

Summary
Aims
Spinal cord injury (SCI) occurs following damage to the spinal column. Following trauma, tissue damage is further exacerbated by a secondary damage due to a SCI‐activated inflammatory process. Control of leukocytes activity is essential to therapeutic inhibition of the spinal cord damage to ameliorate the patient's conditions. The mechanisms that regulate neuroinflammation following SCI, including T‐cell infiltration, have not been completely clarified. Glucocorticoids (GC) are antiinflammatory drugs widely used in therapy, including treatment of SCI. GC efficacy may be linked to many molecular mechanisms that are involved in regulation of leukocytes migration, activation, and differentiation. We have previously shown that the antiinflammatory activity of GC is in part mediated by glucocorticoid‐induced leucine zipper (GILZ). Here, we investigated the role of GILZ in inflammation and spinal cord tissue damage following a spinal trauma.
Methods
We address the role of GILZ in SCI‐induced inflammation and tissue damage using a model of SCI in gilz knockout (gilz KO) and wild‐type (WT) mice.
Results
We found that GILZ deficiency is associated with a strong reduction of SCI‐induced inflammation and a significantly reduced lesion area following SCI.
Conclusion
These results demonstrate that GILZ is involved in induction of neuroinflammation and functional outcomes of spinal cord trauma.
Keywords: Glucocorticoid‐induced leucine zipper, Inflammation, Spinal cord injuries, T lymphocytes
Introduction
The central nervous system (CNS) is sensitive to mechanical injuries, causing permanent functional deficits in patients with spinal cord injury (SCI). The mechanical forces imparted to the spinal cord cause immediate tissue disruption, with a direct axonal and neuronal injury, characterized by death of a number of neurons that can neither be recovered nor regenerated. Moreover, neurons continue to die for hours after SCI as a result of several mechanisms, including excitotoxicity, vascular abnormalities and inflammatory response that contribute to evolution of spinal cord secondary injury 1. Both innate and adaptive immune responses play a key role in gravity and extent of spinal cord injury and repair processes after SCI 2, 3, 4. The degree of accumulation of leukocytes, especially lymphocytes and granulocytes, in the injured spinal cord area correlates with the extent of inflammation and secondary neuronal damage. Lymphocytes, in particular, are believed to be important for the initiation and progression of inflammation following SCI because they contain and release a significant number of inflammatory mediators that may damage neurons. The humoral components of neuroinflammation, such a cytokines, are also found to play an important role in the initiation, maintenance, and resolution of inflammation following SCI 3, 5, 6, 7.
Despite major progress in pharmacological, surgical, and rehabilitative treatment approaches, SCI still remains a very complex medical and psychological challenge, with no curative therapy available 8. The only pharmacological compound that has demonstrated valuable therapeutic efficacy and neuroprotection is the synthetic glucocorticoid (GC) methylprednisolone, when administered at high doses within 3–8 h from the trauma 9, 10, 11. Moreover, recent studies further confirmed the usefulness of such a treatment 12, 13. GC have also been widely used to treat many inflammatory and autoimmune diseases 14, 15, but a number of adverse drug reactions, associated with chronic treatment with GC, represent an important limit that in some circumstances causes the suspension of therapy 16.
Most of the effects mediated by GC depend on the interaction with the GC receptor (GR) and consequent modulation of its transcriptional activity. Glucocorticoid‐induced leucine zipper (GILZ) is a gene rapidly and potently upregulated by GC treatment 17, 18. Several data support a role for GILZ as a mediator of GC‐activated antiinflammatory effect 19, 20. It mediates a number of GC effects such as control of cell proliferation, apoptosis, and differentiation, including in T cells 21, 22, 23, 24. Notably, GILZ is expressed in different areas of the CNS, such as forebrain, spinal cord, and cerebellum 25, 26.
Here, we addressed to the role of GILZ in inflammation and spinal cord tissue damage following a spinal trauma using a mouse model of SCI. We studied the effects of GILZ deficiency in SCI using gilz knockout (KO) mice. We found that lack of GILZ determines a strong reduction of spinal cord tissue damage and of leukocytes infiltration upon SCI. GILZ deficiency is associated with an altered cytokines expression profile upon SCI induction, with an increased production of Th1 cytokines and decreased expression of Th2 and Th17 cytokines, thus suggesting that GILZ is important for the modulation of inflammatory processes occurring upon SCI and may represent a new therapeutic target to achieve a better functional outcome of spinal cord lesions.
Materials and Methods
Mice
Six‐ to 10‐week‐old male gilz KO and wild‐type (WT) mice with C57BL/6J background were used and analyzed for genotype and GILZ expression, as previously described 22. In each experiment, gilz KO mice were compared with WT littermates. Animal care was in compliance with regulations in Italy (DM 116/92 and DL 26/2014) and Europe (2010/63/UE).
Sci
SCI was induced as previously described 27. Briefly, mice were anesthetized using an anesthetic cocktail containing 2% xylazine and 1 mg/mL tiletamine (10 mL/kg body weight). A longitudinal incision of about 5 cm was made on the midline of the back, exposing the paravertebral muscles. These muscles were dissected away exposing T5‐T8 vertebrae. The spinal cord was exposed via a 4‐level T5‐T8 laminectomy, and SCI was produced by a compression model induced at T6‐T7 level using an aneurysm clip with a closing force of 24 g. In all injured groups, the spinal cord was compressed for 1 min. Sham animals were only subjected to cutaneous excision. Following surgery, 1.0 mL of saline was administered subcutaneously to replace the blood volume lost during the surgery. During recovery from anesthesia, the mice were placed on a warm heating pad and covered with a warm towel. The mice were individually housed in a temperature‐controlled room at 27°C for a survival period of 7 days. Food and water were provided to the mice ad libitum. During this time period, the animals' bladders were manually voided twice a day until the mice were able to regain normal bladder function.
Mice were randomly allocated into the following groups: (1) WT SCI group: mice were subjected to SCI; (2) gilz KO SCI group: mice were subjected to SCI; (3) control WT group (sham WT): mice were subjected to a cutaneous excision of about 5 cm, as with the previous groups, without laminectomy so that aneurysm clip was not applied; and (4) control gilz KO group (sham KO): mice were subjected a cutaneous excision of about 5 cm, as with the previous groups, without laminectomy so that the aneurysm clip was not applied. In each experiment, we used five animals per group, unless otherwise indicated. Mice from each group were sacrificed 7 days after SCI to collect samples for the evaluation of the parameters described as follows.
Histology
Spinal cord tissues were taken 7 days following trauma and were fixed for 24 h in paraformaldehyde solution (4% in PBS 0.1 M) at room temperature, dehydrated by graded ethanol, and embedded in Paraplast (Sherwood Medical, Mahwah, NJ, USA). Tissue sections (thickness, 7 μm) were deparaffinized with xylene, stained with hematoxylin and eosin, and studied using light microscopy (LEICA DM 2000 combined with a LEICA ICC50 HD camera). All the histological studies were performed in a blinded fashion.
Immunohistochemistry
At the 24 h after SCI induction, the tissues were fixed in 10% (w/v) PBS‐buffered formaldehyde, and 7‐μm sections were prepared from paraffin‐embedded tissues. After de‐paraffinization, endogenous peroxidase was quenched with 0.3% (v/v) hydrogen peroxide in 60% (v/v) methanol for 30 min. Sections were permeabilized with 0.2% Triton‐X 100, and nonspecific binding sites were subsequently blocked with 10% normal goat serum. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with biotin and avidin (Vector Laboratories, Burlingame, CA, USA), respectively. Sections were incubated overnight at 4°C with anti‐CD4 rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, 1:100 in PBS, v/v), anti‐CD8α rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc., 1:100 in PBS, v/v), anti‐Chymase rabbit polyclonal antibody (Millipore, Milan, Italy, 1:500 in PBS, v/v), anti‐ICAM‐1 rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc., 1:200 in PBS, v/v), and anti‐P‐selectin (Santa Cruz Biotechnology, Inc., 1:100 in PBS). After washing with PBS, sections were incubated with HRP‐conjugated secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 30 min at room temperature, followed by applying DAB regents (Vector Laboratories) for DAB detection. The counterstain was developed with hematoxylin (blue background). To verify the binding specificity, some sections were also incubated with only the primary antibody (no secondary) or with only the secondary antibody (no primary). In these situations, no positive staining was found in the sections, indicating that the immunoreaction was positive in all the experiments carried out.
All sections were obtained using light microscopy (LEICA DM 2000 combined with LEICA ICC50 HD camera), and the quantitative analysis was performed using computer program (Leica Application Suite V4.1; Leica mcrosystems Cambridge/LAS V4.4, Milano, Italy).
Quantitative Real‐time PCR
Total RNA was extracted using TRIzol reagent (Life Technologies, Monza, Italy). RT‐PCR was performed using QuantiTect Reverse Transcription (Qiagen, Milano, Italy). PCR was performed in a final volume of 20 μL containing 0.02 μM cDNA, 0.5 μM sense and antisense primers, 1.5 mM MgCl2, and 1 unit of platinum TaqDNA polymerase (Life Technologies). The PCR was performed according to the manufacturer's instructions. The PCR products were electrophoresed on a 2% agarose gels in the presence of ethidium bromide. Real‐Time quantitative PCR (qPCR) analysis was performed in Applied Biosystems real‐time PCR machine (ABI7300) using TaqMan Gene Expression Master Mix (Life Technologies). The real‐time PCR primers and probes (Life Technologies) used were the following: Il12: Mm00434169; Ifnγ: Mm00801778; Il4: Mm99999154; Il10: Mm01288386; Il17: Mm00439619; Tgfb: Mm00441724; and gilz: Mm00726417. mRNA expression levels were calculated by the Comparative ΔΔC(t) method relative to beta‐actin (Actb) housekeeping gene.
Statistical Analysis
Statistical analysis was performed with Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA). The nonparametric Mann–Whitney U‐test or a two‐tailed unpaired Student's t‐test were used for statistical comparisons.
For immunohistochemical densitometric analysis, multiple group comparisons were conducted by ordinary one‐way analysis of variance (ANOVA) with Bonferroni test. For qPCR analysis, all experiments were run in triplicate and two‐tailed unpaired Student's t‐test were used for statistical comparisons. *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001.
Results
Lack of GILZ Reduces Spinal Cord Damage
To evaluate the role of GILZ in spinal cord damage following a traumatic event, we applied the SCI protocol to gilz KO and WT control mice. Body weight was monitored for the period from 1 day before until day 7 after SCI induction. During this experimental period, both WT SCI and gilz KO SCI groups showed about 20% decrease in body weight compared with sham groups (Figure 1). At the end of follow‐up (7 days), mice were sacrificed and the spinal cords were collected for histological analysis of tissue damage. The spinal cords were sampled in correspondence to the thoracic portion lesion to analyze for histomorphological alterations. Results clearly show major damage at the perilesional area of the spinal cord in WT SCI mice compared with the gilz KO SCI mice (Figure 2C vs. 2D), the latter showing a similar morphology of the sham group (Figure 2A and B). These results show that the absence of GILZ results in a strong reduction of damage following SCI.
Figure 1.
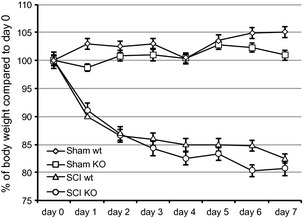
No differences in body weight between WT and gilz KO mice after SCI. Mice were daily weighted starting from the day just prior SCI induction (Day 0). Mice n = 5, 6 per group. Error bars, SD; statistical analysis: two‐tailed Student's t‐test.
Figure 2.
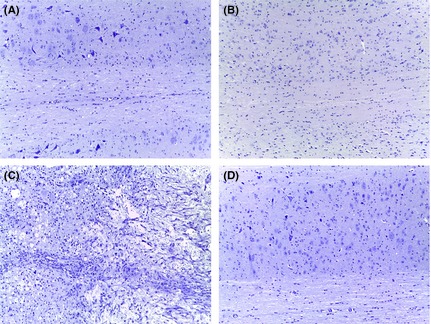
Reduction of tissue damage in gilz KO compared with WT in spinal cord after SCI. Spinal cord histological phenotype of WT and gilz KO mice after SCI. Images of spinal cord histological section from WT and gilz KO mice at 7 days post‐SCI assessed by H&E staining. Scale bars 100 μm, 20X. (A) Sham WT; (B) Sham gilz KO; (C) WT SCI; (D) gilz KO SCI.
Lack of GILZ Inhibits Leukocytes Infiltration in Spinal Cord after SCI
SCI leads to a robust and persistent inflammatory response, which involves cell activation of both innate and adaptive immune system 2, 4, 28. To evaluate possible differences in leukocytes presence in the damaged area, we have first assessed the recruitment of leukocytes to the spinal cord around the site of injury in WT and gilz KO mice by histological examinations. As expected, no leukocytes were observed in spinal cords of sham animals, while mice subjected to SCI revealed a presence of leukocytes in the spinal cords. A significant decrease in the number of CD4 (Figure 3C vs. 3D. See densitometric analysis bottom graph) and CD8 (Figure 3G vs. 3H. See densitometric analysis bottom graph) positive T cells was observed in spinal cords of gilz KO mice compared with controls after SCI induction.
Figure 3.
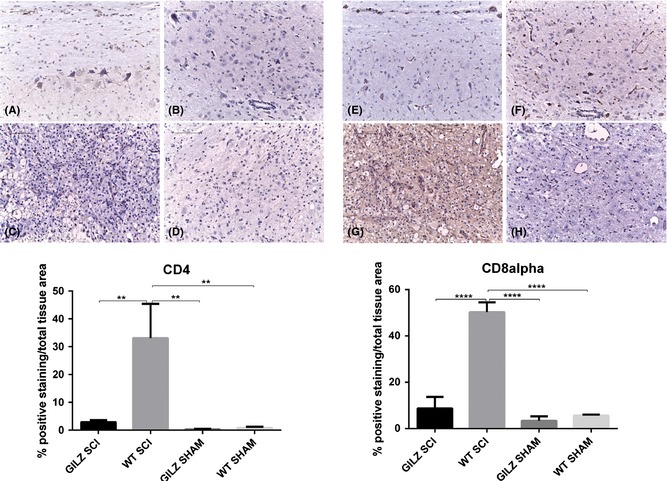
Decreased numbers of CD4 and CD8 positive cells in gilz KO compared with WT in spinal cord after SCI. CD4 expression in spinal cord after SCI. Immunohistochemical analyses show that the number of CD4+ lymphocytes in perilesional area of spinal cord is reduced in gilz KO SCI compared with WT SCI mice. Scale bars 100 μm, 20X. CD4 staining: (A) Sham WT; (B) Sham gilz KO; (C) WT SCI; (D) gilz KO SCI. CD8 staining: (E) Sham WT; (F) Sham gilz KO; (G) WT SCI; and (H) gilz KO SCI. In the bottom graph is represented the densitometric analysis to quantify and highlight significant differences among experimental groups. For each staining, results are expressed as “% of positive staining” calculated on the mean of at least n = 3 acquired IHC image/group. **P < 0.005, ****P < 0.0001, Bonferroni test.
Mast cells are the first line of defense in inflammatory response to tissue injury, including SCI 29. We compared the degree of mast cells infiltration in the spinal cord of WT and gilz KO SCI mice by evaluating the expression of the protein chymase, a peptidase expressed in mast cells and in pathological conditions including inflammation 30. Immunohistochemical analysis revealed a positive chymase staining in spinal cord sections of WT SCI animals and not in sham controls. To the contrary, gilz KO SCI mice appeared negative for chymase staining, suggesting that granulocyte infiltration into spinal cords does not efficiently occur in GILZ‐deficient mice (Figure 4C. See densitometric analysis bottom graph).
Figure 4.
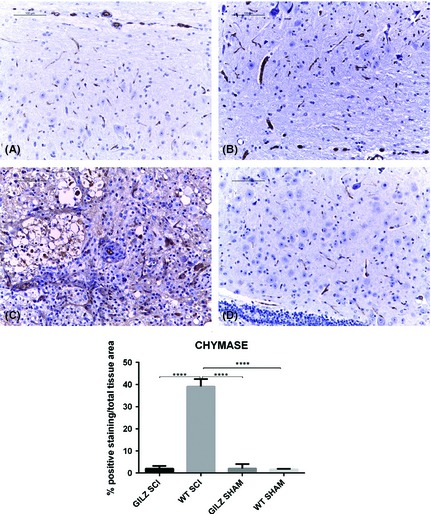
Decreased numbers of granulocytes in gilz KO compared with WT in spinal cord after SCI. Chymase expression in spinal cord after SCI. Immunohistochemical analyses show that the number of granulocytes (chymase+ cells) in perilesional area of spinal cord is reduced in gilz KO SCI compared with WT SCI mice. Scale bars 100 μm, 20X. (A) Sham WT; (B) Sham gilz KO; (C) WT SCI; and (D) gilz KO SCI. In the bottom graph is represented the densitometric analysis to quantify and highlight significant differences among experimental groups. For each staining, results are expressed as “% of positive staining” calculated on the mean of at least n = 3 acquired IHC image/group. ****P < 0.0001, Bonferroni test.
Leukocytes that originate in bone marrow from lymphoid and myeloid precursors circulate in the peripheral blood and migrate to the sites of injury in response to damage. Some authors declare that severe SCI causes leukocyte arrest and transmigration across spinal cord endothelial barrier facilitated by endothelial adhesion molecules P‐selectin and ICAM‐1 exposition 31. Also, according to Farooque et colleagues, ICAM‐1 and P‐selectin knockout mice subjected to SCI show a better functional outcome during an observation period of 14 days 32. In our study, we measured the expression of ICAM‐1 and P‐selectin in spinal cords of WT and gilz KO mice. We detected significant expression of ICAM‐1 and P‐selectin in SCI wt mice (Figure 5C and G, respectively. See densitometric analysis bottom graph) but not in gilz KO SCI mice (Figure 5D and H, respectively. See densitometric analysis bottom graph). Taken together, these data demonstrate that lack of GILZ ameliorates spinal cord functional recovery due to a reduced polymorphonuclear leukocytes infiltration.
Figure 5.
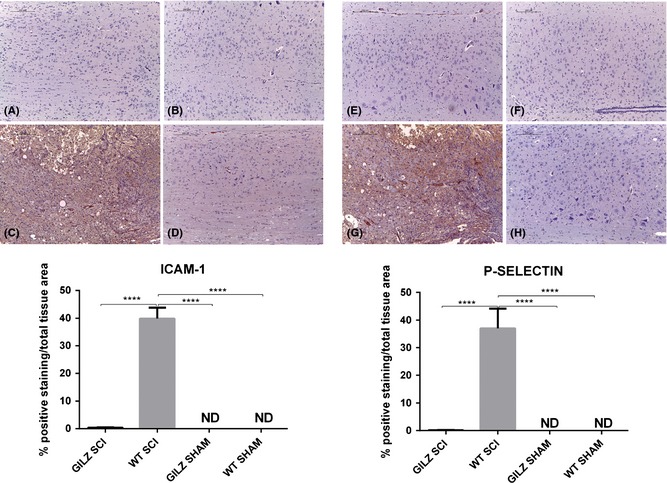
Reduced ICAM‐1 and P‐selectin positive cells in gilz KO compared with WT in spinal cord after SCI. ICAM‐1 and P‐selectin expression in spinal cord after SCI. Immunohistochemical analyses show that both ICAM‐1 and P‐selectin expression in perilesional area of spinal cord is reduced in gilz KO SCI compared with WT SCI mice. Scale bars 200 μm, 10X. ICAM‐1 staining: (A) Sham WT; (B) Sham gilz KO; (C) WT SCI; (D) gilz KO SCI. P‐selectin staining: (E) Sham WT; (F) Sham gilz KO; (G) WT SCI; and (H) gilz KO SCI. In the bottom graphs are represented the densitometric analysis to quantify and highlight significant differences among experimental groups. For each staining, results are expressed as “% of positive staining” calculated on the mean of at least n = 3 acquired IHC image/group. ****P < 0.0001, Bonferroni test.
Lack of GILZ Affects Cytokine Production in Periphery and Spinal Cord after SCI
In addition to induce local neuroinflammation, SCI represents an inflammatory signal for peripheral lymphocyte activation 5. It has been suggested that the T‐cell subsets and activation status influence the degree of neuronal damage and the functional outcome after SCI 1. To characterize the type of the inflammatory response occurring after SCI, we analyzed the expression of proinflammatory cytokines in peripheral lymph nodes of WT and gilz KO mice at day 7 after SCI induction, a time‐point previously shown to be associated with T‐cell activation after SCI induction in animals 5, 6.
We measured the expression of proinflammatory cytokines in peripheral lymph nodes by quantitative PCR analyses. As expected, cytokine production in sham animals was minimal or absent (Figure 6A–F), while it was detectable in WT SCI mice. However, the pattern of cytokine expression after SCI was clearly different in gilz KO mice compared with WT animals. Interestingly, we observed a significant increased expression of Th1‐type cytokines IFNγ and IL‐12 in gilz KO mice (Figure 6A and B) and a decreased expression of Th2 type cytokines IL‐4 and IL‐10 (Figure 6C and D), compared with WT mice. Moreover, the proinflammatory cytokine IL‐17 was not upregulated in gilz KO peripheral lymph nodes following SCI as in occurred in WT after SCI induction (Figure 6E, P value = 0.07). Finally, no differences in TGFβ expression were found between WT and gilz KO lymph nodes after SCI (Figure 6F). We have also not found differences in weight and cell number of spleens and peripheral lymph nodes isolated from WT and gilz KO mice after SCI (Table 1). Interestingly, contrary to what we observed in lymph nodes, we did not detect an increased production of IFNγ in spinal cord of gilz KO mice upon SCI (Figure 6G). Instead, gilz KO mice upon SCI revealed a significant upregulation of IL‐10 levels in spinal cord (Figure 6I).
Figure 6.
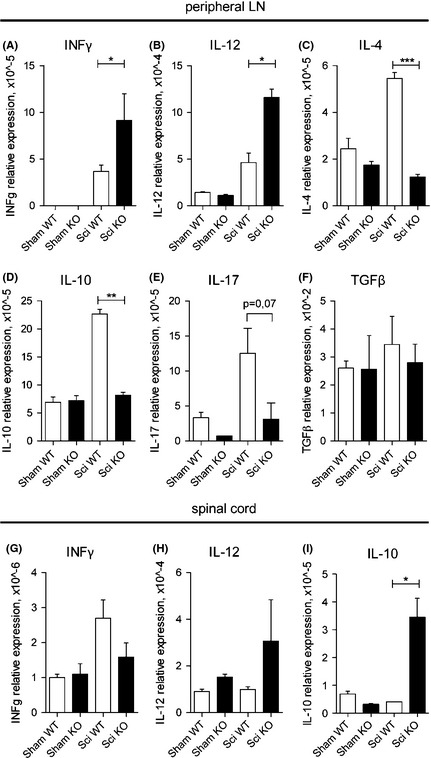
Altered inflammatory cytokines expression in gilz KO peripheral lymph nodes and spinal cord after SCI. (A–F) qPCR analysis in peripheral lymph nodes of IFN γ (A), IL‐12 (B), IL‐4 (C), IL‐10 (D), Il‐17 (E), TGFβ (F), mRNA expression or (G–I) in spinal cord of IFNγ (G), IL‐12 (H), and IL‐10 (I) in mice not subjected to SCI (Sham WT and Sham KO mice) or after SCI (Sci WT and Sci KO). All data are presented relative to the expression of Actb mRNA. Results are shown as means ± SE (error bars); n = 5 mice/group. *P < 0.05; **P < 0.005; ***P < 0.0005, two‐tailed Student's t‐test.
Table 1.
Weight and cell numbers of spleen and peripheral lymph nodes (LN)
| Group | Spleen weight (gr) | Cell count spleen ×106 | Cell count LN ×106 |
|---|---|---|---|
| Sham WT | 88.5 ± 2.1 | 61.2 ± 12.4 | 16.2 ± 8.83 |
| Sham KO | 76.5 ± 27 | 45.6 ± 13.2 | 12.1 ± 1.23 |
| SCI WT | 71.6 ± 14 | 78.5 ± 16.5 | 11.7 ± 4.07 |
| SCI KO | 99.5 ± 30 | 74.8 ± 22.8 | 14.1 ± 7.2 |
These results demonstrate that absence of GILZ leads to the alteration of inflammatory response induced by SCI resulting in a decreased Th17 and Th2 and increased Th1 cytokine production in periphery, and in an increased IL‐10 production in perilesional area of spinal cord associated with neuroprotection.
Conclusion
SCI is a devastating lesion of nervous system that is a frequent cause of motor impairment. SCI causes immediate tissue disruption, with a direct axonal and neuronal injury, inducing the death of a number of neurons. At present, this lesion is irreversible. Moreover, the outcome of SCI depends on the events that follow the initial lesion. Indeed, neurons continue to die for hours after SCI as a result of several mechanisms, including excitotoxicity, vascular abnormalities, and inflammatory response that contribute to the evolution of spinal cord secondary injury.
Despite major progress in pharmacological, surgical, and rehabilitative treatment approaches, SCI still remains a very complex medical and psychological challenge. In the mid‐1960s, the idea of using GC in the treatment of SCI was based on the empirical notion that they would attenuate posttraumatic spinal cord edema. More recently, proposed mechanisms include the inhibition of inflammatory cytokines, preservation of calcium homeostasis, preservation of spinal cord blood flow, and modulation of the inflammatory cells activity 33. The use of GC in the treatment of other CNS inflammatory diseases, such as the Parkinson disease, has been also proposed on the basis of the antiinflammatory properties of GC 34, 35, 36. To date, the only pharmacological compound that has demonstrated “neuroprotective” ability is methylprednisolone that is still administered as the most appropriate therapeutic drug for human SCI 9, 10, 11, 13.
GILZ gene is one of the few genes potently regulated by GR transcriptional activity, and data on human diseases suggest that GILZ mediates a number of GC effects 18, 37, 38. We have previously reported that GILZ modulates the immune response after SCI suggesting a potential role due to therapeutic effects 27. Moreover, GILZ appears to be a marker of response to GC and is basally expressed in central nervous system 25, 26, 39.
In this study, we showed that lack of GILZ determines a reduced damage in spinal cord after SCI, as evidenced by histological analyses of the spinal cord following SCI damage in WT and gilz KO mice. Gilz KO mice present a consistent reduction of leukocytes infiltration into perilesional area of injured spinal cord.
Neuroinflammation plays a central role in the control of damage of injured tissues following primary traumatic damage, and functional outcomes are largely dependent by the inflammatory response of the patient and by the SCI treatment, which includes antiinflammatory drugs such as GC. Inflammatory processes following SCI are controlled not only by innate immunity, such as microglia activation and granulocytes infiltration, but also by adaptive immunity, with the activation and migration of lymphocytes to the damaged area. However, the biological impact of this lymphocyte infiltration remains controversial. Recent studies report that activated microglia might regulate the function and maintenance of activated T cells in the damaged area 40. Moreover, experimental and human studies of brain and spinal cord trauma have revealed the presence of T cells auto‐reactive for cerebral antigens 28. However, whether neuroprotection may be conferred by activated or auto‐reactive T cells is still controversial.
Comparison of expression levels of adhesion molecules in WT and GILZ‐deficient mice revealed strong differences in ICAM‐1 and P‐selectin tissue localization. The lower degree of adhesion molecule expression in this case suggests a reduced leukocyte infiltration in gilz KO mice. This could be due to: (1) a defect in the migration capability of leukocytes normally associated to damage; or (2) modulation of activation of adaptive immune response, or both, resulting in reduced neuroinflammation at the level of spinal cord.
Altogether, in the present study, we found that GILZ deletion in the whole body is associated with spinal postinjury benefit, as measured by alterations in tissue damage and leukocyte infiltration. These results do not apparently fit with previous observations suggesting that GILZ overexpression in T cells also protects from SCI 27. Genetic ablation or activation of a gene in different cell types may provide a complex picture of the temporally and tissue‐specific roles of that gene. Specific GILZ overexpression in T cells 24 h upon SCI mimics the antiinflammatory effects of GC and inhibits early proinflammatory responses after injury 27. In the present article, analysis of SCI in wt and gilz KO mice 7 days postinjury shows a different picture suggesting that genetic ablation of GILZ in all tissues may affect in a different way inflammatory response. It is known that GILZ is involved in the control of activation and differentiation of lymphoid cells and in the activation of immune responses 18, 19, 38, 41. GILZ may have different short and long term effects on cell growth and differentiation or migration of different cell types. For example, it may regulate both innate and adaptive immune responses and in this way contributes to the outcome of SCI. Indeed, we have previously shown that GILZ overexpression favors Th2 over Th1 differentiation 42. Here, we have found altered pattern of cytokines production in peripheral lymph nodes upon SCI induction in gilz KO mice compared with controls. We found an increased production of Th1‐type cytokines, such as IL‐12 and IFNγ, while expression of Th2 cytokines, such as IL‐4 and IL‐10, was decreased in lymph nodes of gilz KO mice, indicating that lack of GILZ promotes the differentiation of T cells to Th1 type in response to damage induced by SCI. This altered cytokine expression profile correlates with reduced damage in spinal cord of gilz KO mice. This is not surprising considering that Th1 immune response might play a neuroprotective role in spinal cord damage by maintaining activated T cells that could provide beneficial effects following SCI 40, 43, 44. The neuroprotective role of Th1 and Th2 type cells following traumatic events in spinal cord is still not well defined 45, 46, 47, 48. To note, it has already been proposed that Th1 cells producing IFNγ controls IL10 production by resident microglia and infiltrating macrophages 49. Here, we found that IL‐10 is upregulated in perilesional spinal cord of gilz KO mice upon SCI compared with controls, suggesting that IL‐10 increased production confers neuroprotective role in spinal cord of gilz KO mice upon SCI. Moreover, we found that IL‐17 is not expressed in gilz KO mice compared with WT after SCI in peripheral lymph nodes. IL‐17 is a proinflammatory cytokine, which is implicated in chronic inflammatory/autoimmune mediated diseases, including neurodegenerative diseases 50, 51, 52. The hypothesis that decreased production of IL‐17 observed in gilz KO mice may contribute to reduction of neuroinflammation is supported by other studies showing that IL‐17 KO mice are protected from inflammatory damage secondary to spinal trauma 53, 54.
Altogether, our results indicate that lack of GILZ results in reduced spinal cord lesions after SCI. This neuroprotective phenotype in GILZ‐deficient mice may be due, at least in part, to the fact that lack of GILZ affects polymorphonuclear leukocytes migration and T‐cells activation, differentiation, and production of inflammatory cytokines. These decreased leukocyte infiltration and unbalanced production of inflammatory cytokines in gilz KO mice are associated with minor signs of spinal cord damage upon SCI induction. Understanding the mechanisms that are implicated in the control of GILZ expression and function is warranted for the development of new and effective therapeutic strategies in the treatment of SCI to reduce tissue damage after the injury.
Disclosure
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by grants from the Italian Ministry of Health (Ricerca Finalizzata, RF‐2009‐1525703) and from Associazione Italiana per la Ricerca sul Cancro to CR (IG‐14291).
The first two authors contributed equally to this work.
References
- 1. Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J 2004;4:451–464. [DOI] [PubMed] [Google Scholar]
- 2. Hawthorne AL, Popovich PG. Emerging concepts in myeloid cell biology after spinal cord injury. Neurotherapeutics 2011;8:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwartz M, Moalem G, Leibowitz‐Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci 1999;22:295–299. [DOI] [PubMed] [Google Scholar]
- 4. Taoka Y, Okajima K, Uchiba M, et al. Role of neutrophils in spinal cord injury in the rat. Neuroscience 1997;79:1177–1182. [DOI] [PubMed] [Google Scholar]
- 5. Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol 2008;209:378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Popovich PG, Stuckman S, Gienapp IE, Whitacre CC. Alterations in immune cell phenotype and function after experimental spinal cord injury. J Neurotrauma 2001;18:957–966. [DOI] [PubMed] [Google Scholar]
- 7. Schnell L, Schneider R, Berman MA, Perry VH, Schwab ME. Lymphocyte recruitment following spinal cord injury in mice is altered by prior viral exposure. Eur J Neurosci 1997;9:1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rossignol S, Schwab M, Schwartz M, Fehlings MG. Spinal cord injury: time to move? J Neurosci 2007;27:11782–11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bracken MB, Shepard MJ, Collins WF Jr, et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1‐year follow‐up data. Results of the second National Acute Spinal Cord Injury Study. J Neurosurg 1992;76:23–31. [DOI] [PubMed] [Google Scholar]
- 10. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal‐cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med 1990;322:1405–1411. [DOI] [PubMed] [Google Scholar]
- 11. Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA 1997;277:1597–1604. [PubMed] [Google Scholar]
- 12. Lee JM, Yan P, Xiao Q, et al. Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury. J Neurosci 2008;28:3141–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsutsumi S, Ueta T, Shiba K, Yamamoto S, Takagishi K. Effects of the Second National Acute Spinal Cord Injury Study of high‐dose methylprednisolone therapy on acute cervical spinal cord injury‐results in spinal injuries center. Spine 2006;31:2992–2996. [DOI] [PubMed] [Google Scholar]
- 14. Hoffman GS. Immunosuppressive therapy for autoimmune diseases. Ann Allergy 1993;70:263–274. [PubMed] [Google Scholar]
- 15. Kofler R. The molecular basis of glucocorticoid‐induced apoptosis of lymphoblastic leukemia cells. Histochem Cell Biol 2000;114:1–7. [DOI] [PubMed] [Google Scholar]
- 16. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids‐new mechanisms for old drugs. N Engl J Med 2005;353:1711–1723. [DOI] [PubMed] [Google Scholar]
- 17. Bruscoli S, Di Virgilio R, Donato V, et al. Genomic and non‐genomic effects of different glucocorticoids on mouse thymocyte apoptosis. Eur J Pharmacol 2006;529:63–70. [DOI] [PubMed] [Google Scholar]
- 18. D'Adamio F, Zollo O, Moraca R, et al. A new dexamethasone‐induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3‐activated cell death. Immunity 1997;7:803–812. [DOI] [PubMed] [Google Scholar]
- 19. Ayroldi E, Riccardi C. Glucocorticoid‐induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J 2009;23:3649–3658. [DOI] [PubMed] [Google Scholar]
- 20. Riccardi C, Bruscoli S, Migliorati G. Molecular mechanisms of immunomodulatory activity of glucocorticoids. Pharmacol Res 2002;45:361–368. [DOI] [PubMed] [Google Scholar]
- 21. Ayroldi E, Migliorati G, Bruscoli S, et al. Modulation of T‐cell activation by the glucocorticoid‐induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood 2001;98:743–753. [DOI] [PubMed] [Google Scholar]
- 22. Bereshchenko O, Coppo M, Bruscoli S, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF‐β signaling. Cell Rep 2014;7:464–475. [DOI] [PubMed] [Google Scholar]
- 23. Bruscoli S, Donato V, Velardi E, et al. Glucocorticoid‐induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti‐myogenic effects of glucocorticoids. J Biol Chem 2010;285:10385–10396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bruscoli S, Velardi E, Di Sante M, et al. Long glucocorticoid‐induced leucine zipper (L‐GILZ) protein interacts with ras protein pathway and contributes to spermatogenesis control. J Biol Chem 2012;287:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ellestad LE, Malkiewicz SA, Guthrie HD, Welch GR, Porter TE. Expression and regulation of glucocorticoid‐induced leucine zipper in the developing anterior pituitary gland. J Mol Endocrinol 2009;42:171–183. [DOI] [PubMed] [Google Scholar]
- 26. Yachi K, Inoue K, Tanaka H, Yoshikawa H, Tohyama M. Localization of glucocorticoid‐induced leucine zipper (GILZ) expressing neurons in the central nervous system and its relationship to the stress response. Brain Res 2007;1159:141–147. [DOI] [PubMed] [Google Scholar]
- 27. Esposito E, Bruscoli S, Mazzon E, et al. Glucocorticoid‐induced leucine zipper (GILZ) over‐expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics 2012;9:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res 1996;45:349–363. [DOI] [PubMed] [Google Scholar]
- 29. Skaper SD, Giusti P, Facci L. Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J 2012;26:3103–3117. [DOI] [PubMed] [Google Scholar]
- 30. Tani K, Ogushi F, Kido H, et al. Chymase is a potent chemoattractant for human monocytes and neutrophils. J Leuk Biol 2000;67:585–589. [DOI] [PubMed] [Google Scholar]
- 31. Farooque M, Isaksson J, Olsson Y. White matter preservation after spinal cord injury in ICAM‐1/P‐selectin‐deficient mice. Acta Neuropathol 2001;102:132–140. [DOI] [PubMed] [Google Scholar]
- 32. Farooque M, Isaksson J, Olsson Y. Improved recovery after spinal cord trauma in ICAM‐1 and P‐selectin knockout mice. NeuroReport 1999;10:131–134. [DOI] [PubMed] [Google Scholar]
- 33. Hulsebosch CE, Hains BC, Waldrep K, Young W. Bridging the gap: from discovery to clinical trials in spinal cord injury. J Neurotrauma 2000;17:1117–1128. [DOI] [PubMed] [Google Scholar]
- 34. Kurkowska‐Jastrzebska I, Litwin T, Joniec I, et al. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson's disease. Int Immunopharmacol 2004;4:1307–1318. [DOI] [PubMed] [Google Scholar]
- 35. Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL‐1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain 2008;131:1880–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato Y, Asoh T, Metoki N, Satoh K. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson's disease. J Neurol Neurosurg Psychiatry 2003;74:574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berrebi D, Bruscoli S, Cohen N, et al. Synthesis of glucocorticoid‐induced leucine zipper (GILZ) by macrophages: an anti‐inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL‐10. Blood 2003;101:729–738. [DOI] [PubMed] [Google Scholar]
- 38. Cannarile L, Zollo O, D'Adamio F, et al. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone‐induced gene. Cell Death Differ 2001;8:201–203. [DOI] [PubMed] [Google Scholar]
- 39. Broccoletti T, Del Giudice E, Cirillo E, et al. Efficacy of very‐low‐dose betamethasone on neurological symptoms in ataxia‐telangiectasia. Eur J Neurol 2011;18:564–570. [DOI] [PubMed] [Google Scholar]
- 40. Hauben E, Butovsky O, Nevo U, et al. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci 2000;20:6421–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cannarile L, Fallarino F, Agostini M, et al. Increased GILZ expression in transgenic mice up‐regulates Th‐2 lymphokines. Blood 2006;107:1039–1047. [DOI] [PubMed] [Google Scholar]
- 42. Cannarile L, Cuzzocrea S, Santucci L, et al. Glucocorticoid‐induced leucine zipper is protective in Th1‐mediated models of colitis. Gastroenterology 2009;136:530–541. [DOI] [PubMed] [Google Scholar]
- 43. Fee D, Crumbaugh A, Jacques T, et al. Activated/effector CD4 + T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol 2003;136:54–66. [DOI] [PubMed] [Google Scholar]
- 44. Kipnis J, Mizrahi T, Yoles E, Ben‐Nun A, Schwartz M. Myelin specific Th1 cells are necessary for post‐traumatic protective autoimmunity. J Neuroimmunol 2002;130:78–85. [DOI] [PubMed] [Google Scholar]
- 45. Jones TB. Lymphocytes and autoimmunity after spinal cord injury. Exp Neurol 2014;258C:78–90. [DOI] [PubMed] [Google Scholar]
- 46. Sato A, Ohtaki H, Tsumuraya T, et al. Interleukin‐1 participates in the classical and alternative activation of microglia/macrophages after spinal cord injury. J Neuroinflammation 2012;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davies AL, Hayes KC, Dekaban GA. Clinical correlates of elevated serum concentrations of cytokines and autoantibodies in patients with spinal cord injury. Arch Phys Med Rehabil 2007;88:1384–1393. [DOI] [PubMed] [Google Scholar]
- 48. Hayes KC, Hull TC, Delaney GA, et al. Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J Neurotrauma 2002;19:753–761. [DOI] [PubMed] [Google Scholar]
- 49. Ishii H, Tanabe S, Ueno M, et al. ifn‐gamma‐dependent secretion of IL‐10 from Th1 cells and microglia/macrophages contributes to functional recovery after spinal cord injury. Cell Death Dis 2013;4:e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kostic M, Dzopalic T, Zivanovic S, et al. IL‐17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol 2014;79:181–186. [DOI] [PubMed] [Google Scholar]
- 51. Lanzilli G, Cottarelli A, Nicotera G, Guida S, Ravagnan G, Fuggetta MP. Anti‐inflammatory effect of resveratrol and polydatin by in vitro IL‐17 modulation. Inflammation 2012;35:240–248. [DOI] [PubMed] [Google Scholar]
- 52. Zhang X, Li QY, Xiao BG. Anti‐inflammatory effect of erythropoietin therapy on experimental autoimmune encephalomyelitis. Int J Neurosci 2012;122:255–262. [DOI] [PubMed] [Google Scholar]
- 53. Hill F, Kim CF, Gorrie CA, Moalem‐Taylor G. Interleukin‐17 deficiency improves locomotor recovery and tissue sparing after spinal cord contusion injury in mice. Neuroscience Lett 2011;487:363–367. [DOI] [PubMed] [Google Scholar]
- 54. Kim CF, Moalem‐Taylor G. Interleukin‐17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain 2011;12:370–383. [DOI] [PubMed] [Google Scholar]