

Summary
Cortistatin (CST)‐14, a neuropeptide that is structurally and functionally related to somatostatin‐14 (SRIF) binds all five somatostatin receptor subtypes (sst1–sst5). Using in vivo microdialysis and telemetry‐based electroencephalographic recordings, we provide the first experimental evidence for anticonvulsive effects of CST‐14 in a pilocarpine‐induced seizure model in rats and mice and for the involvement of sst2 and sst3 receptors in these anticonvulsant actions of CST‐14. Both receptor subtypes are required for the anticonvulsant effects of CST‐14 given that co‐perfusion of a selective sst2 antagonist (cyanamid15486) or a selective sst3 antagonist (SST3‐ODN‐8) reversed anticonvulsant effect of CST‐14, and this, independently of each other. Next, as the ghrelin receptor has been proposed as a target for the biological effects of CST‐14, we used ghrelin receptor knockout mice and their wild type littermates to study the involvement of this receptor in the anticonvulsive actions of CST‐14. Our results show a significant decrease in seizure duration in both genotypes when CST‐14 treated mice were compared with corresponding control animals receiving only pilocarpine. In addition, this CST‐14‐induced decrease was comparable in both genotypes. We here thus provide the first evidence that ghrelin receptors are not involved in mediating anticonvulsant actions of CST‐14 in vivo.
Keywords: Cortistatin, Epilepsy, Ghrelin receptor, Pilocarpine, Somatostatin, sst Receptors
Introduction
Cortistatin (CST) is a neuropeptide that is named after its predominantly cortical expression and its strong similarities with somatostatin 1. Enzymatic cleavage of their respective precursor peptides produces CST‐14 and somatostatin‐14 (SRIF) 2. Although both peptides are encoded by distinct genes, CST‐14 (PCKNFFWKTFSSCK) shares 11 of 14 amino acid residues with SRIF (AGCKNFFWKTFTSC) including the FWKT sequence that is critical for SRIF binding to its receptors 2. SRIF binds with nanomolar affinity to five specific G‐protein‐coupled receptors (GPCRs): sst1‐sst5, of which only sst1‐sst4 are expressed in the hippocampus 3. Given the structural similarities between CST‐14 and SRIF, sst receptors were proposed as potential target sites for the biological effects of CST‐14. Initially, in GH4 rat pituitary tumour cells, synthetic CST‐14 was found to displace iodine labelled SRIF as potently as SRIF itself 2. Subsequently, rat CST‐14 has been shown to displace with nanomolar affinities iodine labelled SRIF binding to each of the five cloned sst receptors expressed in transfected cell lines 4, 5. The binding of CST‐14 to these sst receptors might explain the functional resemblances with SRIF, such as the activation of common receptors and signaling pathways and inhibition of neurons via activation of the M‐current 1. Despite these homologies, CST‐14 also displays a range of biological effects distinct from SRIF including activation of cation‐selective currents not responsive to SRIF, induction of slow‐wave sleep, and reduction of locomotor activity 1. These observations suggest that CST‐14 acts at both sst receptors and at as yet unidentified CST‐14‐specific receptor(s). Although, the existence of such specific CST‐14 receptors has not been demonstrated so far, it has been suggested that the ghrelin and/or the MAS‐related gene receptor (MrgX2) may play such a role 6.
The ghrelin receptor is a GPCR of which acylated ghrelin is an endogenous ligand. Several studies have been performed, investigating the ability of CST‐14 and SRIF to bind to ghrelin receptors and showed that CST‐14, similar to ghrelin but unlike SRIF, displaces ghrelin from its specific binding sites 7, 8. This suggests that CST‐14 could represent another endogenous ligand for the ghrelin receptor. More recently, MrgX2, an orphan GPCR mainly expressed in dorsal root ganglia, has also been reported to bind CST‐14 but neither SRIF nor ghrelin 9. It is noteworthy to mention that the MrgX2 receptor is not expressed in the cerebral cortex, in which CST‐14 is abundantly present. Furthermore, the affinity of CST‐14 for this receptor has been found to be in the micromolar range, which is uncommon for a brain peptide 6. The biological role of CST‐14 binding to MrgX2 therefore remains promiscuous and incompletely characterized and demands further exploration. The latter is however impeded by the fact that no mouse or rat orthologues of MrgX2 have been described so far.
Somatostatin‐14 (SRIF) is known to be a potent anticonvulsant in many rodent models 10, 11. Several studies have been performed investigating the role of sst receptors in the anticonvulsant effects of SRIF. In general, sst2 but not sst1 receptors play a critical role in the anticonvulsant effects of SRIF in rats 12, 13, 14. In addition, we recently hypothesized that both sst3 and sst4 receptors could be functionally coupled to sst2 receptors 14. In mice however, contradictory data have been reported. Inhibitory actions were demonstrated for mice sst1 15, 16 and sst4 receptors 16, 17 although other studies report on a minor role of sst1 receptors 17 and excitatory actions of sst4 receptors 15, 18.
To the best of our knowledge, there is only one report investigating the anticonvulsant actions of CST‐14. In the latter study, intracerebroventricular injection of CST‐14 prevented the kainic acid‐induced seizure activity in rats and exhibited clear neuroprotective effects 19. In the present study, we first used a pilocarpine rat model for limbic seizures to confirm CST‐14‐mediated anticonvulsant actions in rats and demonstrated for the first time that this neuropeptide is also able to protect mice from focally induced seizures. The major rationale of the present investigations was to unveil what possible receptor(s) are involved in the anticonvulsant actions of CST‐14. Given the known homologies in sst receptor distribution between humans and rats 3, we used rats to investigate the involvement of the specific sst receptor subtypes. For this purpose, we used cyanamid154806, a selective sst2 receptor antagonist and SST3‐ODN‐8, a selective sst3 receptor antagonist. The commercial unavailability of selective sst4 antagonists hampered us to further investigate the role of this receptor subtype. Next, to investigate the possible involvement of the ghrelin receptor in the anticonvulsant actions of CST‐14, we compared the CST‐14‐mediated anticonvulsant effects in ghrelin receptor knockout mice versus their wild‐type littermates.
Materials and methods
Animals
All experiments were carried out according to the national guidelines on animal experimentation and were approved by the Ethical Committee for Animal Experiments of the Faculty of Medicine and Pharmacy of the Vrije Universiteit Brussel (Brussels, Belgium). The animals were kept under standard laboratory conditions, received food and water ad libitum and were kept on a 10/14 h dark/light cycle.
For the experiments investigating the role of sst receptor subtypes in the anticonvulsant actions of CST‐14, male albino Wistar rats (250–320 g) were used (Charles River, Chatillon‐sur‐Chalaronne, France). The involvement of the ghrelin receptor was investigated by using male ghrelin receptor−/− and ghrelin receptor+/+ littermate mice (25–35 g) bred in the animal facilities of the Vrije Universiteit Brussel. These mice are descendants of the strain developed by Janssen Pharmaceutica (Beerse, Belgium) in collaboration with Lexicon Genetics, Inc (The Woodlands, TX, USA) on a C57Bl/6 background, as previously described 20. Genotypes were confirmed by real‐time reverse transcription polymerase chain reaction using DNA isolated from mouse tail biopsy samples.
Drugs and Reagents
CST‐14 was supplied by NeoMPS (Strasbourg, France). Cyanamid15486 was purchased from Tocris Bioscience (Bristol, UK) and pilocarpine. HCl from Sigma (St Louis, MO, USA). SST3‐ODN‐8 was a generous gift, donated by Prof. J. Rivier, Salk Institute for Biological Studies (La Jolla, CA, USA). All other chemicals were analytical reagent grade and were obtained from Merck (Darmstadt, Germany). Aqueous solutions were prepared in fresh water purified by an arium pro UV system (Sartorius Stedim Biotech GmbH, Goettingen, Germany) and filtered through a membrane filter (0.2 μm diameter). For all compounds, stock solutions were prepared in ultra purified water, diluted with modified Ringer's solution and directly delivered into the hippocampus through perfusion of the microdialysis probe. The final perfusion concentrations of the selective sst2 and sst3 antagonists were set at 100 nM and were calculated in function of their Ki values and probe recovery as previously described 14.
Surgery
All animals received 4 mg/kg ketoprofen subcutaneously to prevent postoperative pain.
Rats
Rats were anesthetized with a mixture of ketamine:diazepam (start dose = 90.5:4.5 mg/kg, intraperitoneally, i.p.). A cannula with replaceable inner guide (CMA Microdialysis, Solna, Sweden) was implanted stereotactically (3 mm) above the right hippocampus at flat skull coordinates of +4.6 mm medial‐lateral, −5.6 mm anterior‐posterior and −4.6 mm ventral‐dorsal relative to bregma. In addition, at least two rats of each group were i.p. implanted with a sterilised radiotelemetric rat transmitter (F20‐EET; Data Sciences International®, Tilburg, the Netherlands) to verify behavioural seizure activity (cf. further: “Seizure Severity Assessment”). The measuring electrode and the reference electrode, both with a stainless steel screw attached at the end, were subcutaneously tunnelled to the skull. The measuring electrode was stereotactically positioned above the hippocampus (contralateral site of probe insertion) and the reference electrode above the cerebellum (1 mm anterior according to lambda).
Mice
Mice were anesthetized with 2.5% isoflurane (Florene®; Abott, Kent, UK) in 100% oxygen. After induction, anesthesia was maintained during the entire duration of the surgery by 1.5% isoflurane in 100% oxygen via a facemask. A microdialysis guide (CMA Microdialysis) was implanted, 2 mm above the hippocampus with the following coordinates relative to bregma: +3.0 mm medial‐lateral, −2.7 mm anterior‐posterior and −1.5 mm ventral‐dorsal. Due to unclear behaviour in mice (cf. further: “Seizure Severity Assessment”), seizure severity in these animals was only examined by means of total seizure duration, as calculated from the electrocorticography (ECoG) recordings. Therefore, all mice were implanted with a sterilised radiotelemetric mouse transmitter (TA10EA‐F20; Data Sciences International®), as described above.
In vivo Microdialysis
Immediately after surgery, the inner guide was replaced with a microdialysis probe (CMA Microdialysis) with a membrane length of 3 mm for rats and 2 mm for mice. This probe was continuously perfused with a modified Ringer's solution (147 mM NaCl; 4 mM KCl; 2.3 mM CaCl2) at a constant flow rate of 2 μL/min (CMA 100 microdialysis pump, CMA Microdialysis). The animals were then allowed to recover from surgery overnight.
In general, every microdialysis experiment began the following day with perfusion of the probe with modified Ringer's solution alone (“baseline” conditions) for at least 2 h, and seizures were always evoked by intrahippocampal administration of pilocarpine (12 mM) during 40 min. Three types of experiments were performed and protocols differed between these experimental groups as depicted in the upper panels of each figure.
First, pilocarpine control experiments were carried out in both rats and mice. In these experiments, the perfusion of Ringer's solution was immediately followed by the pilocarpine administration. Secondly, the anticonvulsant actions of different doses of CST‐14 were investigated in both rats and mice. In these experimental groups, the perfusion of Ringer's solution was followed by 2 h of CST‐14 administration, prior to the pilocarpine perfusion. Detailed protocols for these experimental groups are illustrated in Figure 2A upper panel (rats) and Figure 4 upper panel (mice). Finally, rat experiments were performed studying the involvement of sst2 and sst3 receptor subtypes in the anticonvulsant actions of CST‐14. During these latter experiments, basal Ringer's perfusion was followed by 1 h of sst2‐ or sst3 antagonist administration. Hereafter, CST‐14 was co‐perfused with the antagonists for 2 h, followed by the pilocarpine administration (Protocol shown in Figure 2B upper panel).
Figure 2.
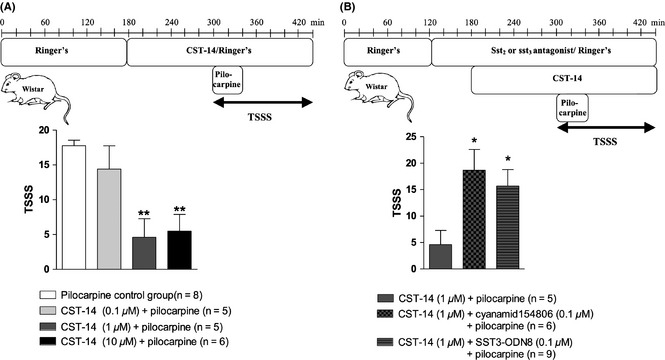
Experimental microdialysis protocol (A upper panel) and seizure severity scores (A lower panel) of dose response experiments of CST‐14 against pilocarpine‐induced seizures in rats. Experimental microdialysis protocol (B upper panel) and seizure severity scores (B lower panel) of experiments where the involvement of sst2 and sst3 receptors in the anticonvulsant actions of CST‐14 was tested in rats. The seizure severity of each set of experiments is expressed as total seizure severity score (TSSS) (mean ± SEM). Statistics: one‐way ANOVA, followed by the Bonferroni's posthoc test (*P < 0.05; **P < 0.01).
Figure 4.
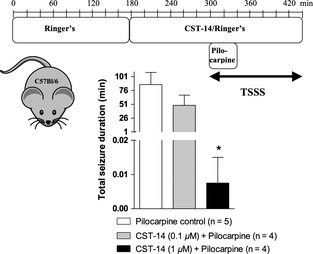
Experimental microdialysis protocol of pilocarpine control experiments and experiments where the effect of CST‐14 was tested in C57Bl/6 mice (upper panel). Effects of intrahippocampal CST‐14 (0.1–1 μM) administration on the severity of the pilocarpine‐evoked seizures (lower panel). The seizure severity of each set of experiments is expressed as the total seizure duration (mean ± SEM). The total seizure duration for each mouse was calculated as the sum of the duration of all individual seizures. Statistics: one‐way ANOVA, followed by the Bonferroni's posthoc test (*P < 0.05).
In all these experimental groups, the 40 min perfusion of pilocarpine was followed by 100 min perfusion of Ringer's, CST‐14 or co‐perfusion of sst antagonist with CST‐14, respectively, as depicted in the protocols (Figures 1, 2, 3, 4, 5).
Figure 1.
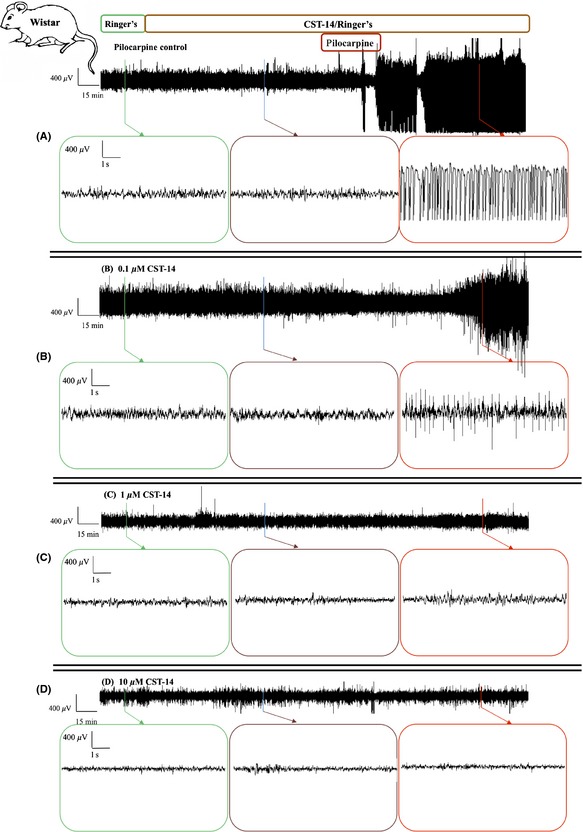
The sample time courses of in vivo ECoG recordings in rats. Representative ECoG recordings of (A) a control experiment, an experiment where the effect of (B) 0.1 μM CST‐14, (C) 1 μM CST‐14 and (D) 10 μM CST‐14 was examined. Each ECoG recording represents 30 min of baseline, 2 h of CST‐14 or Ringer's (pilocarpine control) perfusion, 40 min of co‐perfusion with pilocarpine and again 100 min of CST‐14 or Ringer's perfusion alone.
Figure 3.
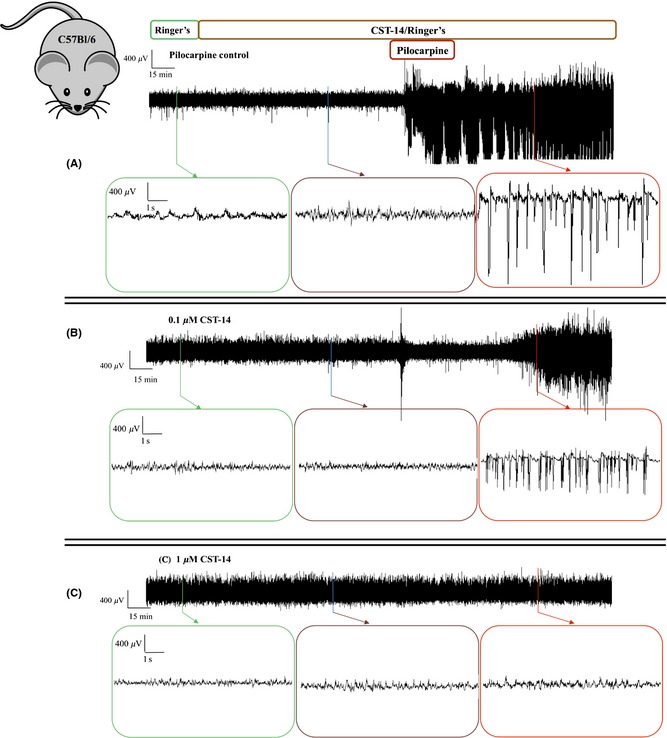
The sample time courses of in vivo ECoG recordings in C57Bl/6 mice. Representative ECoG recordings of (A) a control experiment, an experiment where the effect of (B) 0.1 μM CST‐14 and (C) 1 μM CST‐14 was examined. Each ECoG recording represents 30 min of baseline, 2 h of CST‐14 or Ringer's (pilocarpine control) perfusion, 40 min of co‐perfusion with pilocarpine and again 100 min of CST‐14 or Ringer's perfusion alone.
Figure 5.
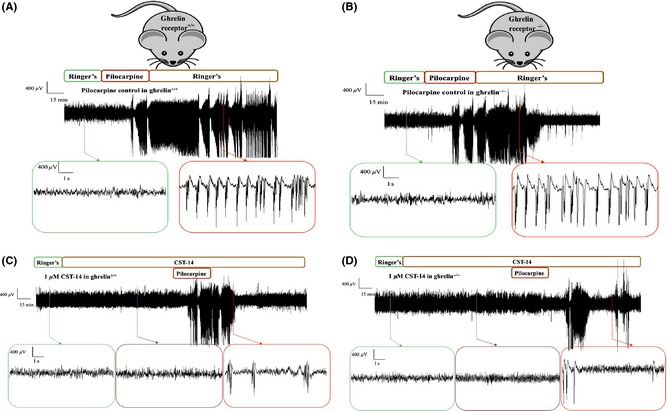
Representative ECoG recordings of a control experiment in (A) ghrelin receptor+/+ and (B) ghrelin receptor−/− mice. Each ECoG recording represents 30 min of baseline, 40 min of pilocarpine perfusion and again 100 min of Ringer's perfusion. Representative ECoG recordings of an experiment where the effect of 1 μM CST‐14 was examined in (C) ghrelin receptor+/+ and (D) ghrelin receptor−/− mice. Each ECoG recording represents 30 min of baseline, 2 h of CST‐14 perfusion, 40 min of co‐perfusion with pilocarpine and again 100 min of CST‐14 perfusion alone.
Seizure Severity Assessment
Rats
In rats, seizure severity was assessed using a scoring system derived from Racine's scale 21. This system is based on the typical behavioural changes associated with pilocarpine‐induced seizures and consists of six stages which correspond to the successive developmental seizure stages: (0) normal activity; (1) mouth and facial movements, hyperactivity, grooming, sniffing, scratching, wet dog shakes; (2) head nodding, staring, tremor; (3) forelimb clonus, forelimb extension; (4) rearing, salivating; (5) falling. Corresponding to these behavioural stages, every 20 min, the highest seizure severity score (SSS) was assigned during 140 min, starting from the pilocarpine administration. The total seizure severity score (TSSS) for each animal was then calculated as the sum of these 7 SSSs and was used as a measure of seizure severity. This behavioural scoring method was previously validated using ECoG 22. We routinely use at least 2–3 control rats within every set of new experiments to confirm the seizure severity using ECoG. Moreover, behavioural seizure severity assessment was verified with ECoG in at least 2 rats treated with each dose of CST‐14.
Mice
The behavioural scoring system, as described above for rats, could not be applied to mice due to a very unclear behaviour in these animals. Therefore, we here used ECoG recordings only to compare seizure severity (by means of total seizure duration) in ghrelin receptor−/− and ghrelin receptor+/+ mice. The total seizure duration for each mouse was calculated as the sum of the durations of all individual seizures, displayed on the ECoG recordings. Calculation of one seizure started from the time of identifiable change of ECoG activity to the end of ictal ECoG activity.
ECoG Recordings
The animals were placed in a video‐electroencephalographic monitoring unit equipped with the radiotelemetric receiver and coupled to Notocord‐hem Evolution® acquisition software (Notocord®, Croissy‐sur‐Seine, France). The ECoG was sampled with a frequency of 100 Hz during the microdialysis experiments. Behavioural seizure activity was correlated with ECoG activity in the rats. In mice, the ECoG recordings were used to calculate the total seizure duration.
Statistical Analysis
All mouse experiments were performed by the experimenter blinded to the genotype of the mice. All in vivo mouse and rat experiments were performed in a randomized fashion.
Statistical analyses were performed using GraphPad Prism 4.0 (GraphPad Software, Inc., La Jolla, CA, USA) software package for Windows. Data are expressed as the mean value ± SEM. One‐way ANOVA, followed by the Bonferroni's posthoc test was used to compare the mean TSSSs obtained from the pilocarpine control experiments with those of the treatment groups. The same statistical test was used in C57Bl/6 mice to compare the total seizure duration of the control versus CST‐14‐treated experimental groups. The effect of genotype on the CST‐14‐mediated anticonvulsant actions was assessed by two‐way ANOVA followed by Bonferroni's posthoc test. For all statistical analysis, α was set at 0.05.
Results
Intrahippocampal CST‐14 Administration Attenuates Pilocarpine‐Induced Seizures in Rats
Intrahippocampal administration of 12 mM pilocarpine evoked a host of behavioural changes with characteristic ECoG pattern (Figure 1A), as previously described 14. The mean TSSS in the pilocarpine control group was 17.8 ± 0.8, n = 8 (Figure 2A lower panel).
We investigated whether intrahippocampal administration of CST‐14 changes seizure severity in our rat limbic seizure model. Pilocarpine‐induced seizures were not significantly attenuated by intrahippocampal perfusion of 0.1 μM CST‐14. As monitored by ECoG, 0.1 μM CST‐14 seems to induce an increased latency to first seizure (Figure 1B), however, no significant decrease was observed in TSSS. The mean TSSS of the CST‐14 (0.1 μM)‐treated animals was 14.4 ± 3.3 (n = 5, Figure 2A lower panel) and was not significantly different from the TSSS of the pilocarpine control group (17.8 ± 0.8, n = 8). Intrahippocampal perfusion of 1 and 10 μM CST‐14 abolished full‐blown pilocarpine‐induced seizures (Figure 1C,D). The mean TSSS was 4.6 ± 2.7 for the 1 μM CST‐14‐treated group (n = 5) and 5.5 ± 2.4 for the rats receiving 10 μM CST‐14 (n = 6). These TSSSs were significantly lower (P < 0.01) from the TSSS of the control group (17.8 ± 0.8, n = 8; Figure 2A lower panel).
Anticonvulsant Actions of CST‐14 are Reversed by Selective sst2 or sst3 Receptor Antagonism
To examine the possible involvement of the sst2 and the sst3 receptor subtypes in the anticonvulsant actions of CST‐14, we used combinations of CST‐14 with selective sst2 or sst3 receptor antagonists in our rat model. Perfusion of the selective sst2 or sst3 antagonist alone did not alter the pilocarpine‐induced seizure severity (data not shown), as previously demonstrated 14.
We here show that selective blockade of hippocampal sst2 receptors with 100 nM cyanamid15486 reversed the anticonvulsant actions of CST‐14. Indeed, co‐perfusion of the sst2 receptor antagonist cyanamid15486 with 1 μM CST‐14 abolished the anticonvulsant effect of CST‐14 (Figure 2B lower panel). The mean TSSS of the CST‐14/cyanamid15486‐treated rats (18.7 ± 3.9, n = 6) was significantly higher (P < 0.05) than the TSSS for rats receiving only CST‐14 before pilocarpine (4.6 ± 2.7, n = 5). Co‐perfusion of the sst3 receptor antagonist SST3‐ODN‐8 (100 nM) with CST‐14 (1 μM) also clearly reversed the anticonvulsant actions of CST‐14 (Figure 2B lower panel). The mean TSSS of the CST‐14/SST3‐ODN‐8 group (15.7 ± 3.1, n = 9) was significantly higher (P < 0.05) compared with the rats receiving only CST‐14 before pilocarpine (4.6 ± 2.7, n = 5).
Absence of the Ghrelin Receptor does not Influence the Anticonvulsant Effect of CST‐14
To investigate whether the ghrelin receptor plays a role in mediating the anticonvulsant actions of CST‐14, we locally perfused a threshold anticonvulsant dose of CST‐14, as obtained in this study, into the hippocampus of both ghrelin receptor−/− mice and their ghrelin receptor+/+ littermates and compared the total seizure duration of both genotypes. First, we determined the threshold anticonvulsant dose of CST‐14 in mice. C57Bl/6 mice were used to stay in line with the genetic background of the ghrelin receptor−/− animals. Representative in vivo ECoG recordings of these dose–response experiments are depicted in Figure 3. We showed, similar to the previous rat experiments that 0.1 μM CST‐14 administration seems to induce an increased latency to the first seizure (Figure 3B), however, also in mice, no significant decrease was observed when the total seizure duration in the control animals (86.7 min ± 22.1, n = 5) was compared with the total seizure duration observed in the CST‐14 (0.1 μM) treated animals (49.1 min ± 18.5, n = 4; Figure 4 lower panel). Perfusion of 1 μM CST‐14 resulted in a clear significant decrease (P < 0.05) of the total seizure duration (0.008 min ± 0.008) when compared to the control mice (Figure 4 lower panel).
Secondly, pilocarpine control experiments were also carried out in ghrelin receptor−/− mice and ghrelin receptor+/+ mice and finally the threshold anticonvulsant dose of CST‐14 (1 μM) was tested in both genotypes. Representative in vivo ECoG recordings in ghrelin receptor+/+ mice and ghrelin receptor−/− mice are depicted in Figure 5. These experiments revealed both genotype‐ and treatment‐dependent alterations (Figure 6A). Indeed, by means of two‐way ANOVA, we showed that ghrelin receptor deletion induces a clear overall decrease in total seizure duration (P < 0.01). The same statistical test shows that, although not resulting into complete seizure protection, administration of 1 μM CST‐14 significantly decreased the total seizure duration in both ghrelin receptor+/+ mice and ghrelin receptor−/− mice. This decrease was however similar (~50%) in both genotypes (Figure 6B). These observations suggest that, although ghrelin receptor−/− mice are less susceptible to the pilocarpine‐induced seizures, the overall anticonvulsant effect of CST‐14 remains unchanged in both genotypes and thus, ghrelin receptors seem not to play a role in the anticonvulsant actions of CST‐14.
Figure 6.
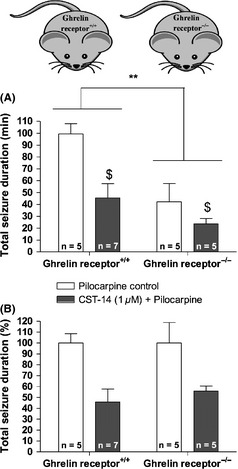
The effect of ghrelin receptor deletion and CST‐14 (1 μM) treatment in ghrelin receptor+/+ and ghrelin receptor−/− mice. (A) The total seizure duration in each genotype is expressed as minutes (mean values ± SEM) (**P < 0.01: Effect of genotype; $P < 0.05 effect of CST‐14 [1 μM] treatment). The total seizure duration for each mouse was calculated as the sum of the duration of all individual seizures. (B) For clarity of presentation, the overall decrease in seizure duration following CST‐14 (1 μM) perfusion was expressed as the percentage of the mean seizure duration obtained in the corresponding control mice.
Discussion
Selective sst2 or sst3 Receptor Antagonism Abolishes the Anticonvulsant Actions of CST‐14, Comparison with SRIF
Studies investigating the role of sst receptor subtypes in the anticonvulsant effects of CST‐14 are currently lacking. In this study, we demonstrate for the first time that anticonvulsant actions of CST‐14 are mediated by sst receptor activation in vivo. Indeed, we here show that CST‐14 elicits potent anticonvulsant actions and that these effects can be reversed in the presence of a selective sst2 or a selective sst3 receptor antagonist and this, independently of each other. These observations indicate that sst3 receptor activation is yet as important as sst2 receptor activation when it comes to the CST‐14‐induced anticonvulsant effects. These novel findings confirm once again that, despite many similarities, CST‐14 and SRIF can act differently. For SRIF, we indeed recently showed that co‐perfusion of this peptide with the sst2 ‐but not with the sst3‐ receptor antagonist was able to reverse the SRIF‐mediated anticonvulsant actions in the same focal rat model for limbic seizures 14. These findings suggested that in rats, mainly sst2 receptors are indispensably involved in the anticonvulsant effects of SRIF. Although CST‐14 is able to bind all sst receptors with comparable affinity as SRIF itself, functional differences between CST‐14 and SRIF signalling can occur 1, 6, 23. These differences might possibly be explained by sst receptor heterodimerisation 6, 23. Indeed, the complexity of sst receptors and their signalling is increased by their ability to form both homo‐ and heterodimers 24, 25. The effects of CST‐14 and SRIF have so far not been compared at dimeric receptors, but it is possible that the peptides may induce the formation of different dimers, which could result in different pharmacological features for CST‐14 compared with SRIF 6. This dimerisation of sst receptors however has only been observed in vitro, and whether this concept can be extended to native systems remains unclear. In addition, the presence of receptor‐activity‐modifying proteins (RAMPs) may also explain functional differences between CST‐14 and SRIF signalling 6, 23. Indeed, these RAMPs could interact with sst receptors to affect their CST‐14/SRIF selectivity or the result of ligand‐binding 1. To the best of our knowledge, there are however no data available that describe the interaction of such RAMPs with sst receptors, so far.
Ghrelin Receptors are not Involved in the Anticonvulsant Actions of CST‐14
In 2001, Deghenghi and co‐workers showed that CST‐14, but not SRIF, was able to bind ghrelin receptors with similar affinity as acylated ghrelin, a natural ligand for the ghrelin receptor 7. Ghrelin receptors were therefore proposed as a possible CST‐14‐specific receptor. In the current study, we demonstrate for the first time that CST‐14 is also able to exert potent anticonvulsant effects in a mouse model for limbic seizures, and we show that these anticonvulsant actions were not influenced by a deletion of ghrelin receptors. Indeed, we showed a clear decrease in total seizure duration when CST‐14 treated mice were compared with control animals. Furthermore, the CST‐14‐induced decrease was comparable in ghrelin receptor+/+ mice and ghrelin receptor−/− mice. We here thus provide the first evidence that ghrelin receptors are not involved in mediating anticonvulsant actions of CST‐14. Notably, the ghrelin receptor−/− mice showed lower susceptibility to pilocarpine‐induced seizures, compared with their wild type littermates as a significant overall decrease in total seizure duration was caused by the absence of ghrelin receptors. These observations can be explained when having a closer look at ghrelin and at the pharmacology of the ghrelin receptor. Indeed, although anticonvulsant actions of ghrelin have already clearly been demonstrated in rodents 26, 27, we recently showed that complete deletion of ghrelin receptors, inverse agonism or agonism followed by desensitization/internalization of ghrelin receptors attenuate limbic seizures 28. An answer for these, at first sight paradoxal observations, can be found in the complex pharmacology of the ghrelin receptor. This receptor is one of the few known GPCRs having a relatively high constitutive activity of approximately 50% 29. In general, inactivation of this constitutively active receptor, including deletion such as in the ghrelin receptor−/− mice, is necessary to exert seizure‐attenuating effects in several rodent models for limbic seizures 28 and explains why ghrelin receptor−/− mice show a lower seizure susceptibility when compared with ghrelin receptor+/+ mice.
Conclusion
In the current study, we show that CST‐14‐mediated anticonvulsant actions are reversed by sst2 or sst3 receptor antagonism. Ghrelin receptors seem not to modulate CST‐14‐induced anticonvulsive effects. To the best of our knowledge, this is the first study investigating the role of the major receptors CST‐14 shows high affinity for, in the anticonvulsant actions of this neuropeptide.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors wish to thank Mr. G. De Smet and Mr. A. De Baerdemaeker for the technical assistance. We would also like to acknowledge Prof. Jean Rivier for providing us with the sst3 antagonist and Prof. R. Buyl for his contribution in processing the statistical data. I. Smolders and G. Di Giovanni are indebted to the EU COST Action CM1103 “Structure‐based drug design for diagnosis and treatment of neurological diseases: dissecting and modulating complex function in the monoaminergic systems of the brain” for supporting their international collaboration. This work was supported by funding from the FWO‐Vlaanderen (G.0163.10N) and the Research Council of the Vrije Universiteit Brussel.
References
- 1. Spier AD, de Lecea L. Cortistatin: A member of the somatostatin neuropeptide family with distinct physiological functions. Brain Res Brain Res Rev 2000;33:228–241. [DOI] [PubMed] [Google Scholar]
- 2. de Lecea L, Criado JR, Prospero‐Garcia O, et al. A cortical neuropeptide with neuronal depressant and sleep‐modulating properties. Nature 1996;381:242–245. [DOI] [PubMed] [Google Scholar]
- 3. Csaba Z, Dournaud P. Cellular biology of somatostatin receptors. Neuropeptides 2001;35:1–23. [DOI] [PubMed] [Google Scholar]
- 4. Fukusumi S, Kitada C, Takekawa S, et al. Identification and characterization of a novel human cortistatin‐like peptide. Biochem Biophys Res Commun 1997;232:157–163. [DOI] [PubMed] [Google Scholar]
- 5. Vasilaki A, Lanneau C, Dournaud P, De Lecea L, Gardette R, Epelbaum J. Cortistatin affects glutamate sensitivity in mouse hypothalamic neurons through activation of sst2 somatostatin receptor subtype. Neuroscience 1999;88:359–364. [DOI] [PubMed] [Google Scholar]
- 6. Siehler S, Nunn C, Hannon J, Feuerbach D, Hoyer D. Pharmacological profile of somatostatin and cortistatin receptors. Mol Cell Endocrinol 2008;286:26–34. [DOI] [PubMed] [Google Scholar]
- 7. Deghenghi R, Papotti M, Ghigo E, Muccioli G. Cortistatin, but not somatostatin, binds to growth hormone secretagogue (GHS) receptors of human pituitary gland. J Endocrinol Invest 2001;24:RC1–RC3. [DOI] [PubMed] [Google Scholar]
- 8. Muccioli G, Papotti M, Locatelli V, Ghigo E, Deghenghi R. Binding of 125I‐labeled ghrelin to membranes from human hypothalamus and pituitary gland. J Endocrinol Invest 2001;24:RC7–RC9. [DOI] [PubMed] [Google Scholar]
- 9. Broglio F, Papotti M, Muccioli G, Ghigo E. Brain‐gut communication: Cortistatin, somatostatin and ghrelin. Trends Endocrinol Metab 2007;18:246–251. [DOI] [PubMed] [Google Scholar]
- 10. Vezzani A, Hoyer D. Brain somatostatin: A candidate inhibitory role in seizures and epileptogenesis. Eur J Neurosci 1999;11:3767–3776. [DOI] [PubMed] [Google Scholar]
- 11. Tallent M, Qiu C. Somatostatin: An endogenous antiepileptic. Mol Cell Endocrinol 2008;286:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. J Neurosci 1997;17:4066–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tallent M, Siggins G. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol 1999;81:1626–1635. [DOI] [PubMed] [Google Scholar]
- 14. Aourz N, De Bundel D, Stragier B, et al. Rat hippocampal somatostatin sst3 and sst4 receptors mediate anticonvulsive effects in vivo: Indications of functional interactions with sst2 receptors. Neuropharmacology 2011;61:1327–1333. [DOI] [PubMed] [Google Scholar]
- 15. Cammalleri M, Cervia D, Langenegger D, et al. Somatostatin receptors differentially affect spontaneous epileptiform activity in mouse hippocampal slices. Eur J Neurosci 2004;20:2711–2721. [DOI] [PubMed] [Google Scholar]
- 16. Cammalleri M, Martini D, Timperio AM, Bagnoli P. Functional effects of somatostatin receptor 1 activation on synaptic transmission in the mouse hippocampus. J Neurochem 2009;111:1466–1477. [DOI] [PubMed] [Google Scholar]
- 17. Qiu C, Zeyda T, Johnson B, Hochgeschwender U, de Lecea L, Tallent M. Somatostatin receptor subtype 4 couples to the M‐current to regulate seizures. J Neurosci 2008;28:3567–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moneta D, Richichi C, Aliprandi M, et al. Somatostatin receptor subtypes 2 and 4 affect seizure susceptibility and hippocampal excitatory neurotransmission in mice. Eur J Neurosci 2002;16:843–849. [DOI] [PubMed] [Google Scholar]
- 19. Braun H, Schulz S, Becker A, Schröder H, Höllt V. Protective effects of cortistatin (CST‐14) against kainate‐induced neurotoxicity in rat brain. Brain Res 1998;803:54–60. [DOI] [PubMed] [Google Scholar]
- 20. Verhulst PJ, De Smet B, Saels I, et al. Role of ghrelin in the relationship between hyperphagia and accelerated gastric emptying in diabetic mice. Gastroenterology 2008;135:1267–1276. [DOI] [PubMed] [Google Scholar]
- 21. Racine R. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32:281–294. [DOI] [PubMed] [Google Scholar]
- 22. Meurs A, Clinckers R, Ebinger G, Michotte Y, Smolders I. Substantia nigra is an anticonvulsant site of action of topiramate in the focal pilocarpine model of limbic seizures. Epilepsia 2006;47:1519–1535. [DOI] [PubMed] [Google Scholar]
- 23. de Lecea L. Cortistatin–functions in the central nervous system. Mol Cell Endocrinol 2008;286:88–95. [DOI] [PubMed] [Google Scholar]
- 24. Pfeiffer M, Koch T, Schröder H, et al. Homo‐ and heterodimerization of somatostatin receptor subtypes. Inactivation of sst(3) receptor function by heterodimerization with sst(2A). J Biol Chem 2001;276:14027–14036. [DOI] [PubMed] [Google Scholar]
- 25. Rocheville M, Lange DC, Kumar U, Sasi R, Patel RC, Patel YC. Subtypes of the somatostatin receptor assemble as functional homo‐ and heterodimers. J Biol Chem 2000;275:7862–7869. [DOI] [PubMed] [Google Scholar]
- 26. Aslan A, Yildirim M, Ayyildiz M, Güven A, Agar E. The role of nitric oxide in the inhibitory effect of ghrelin against penicillin‐induced epileptiform activity in rat. Neuropeptides 2009;43:295–302. [DOI] [PubMed] [Google Scholar]
- 27. Obay BD, Tasdemir E, Tümer C, Bilgin HM, Sermet A. Antiepileptic effects of ghrelin on pentylenetetrazole‐induced seizures in rats. Peptides 2007;28:1214–1219. [DOI] [PubMed] [Google Scholar]
- 28. Portelli J, Thielemans L, Ver Donck L, et al. Inactivation of the constitutively active ghrelin receptor attenuates limbic seizure activity in rodents. Neurotherapeutics 2012;9:658–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor–identification of a potent inverse agonist. Mol Endocrinol 2003;17:2201–2210. [DOI] [PubMed] [Google Scholar]