident (as they lack the predominant contacts necessary for stabilizing the structure). For example, the salt bridge between D23 and K28 is totally removed by the interaction with 2a (K28 and/or D23 are targeted during all the simulation) as the distance between these residues is increased and is not compatible with any polar contact (Figure S3). A big amount of intramolecular H‐bonds between the β‐sheets backbone that stabilize the overall structure are also sensibly reduced by the interaction with 2a. This event leads to an increase of RMSF proportional to destabilization of the structure (Figure S4). Moreover, the distances between the residues from the same fibrils also undergo a dramatic change, further contributing to the disruption of the Aβ fibrils assembly. In fact, as reported in Figure S5, the distances measured between selected residues (A21 from chain A with V36 chain A and V36 chain B) during simulation display very large differences between the starting structure and the structure at the end of the MD (e. g. the distance of A21 and V36A and V36B measures 9.4 Å and 6.2 Å, respectively, at the beginning, while at the end the MD it becomes of 23.6 Å and 20.7 Å, Figure S6).
Summing up, the computational procedure herein discussed provided the explanation of the biological events encountered with 2a binding the peptide Aβ 1–42 (that ultimately inhibits oligomer formation and fibrillization), as well as its role in promoting Aβ fibrils disruption. These data confirm the reliability of the computational approaches for investigating complex molecular mechanisms that to date can hardly be studied experimentally (in solution).
Toxicity Studies on NIH3T3 and WRL‐68 Cells
Cytotoxicity assays were performed to establish the effect of compounds 2a–c on the mouse fibroblasts NIH3T3 cells and on the human liver embryo WRL‐68 cell line (Table 2).
Cytotoxicity data for the NIH3T3 cells demonstrate that tacrine, at low doses (1 μM), has almost no toxicity. Afterward, toxicity dose‐dependently increases for tacrine and 2a (being this latter more toxic than the reference tacrine). On the contrary, analogues 2b,c did not show relevant toxicity on these cells up to 100 μM. In the same test, 2b showed the best profile among the tested compounds.
Moreover, in line with the hepatotoxicity of tacrine 15, the toxicity potential of 2a–c on the human liver embryo WRL‐68 cells was assessed. The results evidence that 2a was more toxic than tacrine already at 1 μM, while 2b,c displayed comparable or lower toxicity with respect to tacrine. Again, 2b shows the best profile within the series. These data might be ascribable to the presence of a protonatable N in the spacer, which may account for a lower cell permeability of 2b,c. However, the anti‐ChE activity of 2a–c is in the low nM range (Table 1), whereas comparing antifibrillogenic activity (IC50 Aβ 1–42 aggregation between 51 and 82 μM, Table 1) with NIH3T3 toxicity (detectable from 30 to 300 μM, Table 2), we could identify a narrow therapeutic window for all three compounds 2a–c. Toxicity data, although not favorable on hepatic cells, combined with the IC50 values justified further evaluation on human neuronal cells to assess their potential neuroprotective effect against Aβ‐mediated neurotoxicity. Accordingly, we selected 2a as the most studied analogue, and 2b as the least toxic of the series. As shown in Figure 1 D–E and Table S1, 2a demonstrated potential neuroprotective activity at 20 μM.
Taken together, toxicity data suggest that the acridine system of 2a–c should be replaced to allow the development of multipotent compounds with limited hepatotoxicity.
Conclusion
Combination of biological data and in silico data led to the explanation of the mechanism governing the activity of 2a on Aβ oligomers formation and against the Aβ 1–42 preformed fibrils. Studies on human neuroblastoma cells confirmed the potential neuroprotective role of the lead compound 2a. Furthermore, the definition of the molecular basis of the interaction with Aβ combined to the evaluation of the Aβ‐mediated toxicity profile highlighted the possibility of developing multipotent compounds with acceptable safety.
Conflict of Interest
The authors declare no competing financial interests.
Supporting information
Figure S1. Plot of peak B normalized area % versus elapsed time from solubilization (each experimental point is in triplicate).
Figure S2. (A) Root mean Square deviation (RMSD) of the Aβ 1–42 helix backbone. Root mean square deviation was calculated between the final conformation and the starting conformation through the 100 ns MD simulations. (B) RMSF of all residues of the Aβ helix. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S3. Plot for the distance between D23 and K28 from Aβ 1–42 fibrils (chain A). The picture was generated by Simulation Event Analysis implemented in Desmond.
Figure S4. A) Root mean square deviation (RMSD) of the Aβ fibrils backbone. Root mean square deviation were calculated between the final conformation and the starting conformation through the 100 ns MD simulations. (B) RMSF of all residues of the Aβ fibrils. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S5. (A) Plot for the distance between A21 and V36 from Aβ fibrils (chain A). (B) Plot for the distance between A21 from chain A and V36 from chain B. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S6. (A) Aβ fibrils (cyan cartoon) distances between A21 from chain A and V36 from chain A and B measured before the Molecular Dynamics simulation. The residues are reported as stick. (B) Aβ fibrils (green cartoon) distances between A21 from chain A and V36 from chain A and B measured after 100 ns of the Molecular Dynamics simulation. The measures are reported in Å, the picture is generated by PyMOL, all the residues with exclusion of those selected (A21 chain A, V36 chain A and B) and nonpolar hydrogen atoms were omitted for the sake of clarity.
Table S1: Cell viability on SH‐SY5Y neuroblastoma cells expressed as % of untreated cells for (a) different concentrations of 2a and 2b, (b) 10 μM Aβ 1–42 alone and 10 μM Aβ 1–42 co‐incubated with 2a and 2b, at t0 (immediately after solubilization), (c) 10 μM Aβ 1–42 alone and 10 μM Aβ 1–42 peptide co‐incubated with 2a at t 48 h (48 h after solubilization). Data are normalized as % control (1.5% ethanol in phosphate buffer).
Acknowledgment
The authors thank COST CM1103, BioSolveIT GmbH for providing academic license of PoseView.
References
- 1. Sadowski M, Wisniewski T. Disease modifying approaches for Alzheimer's pathology. Curr Pharm Des 2007;13:1943–1954. [DOI] [PubMed] [Google Scholar]
- 2. Mount C, Downton C. Alzheimer disease: Progress or profit? Nat Med 2006;12:780–784. [DOI] [PubMed] [Google Scholar]
- 3. McShane R, Areosa SA, Minakaran N, Memantine for dementia. Cochrane Database Syst Rev 2006; CD003154. [DOI] [PubMed] [Google Scholar]
- 4. Cummings JL. Treatment of Alzheimer's disease: Current and future therapeutic approaches. Rev Neurol Dis 2004;1:60–69. [PubMed] [Google Scholar]
- 5. Standridge JB. Pharmacotherapeutic approaches to the prevention of Alzheimer's disease. Am J Geriatr Pharmacother 2004;2:119–132. [DOI] [PubMed] [Google Scholar]
- 6. Alvarez A, Opazo C, Alarcon R, Garrido J, Inestrosa NC. Acetylcholinesterase promotes the aggregation of amyloid‐beta‐peptide fragments by forming a complex with the growing fibrils. J Mol Biol 1997;272:348–361. [DOI] [PubMed] [Google Scholar]
- 7. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature 2009;457:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolognesi ML, Cavalli A, Valgimigli L, et al. Multi‐target‐directed drug design strategy: From a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer's disease. J Med Chem 2007;50:6446–6449. [DOI] [PubMed] [Google Scholar]
- 9. Pang YP, Quiram P, Jelacic T, Hong F, Brimijoin S. Highly potent, selective, and low cost bis‐tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer's disease. J Biol Chem 1996;271:23646–23649. [DOI] [PubMed] [Google Scholar]
- 10. Butini S, Campiani G, Borriello M, et al. Exploiting protein fluctuations at the active‐site gorge of human cholinesterases: Further optimization of the design strategy to develop extremely potent inhibitors. J Med Chem 2008;51:3154–3170. [DOI] [PubMed] [Google Scholar]
- 11. Butini S, Brindisi M, Brogi S, et al. Multifunctional cholinesterase amyloid beta fibrillization modulators. Synthesis and biological investigation. ACS Med Chem Lett 2013;4:1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minarini A, Milelli A, Tumiatti V, et al. Cystamine‐tacrine dimer: A new multi‐target‐directed ligand as potential therapeutic agent for Alzheimer's disease treatment. Neuropharmacology 2012;62:997–1003. [DOI] [PubMed] [Google Scholar]
- 13. The PyMOL molecular graphics system, version 1.6‐alpha. New York: Schrödinger, LLC, 2013. [Google Scholar]
- 14. Stierand K, Rarey M. Consistent two‐dimensional visualization of protein‐ligand complex series. J Cheminform 2011;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lagadic‐Gossmann D, Rissel M, Le Bot MA, Guillouzo A. Toxic effects of tacrine on primary hepatocytes and liver epithelial cells in culture. Cell Biol Toxicol 1998;14:361–373. [DOI] [PubMed] [Google Scholar]
- 16. Anzini M, Di Capua A, Valenti S, et al. Novel analgesic/anti‐inflammatory agents: 1,5‐diarylpyrrole nitrooxyalkyl ethers and related compounds as cyclooxygenase‐2 inhibiting nitric oxide donors. J Med Chem 2013;56:3191–3206. [DOI] [PubMed] [Google Scholar]
- 17. Cappelli A, Manini M, Valenti S, et al. Synthesis and structure‐activity relationship studies in serotonin 5‐HT(1A) receptor agonists based on fused pyrrolidone scaffolds. Eur J Med Chem 2013;63:85–94. [DOI] [PubMed] [Google Scholar]
- 18. Gemma S, Camodeca C, Brindisi M, et al. Mimicking the intramolecular hydrogen bond: Synthesis, biological evaluation, and molecular modeling of benzoxazines and quinazolines as potential antimalarial agents. J Med Chem 2012;55:10387–10404. [DOI] [PubMed] [Google Scholar]
- 19. Butini S, Gemma S, Brindisi M, et al. Non‐nucleoside inhibitors of human adenosine kinase: Synthesis, molecular modeling, and biological studies. J Med Chem 2011;54:1401–1420. [DOI] [PubMed] [Google Scholar]
- 20. Gemma S, Brogi S, Patil PR, et al. From (+)‐epigallocatechin gallate to a simplified synthetic analogue as a cytoadherence inhibitor for P. falciparum. RSC Adv 2014;4:4769–4781. [Google Scholar]
- 21. Sabella S, Quaglia M, Lanni C, et al. Capillary electrophoresis studies on the aggregation process of beta‐amyloid 1‐42 and 1‐40 peptides. Electrophoresis 2004;25:3186–3194. [DOI] [PubMed] [Google Scholar]
- 22. Colombo R, Carotti A, Catto M, et al. CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity. Electrophoresis 2009;30:1418–1429. [DOI] [PubMed] [Google Scholar]
- 23. Crescenzi O, Tomaselli S, Guerrini R, et al. Solution structure of the Alzheimer amyloid beta‐peptide (1‐42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem 2002;269:5642–5648. [DOI] [PubMed] [Google Scholar]
- 24. Wang Y, Xia Z, Xu JR, et al. Alpha‐mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates beta‐amyloid oligomers‐induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology 2012;62:871–881. [DOI] [PubMed] [Google Scholar]
- 25. Peters C, Fernandez‐Perez EJ, Burgos CF, et al. Inhibition of amyloid beta‐induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol Aging 2013;34:2805–2814. [DOI] [PubMed] [Google Scholar]
- 26. Yang C, Zhu X, Li J, Shi R. Exploration of the mechanism for LPFFD inhibiting the formation of beta‐sheet conformation of A beta(1‐42) in water. J Mol Model 2010;16:813–821. [DOI] [PubMed] [Google Scholar]
- 27. Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol 1995;245:43–53. [DOI] [PubMed] [Google Scholar]
- 28. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–748. [DOI] [PubMed] [Google Scholar]
- 29. Prime, version 3.0. New York: Schrödinger, LLC, 2011. [Google Scholar]
- 30. Desmond molecular dynamics system, version 3.0. New York, NY: D. E: Shaw Research, 2011. [Google Scholar]
- 31. Maestro‐desmond interoperability tools, version 3.0. New York, NY: Schrodinger, 2011. [Google Scholar]
- 32. Yang C, Li J, Li Y, Zhu X. The effect of solvents on the conformations of Amyloid β‐peptide (1–42) studied by molecular dynamics simulation. J Mol Struct 2009;895:1–8. [Google Scholar]
- 33. Roychaudhuri R, Yang M, Deshpande A, et al. C‐terminal turn stability determines assembly differences between Abeta40 and Abeta42. J Mol Biol 2013;425:292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Streltsov VA, Varghese JN, Masters CL, Nuttall SD. Crystal structure of the amyloid‐beta p3 fragment provides a model for oligomer formation in Alzheimer's disease. J Neurosci 2011;31:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Urbanc B, Betnel M, Cruz L, Bitan G, Teplow DB. Elucidation of amyloid beta‐protein oligomerization mechanisms: Discrete molecular dynamics study. J Am Chem Soc 2010;132:4266–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bruce NJ, Chen D, Dastidar SG, et al. Molecular dynamics simulations of Abeta fibril interactions with beta‐sheet breaker peptides. Peptides 2010;31:2100–2108. [DOI] [PubMed] [Google Scholar]
- 37. Sgarbossa A, Monti S, Lenci F, et al. The effects of ferulic acid on beta‐amyloid fibrillar structures investigated through experimental and computational techniques. Biochim Biophys Acta 2013;1830:2924–2937. [DOI] [PubMed] [Google Scholar]
- 38. Chen D, Martin ZS, Soto C, Schein CH. Computational selection of inhibitors of Abeta aggregation and neuronal toxicity. Bioorg Med Chem 2009;17:5189–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verma S, Singh A, Mishra A. The effect of fulvic acid on pre‐ and postaggregation state of Abeta(17‐42): Molecular dynamics simulation studies. Biochim Biophys Acta 2013;1834:24–33. [DOI] [PubMed] [Google Scholar]
- 40. Zhao JH, Liu HL, Elumalai P, et al. Molecular modeling to investigate the binding of Congo red toward GNNQQNY protofibril and in silico virtual screening for the identification of new aggregation inhibitors. J Mol Model 2013;19:151–162. [DOI] [PubMed] [Google Scholar]
- 41. Lemkul JA, Bevan DR. Destabilizing Alzheimer's Abeta(42) protofibrils with morin: Mechanistic insights from molecular dynamics simulations. Biochemistry 2010;49:3935–3946. [DOI] [PubMed] [Google Scholar]
- 42. Hochdorffer K, Marz‐Berberich J, Nagel‐Steger L, et al. Rational design of beta‐sheet ligands against Abeta42‐induced toxicity. J Am Chem Soc 2011;133:4348–4358. [DOI] [PubMed] [Google Scholar]
- 43. Luhrs T, Ritter C, Adrian M, et al. 3D structure of Alzheimer's amyloid‐beta(1‐42) fibrils. Proc Natl Acad Sci USA 2005;102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kassler K, Horn AH, Sticht H. Effect of pathogenic mutations on the structure and dynamics of Alzheimer's A beta 42‐amyloid oligomers. J Mol Model 2010;16:1011–1020. [DOI] [PubMed] [Google Scholar]
- 45. Rosenman DJ, Connors CR, Chen W, Wang C, Garcia AE. Abeta monomers transiently sample oligomer and fibril‐like configurations: Ensemble characterization using a combined MD/NMR approach. J Mol Biol 2013;425:3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Plot of peak B normalized area % versus elapsed time from solubilization (each experimental point is in triplicate).
Figure S2. (A) Root mean Square deviation (RMSD) of the Aβ 1–42 helix backbone. Root mean square deviation was calculated between the final conformation and the starting conformation through the 100 ns MD simulations. (B) RMSF of all residues of the Aβ helix. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S3. Plot for the distance between D23 and K28 from Aβ 1–42 fibrils (chain A). The picture was generated by Simulation Event Analysis implemented in Desmond.
Figure S4. A) Root mean square deviation (RMSD) of the Aβ fibrils backbone. Root mean square deviation were calculated between the final conformation and the starting conformation through the 100 ns MD simulations. (B) RMSF of all residues of the Aβ fibrils. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S5. (A) Plot for the distance between A21 and V36 from Aβ fibrils (chain A). (B) Plot for the distance between A21 from chain A and V36 from chain B. The pictures were generated by Simulation Event Analysis implemented in Desmond.
Figure S6. (A) Aβ fibrils (cyan cartoon) distances between A21 from chain A and V36 from chain A and B measured before the Molecular Dynamics simulation. The residues are reported as stick. (B) Aβ fibrils (green cartoon) distances between A21 from chain A and V36 from chain A and B measured after 100 ns of the Molecular Dynamics simulation. The measures are reported in Å, the picture is generated by PyMOL, all the residues with exclusion of those selected (A21 chain A, V36 chain A and B) and nonpolar hydrogen atoms were omitted for the sake of clarity.
Table S1: Cell viability on SH‐SY5Y neuroblastoma cells expressed as % of untreated cells for (a) different concentrations of 2a and 2b, (b) 10 μM Aβ 1–42 alone and 10 μM Aβ 1–42 co‐incubated with 2a and 2b, at t0 (immediately after solubilization), (c) 10 μM Aβ 1–42 alone and 10 μM Aβ 1–42 peptide co‐incubated with 2a at t 48 h (48 h after solubilization). Data are normalized as % control (1.5% ethanol in phosphate buffer).