

Summary
Aims
The purpose of this study was to evaluate the energy metabolism and mitochondrial function in skeletal muscle from patients with Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) or chronic progressive external ophthalmoplegia (CPEO) using phosphorus magnetic resonance spectroscopy (31P‐MRS), to determine whether abnormally increasing cytochrome c oxidase (COX), as detected in muscle biopsy, could be a cause for MELAS.
Methods
31P‐MRS was performed on the quadriceps femoris muscle of 12 healthy volunteers and 11 patients diagnosed as MELAS or CPEO by muscle biopsy and genetic analysis. All subjects experienced a state of rest, 5‐min exercise, and 5‐min recovery protocol in a supine position.
Results
Compared to CPEO, MELAS patients typically exhibited COX‐positive ragged‐red fibers (RRFs) as well as strongly SDH‐positive blood vessels (SSVs). However, based on 31P‐MRS results, MELAS showed a higher inorganic phosphate (Pi)/phosphocreatine (PCr) ratio and lower ATP/PCr ratio during exercise and delayed Pi/PCr and ATP/PCr recovery to normal.
Conclusions
This study suggests that high COX expression contributes to severe skeletal energy failure by 31P‐MRS spectroscopy in MELAS.
Keywords: 31P‐MRS; Cytochrome c oxidase; Energy metabolism; Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes; Muscle biopsy
Introduction
Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) is the most common subtype of mitochondrial disorders associated with severe mitochondrial dysfunction and energy abnormalities. The typical pathological characteristics of MELAS are massive mitochondrial proliferation and defects of respiratory chain enzymes. Until now, the pathogenesis of MELAS has been unclear. Previous studies proposed mtDNA mutation‐induced mitochondrial neuropathy as the underlying cause, but this theory cannot fully explain the clinical phenotype and the molecular defect relationship.
An alternative explanation suggested cytochrome c oxidase (COX), the fourth complex of respiratory chain, as a specific pathology seen in MELAS 1. This theory is based on two aspects: (1) an increased COX activity compared to other mitochondrial encephalopathy such as chronic progressive external ophthalmoplegia (CPEO), in which COX activity was significantly absent, and (2) an observed hypocitrullinemia in MELAS patients. Citrulline is a substrate donor for nitric oxide (NO) generation. It has been proposed that excessive COX activity in RRFs could lead to dysfunction of NO catabolism and subsequent low plasma citrulline levels 2. However, this interesting hypothesis lacks direct and efficient evidence to assess muscle weakness and exercise intolerance, that is to say, the skeletal muscle energy metabolism itself.
31P‐MRS is a sensitive and specific method to assess skeletal energy metabolism in vivo. 31P‐MRS allows for noninvasive quantification of high‐energy phosphate compounds. A typical 31P‐MRS from muscles displays seven major peaks representing inorganic phosphate (Pi), phosphocreatine (PCr), phosphomonoester (PME), phosphodiester (PDE), and three peaks of adenosine triphosphate (α‐ATP, β‐ATP, γ‐ATP). Several 31P‐MRS studies have attempted to investigate abnormal skeletal muscle energy metabolism in patients with mitochondrial encephalomyopathy 3, 4. However, few studies have compared the energy metabolism between MELAS and CPEO patients.
This study attempts to assess energy metabolism and mitochondrial function by 3.0T 31P‐MRS during a rest‐exercise‐recovery protocol in MELAS and CPEO patients. These results allow this study to: (1) address the relationship among energy metabolism, respiratory defect (revealed by COX), and mitochondrial proliferation (revealed by RRFs and SSVs), and (2) investigate whether the abnormal COX activity could result in impaired energy metabolism in MELAS patients.
Materials and methods
Patient Selection
From January 2010 to December 2012, eleven patients with the mitochondrial encephalomyopathy were recruited from Drum Tower Hospital of Nanjing University Medical School, Jiangsu Province Hospital of Nanjing Medical University and Nanjing General Hospital of Nanjing Military Command. Twelve healthy control volunteers were recruited and matched in age, but not in gender. The studies were approved by the local ethics committee, and all subjects gave written informed consent.
Mitochondrial Disease Criteria Score
Mitochondrial disease criteria (MDC) score is an improved consensus scoring system as established in Europe in 2006 5. The MDC score is based on an evaluation that includes clinical signs and symptoms (I) (muscle presentation IA, central nervous system (CNS) abnormalities IB, multisystem involvement IC), metabolic/imaging studies (II), and muscle histology (III) (Table S1). Patients in the current study had multiple laboratory examinations including electrocardiography (EKG), electromyography (EMG), electroencephalography (EEG), serum lactate/alanine or cerebrospinal fluid (CSF), lactate/pyruvate ratio and magnetic resonance imaging (MRI) scans, and additional magnetic resonance spectroscopy (MRS). The data from patients' clinical, biochemical, and histological examinations were reviewed and converted into MDC scores, then were compared between MELAS and CPEO groups.
Pathological Study (Muscle Biopsy)
The quadriceps femoris muscle (vastus lateralis) was chosen as the most suitable muscle for biopsy because: (1) a relatively large sample can be acquired without damaging major nerves or blood vessels, and (2) most mitochondrial encephalomyopathy patients have proximal muscle fatigue. The surgical muscle biopsy was performed at the Pathology Department in Drum Tower Hospital. Serial cryostat sections of fresh frozen tissue were stained with routine histochemical reactions. These were generally modified stains including hematoxylin–eosin (HE), adenosine triphosphatase (ATPase pH 9.4, pH 4.6), modified Gomori trichrome (MGT), periodic acid‐Schiff reaction (PAS), oil red O, nicotinamide adenine dinucleotide‐tetrazolium reductase (NADH‐TR), COX, and succinate dehydrogenase (SDH). For MELAS, COX oxidative enzymatic stains were essential.
Molecular Analysis
All patients received gene confirmation. Total mitochondrial DNA was extracted (eight skeletal muscle samples, three urinary sediment samples) using commercial DNA purification kits (Promega Corporation., Madison, WI, USA). Genotype was detected by polymerase chain reaction (PCR) amplification and sequencing to determine the presence or absence of mtDNA mutation as previously described 6. The gene mutation in MELAS syndrome is mtDNA 3243 A>G, and in CPEO syndrome, it is a 4977 bp large‐scale mtDNA deletion.
Maximum Voluntary Isometric Contraction
To apply an individually adjusted exercise load, the maximum voluntary isometric contraction (MVC) of the quadriceps femoris was measured prior to MRS as previously described 7. A sandbag load was fastened to the right ankle. Each subject was instructed to lie in a supine position and encouraged to raise the legs to maximize the effect of the ankle load. They were asked not to move the knee and maintain it straight in order to perform maximal quadriceps femoris contraction. Each contraction was maintained for 3 seconds, and the process was repeated after a resting period of 30 seconds. MVC was quantified in three repeated exercises and defined as the average of the three peak measurements 8.
31P‐MRS Acquisition
Measurements were performed on a 3.0T MRI scanner (Achieva 3.0T TX; Philips Medical Systems, Eindhoven, the Netherlands) 9. The subjects were placed in a supine position in the magnet bore. They were in a state of rest, exercise, and recovery protocol. For exercise, a weight of 25% MVC was loaded to the right ankle and subjects were asked to kick the ankle to contract the quadriceps muscle as mentioned above. Spectra measurements of Pi, PCr, and α‐, β‐, γ‐ATP were acquired from a 50‐mm‐diameter surface coil on the surface of the quadriceps femoris. 31P‐MRS parameters included 31P‐SV, TR = 5000 mseconds, TE = 0.1 mseconds, volume of interest (VOI) = 30 × 30 × 120 mm, NSA 8, Dyn. Scan time for each spectrum was 50 seconds. Spectra were analyzed using a commercially available postprocessing workstation (Extended Workspace [EWS], Philips Medical Systems). Each MR investigation lasted nearly 14 min and comprised three phases: Phase 1: 100 seconds of resting measurements (baseline) of the unloaded resting muscle; Phase 2: 5 min of dynamic loaded with a 25% MVC in resistance; and Phase 3: 5 min of recovery measurements. PME and PDE data were not acquired as they were regarded to play no role in skeletal muscle energy metabolism 10. The ratios of Pi/PCr and ATP/PCr were calculated at rest, 5‐min exercise, and 5‐min recovery time phases. Pi/PCr ratio was considered a sensitive indicator of efficiently generating and maintaining high‐energy phosphate compounds 11. This ratio up to the control level indicated a defect in production and utilization of energy. ATP/PCr ratio was considered a direct evidence of energy generation and maintenance. The ATP value was equal to the sum of the α‐ATP, β‐ATP, and γ‐ATP, three different subtypes of ATP.
Statistical Analyses
The data were expressed as the mean ± SEM, and the statistical analyses were performed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Comparison of quantitative parameters between the two groups was evaluated using Student's t‐test, and multiple group comparisons were analyzed by one‐way analysis of variance (ANOVA), followed by least significant difference (LSD) or Student–Newman–Keuls (SNK) comparisons. P‐value below 0.05 was considered significant.
Results
Clinical Information and MDC Scores
According to muscle biopsy and gene detection, six patients were diagnosed as MELAS and five were CPEO. The clinical information and MDC score evaluations of these patients are shown in Table 1. Common symptoms were proximal muscle weakness, exercise intolerance, and muscular atrophy, as well as other secondary manifestations including numbness, bulbar palsy, intention tremor, depression, ataxia, and cardiac conduction defects cardiomyopathy. The average age was 24.91 ± 6.05 years. One patient died during this study. The control group consisted of 12 matched volunteers (25.00 ± 2.45 years). There were no significant differences (P > 0.05) between the total scores of the MELAS group (9.17 ± 0.61) and the CPEO group (8.80 ± 0.86), indicating the two groups had similar disease severity.
Table 1.
Clinical characteristics of MELAS and CPEO patient groups
| Patients (N = 11) | Age of onset/sex | Clinical manifestation | MDC score | Diagnostic phenotype | mtDNA mutation |
|---|---|---|---|---|---|
| 1 | 29/F | Short stature, mildly mentally retarded, headache, cognitive regression, developmental delay, dysphagia | 8 | MELAS | mt.3243 A>G |
| 2 | 26/M | Moderate mental retardation, developmental delay, migraine, generalized seizure, blurred vision, family history (diabetes mellitus) | 10 | MELAS | mt.3243 A>G |
| 3 | 20/M | Shortness of breath after mild exercise, mild proximal limb weakness, predominantly in the legs, migraine, family history (diabetes mellitus) | 7 | MELAS | mt.3243 A>G |
| 4 | 19/F | Muscle weakness, abnormal EMG, seizure, myoclonus, blurred vision, family history (diabetes mellitus) | 11 | MELAS | mt.3243 A>G |
| 5 | 30/M | Facies myopathy, migraine, blurred vision, exercise intolerance, and myalgia during walking, myoclonus, blurred speech | 10 | MELAS | mt.3243 A>G |
| 6 | 28/M | Short stature, slight headache, abnormal behavior, mentally retarded, developmental delay | 9 | MELAS | mt.3243 A>G |
| 7 | 25/F | Progressive heart complaint, cardiomyopathy, ophthalmoplegia, ventricular conduction abnormalities | 6 | CPEO | mtDNA depletion |
| 8 | 15/M | Progressive ptosis, blurred vision for 2 years, 1 year ago slurred speech, 2 months ago involuntary movement in hands, gait difficulties, slight intension tremor, limited eye movement, abnormal EMG, died suddenly at 17 years old | 9 | CPEO/KSS | mtDNA depletion |
| 9 | 30/F | Progressive ptosis for 10 years, diplopia for 5 years, limited eye motion for 4 years, proximal weakness, abnormal EMG, family history (ophthalmoplegia) | 11 | CPEO | mtDNA depletion |
| 10 | 18/M | Progressive ptosis for 3 years, ophthalmoplegia, limited eye motion, vision problem, muscle weakness, exercise intolerance, abnormal EMG | 8 | CPEO | mtDNA depletion |
| 11 | 34/F | Progressive ptosis for 8 years, ophthalmoplegia, limited eye movement, abnormal EMG, hearing problem, blurred vision | 10 | CPEO | mtDNA depletion |
F, female; M, male; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes; CPEO, chronic progressive external ophthalmoplegia; KSS, Kearns–Sayre syndrome.
Muscle Histopathology in MELAS and CPEO
The morphological anomalies of muscle biopsy associated with mtDNA alteration brought a unique opportunity to investigate the relationship between mitochondrial anomalies and respiratory activity. On the one hand, the abnormal mitochondrial changes included the appearance of ragged‐red fibers (RRFs) on muscle biopsies in both MELAS and CPEO. The reason for pathological RRFs was an abundance of proliferated mitochondrial. As illustrated in Figure 1A, HE staining showed variations in myofiber size and several fibers with basophilic sarcoplasmic masses indicative of RRFs. Gomori staining clearly demonstrated RRFs which were irregular in shape and intensely red in color, whereas normal myofibrils are green. Similarly, reflective of compensatory mitochondrial proliferation, specifically in MELAS, was that the same fibers appear “ragged‐blue” with the SDH stain and several SSVs, which were not observed in CPEO.
Figure 1.
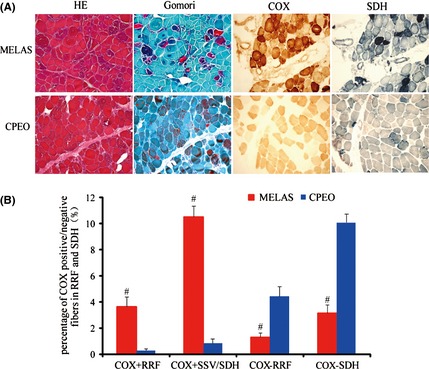
(A). Histopathological features of mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) (patient #1) and chronic progressive external ophthalmoplegia (CPEO) (patient #7) syndrome. (a) HE staining showed scattered vacuolated muscle fibers containing many small basophilic inclusions (RRFs) in both MELAS and CPEO. (b) Gomori staining demonstrated clearly RRFs as well. (c) In MELAS, the SDH staining exhibited intense activity in the subsarcolemmal zones corresponding to RRFs and blood vessel walls (SSVs). In CPEO, there was a complete absence of SSVs and less “ragged‐blue” fibers with the SDH stain. (d) Cytochrome c oxidase (COX) staining exhibited increased staining in MELAS, but significantly decreased staining in CPEO. Frozen sections (10 μm thickness). Scale bar = 50 μm. (B). Percentage of COX+ and COX‐ fibers in RRF and SDH in biopsy of patients with MELAS and CPEO. In MELAS, RRFs and SSVs were typically COX positive, while in CPEO, RRFs were strikingly COX negative. # P < 0.05 compared to CPEO group.
On the other hand, oxidative enzymes were specific markers of respiratory chain complex. COX, the complex IV of the respiratory chain, was only present in mitochondria 12. Cytochrome c oxidase activity, therefore, was regarded as an important indicator to demonstrate respiratory chain function. In CPEO with large mtDNA depletion, the damaged RRFs were often deficient for COX histochemical staining, indicative of severely impaired respiratory chain. However, in MELAS associated with mtDNA 3243 A>G mutation, the RRFs and SSVs were typically COX positive and the percentage of COX+ fibers was significantly higher than that in CPEO (P < 0.05) as shown in Figure 1B. These observations indicate that MELAS patients may have more residual respiratory chain activity relative to CPEO patients, although both MELAS and CPEO had similar mitochondrial proliferation status.
31P‐MRS of Quadriceps Femoris Muscle
Before MRS studies, maximal voluntary contraction (MVC) load was estimated as previously described in the healthy volunteers and patients groups. To maintain consistent exercise intensity, 25% MVC was applied to each person. As illustrated in Figure 2A, the MVC of the plantar flexion exercise in patients with MELAS and CPEO was significantly lower than that in healthy volunteers, while there was no significant difference between the MELAS and CPEO groups.
Figure 2.
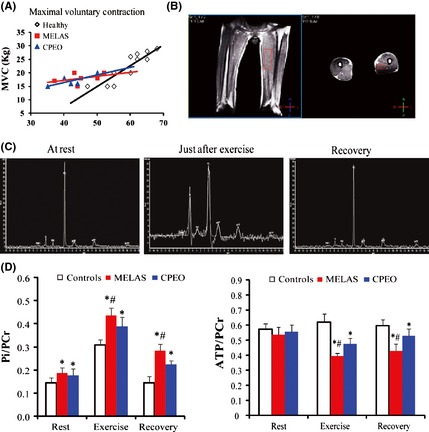
(A). The mean MVC values in the three groups. MVC values in mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) and chronic progressive external ophthalmoplegia (CPEO) were significantly higher than those in control groups. (B). The location of quadriceps on 3.0T MRI. (C). 31P‐MRS obtained from a healthy control at rest, just after 25% MVC exercise and at end recovery. Just after exercise, Pi significantly increased and PCr decreased. At end recovery, PCr returned to its resting phase level. ATP peaks remained constant throughout the exercise protocol. (D). Scheme of the variations of Pi/PCr and ATP/PCr during a rest‐exercise‐recovery protocol. MELAS group had a higher Pi/PCr at exercise and lower ATP/PCr at recovery compared to CPEO and control groups. *P < 0.05 compared to control group, # P < 0.05 compared to CPEO group.
The location of quadriceps on 3.0T MRI is shown in Figure 2B. The quadriceps muscle was chosen because it can produce prominent high‐energy phosphate and has high PCr and ATP peaks. A serial 31P‐MRS procedure at rest, exercise, and recovery phases from a healthy volunteer is shown in Figure 2C. Just after exercise, the Pi peak increased and the PCr peak decreased compared to rest state. During the recovery phase, the Pi gradually decreased and the PCr increased, both of them returned to the normal value of resting phase.
Similar 31P‐MRS phase information from MELAS and CPEO patients is shown in Figure 2D, including:
At rest, compared to healthy controls (0.144 ± 0.021), both MELAS (0.185 ± 0.023) and CPEO (0.176 ± 0.027) patients showed an elevated Pi/PCr ratio (P < 0.05) due to lower oxidative capacity, while there was no significant difference (P > 0.05) in Pi/PCr ratio between the MELAS and CPEO groups.
Just after exercise (25% MVC), all subjects displayed an elevated Pi/PCr ratio. MELAS patients (0.435 ± 0.033) acquired an even higher Pi/PCr ratio, significantly different from controls (0.307 ± 0.023) and CPEO patients (0.388 ± 0.039) (P < 0.05), including MELAS may have more increased nonoxidative glycolytic activity and more impairment in mitochondrial ATP production than CPEO.
During recovery, 31P‐MRS is regarded as the most sensitive and specific method to assess skeletal muscle mitochondrial rate of ATP production 13. In this study, Pi/PCr ratio began to decrease in all subjects from the end of exercise to recovery. This ratio dropped to nearly the resting phase value in healthy controls (0.144 ± 0.026), while it returned slowly in both MELAS (0.283 ± 0.029) and CPEO (0.224 ± 0.016) patients. Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes patients showed the slowest decline (P < 0.05), suggestive of a more prolonged rate of Pi/PCr recovery than CPEO. All these observations indicate a more impaired high‐energy phosphate transfer in proximal muscles and an even worse mitochondrial function in MELAS.
In healthy controls, ATP/PCr ratio had no significant changes during the rest‐exercise‐recovery protocol (P > 0.05), because the ATP could be a continuous resynthesis by mitochondrial aerobic metabolism. However, for both MELAS and CPEO patients, the ATP/PCr ratio declined rapidly after 25% MVC exercise and recovered slowly compared to the control group (P < 0.05). In MELAS, this ratio decreased more dramatically than CPEO during exercise (P < 0.05) and did not return to mean value of resting phase at 5‐min recovery, indicative of a worse energy metabolism and less supplement of ATP.
Discussion
A characteristic of MELAS point mutation (mtDNA 3243 A>G) is the occurrence of mitochondrial proliferation in fibers with increased COX activity. The presence of massive mitochondrial proliferation (revealed by RRFs and SSVs) may allow the total COX activity to surpass that seen in unaffected individual fibers. It differs from the CPEO with large deletion mutation (4977 bp mtDNA deletion) where mitochondrial proliferation is commonly seen in COX‐negative fibers 14. Therefore, analysis of the two different genetic subtypes of mitochondrial encephalopathy allowed investigation of the influence of abnormal COX activity in MELAS pathogenesis.
In general, COX histochemistry has proven extremely useful for the diagnosis of mitochondrial diseases 15. Previous studies showed that COX staining may be decreased, normal, or increased in ragged‐red fibers from MELAS patients; however, in CPEO patients, COX activity was significantly absent. This study indicated high expression of COX in six MELAS patients. So compared to CPEO, COX activity was relatively increased, which indicated preserved respiratory chain function in MELAS.
There is a lot of difference between MELAS and CPEO about energy metabolism, because they had different mutation types (generally 3243A>G point mutation in MELAS, mtDNA deletion in CPEO). In vivo 31P‐MRS has been proposed as a potential tool to evaluate high‐energy phosphate compounds, and it has been successfully applied to study mitochondrial dysfunction. It is more sensitive for assessing mitochondrial function than clinical manifestations 16. Until now, there has been very little research utilizing 31P‐MRS to explore the energy status between the MELAS and CPEO. This study found that MELAS patients had a higher Pi/PCr ratio and a lower ATP/PCr ratio compared to CPEO, suggesting less direct energy source, delayed energy recovery, and worse energy metabolism 16, 17. These observations were opposed to more resident COX expression in MELAS that represented less respiratory defect. In other words, when COX activity increases beyond normal levels, it may become harmful to the MELAS patient. Therefore, at least in patients evaluated in the current study, abnormally increasing COX expression appears to be an important cause in the pathogenesis of MELAS.
The inherent contradiction in MELAS between the presence of residual enzyme activity (relative to CPEO) and its more severe clinical phenotype, which also called “MELAS paradox,” has been reported previously 2. 31P‐MRS in the current study demonstrates that MELAS patients with resident COX activity display a worse energy status compared with CPEO, which further proves the “MELAS paradox” phenomenon. The mechanisms, however, are not clear. An explanation for this is that abnormally increasing COX may lead to dysfunction of NO catabolism. NO plays an integral role in controlling smooth muscle tone and mediating vasodilation and cerebral perfusion. In MELAS, excessive COX binding to NO might cause a relative shortage of circulating NO, which induces endothelial dysfunction and vasodilation retardation, while in CPEO with deficient COX activity, vasodilation will occur normally. Therefore, increased residual COX expression may link to severe skeletal energy failure in MELAS disorders.
Currently, because the clinical manifestations are variable and nonspecific, definite diagnosis of mitochondrial encephalomyopathy still depends on muscle biopsy or gene detection 18. However, genetic classifications are inevitably incomplete because the responsible mutation is identified in only 20–25% of pediatric patients using routine diagnostic tests 19. 31P‐MRS data can be more helpful to elucidate the complex relationship between genotype and phenotype 20. In addition, 31P‐MRS is capable of providing quantitative markers to assess the metabolic status of tissues and monitor progression of the disease or treatment response in vivo 21. For example, 31P‐MRS has proved particularly useful in the therapeutic follow‐up of coenzyme Q treatment of mitochondrial disorders 22. As 31P‐MRS is entirely noninvasive, it is apparently superior to muscle biopsy or gene detection 18, 23.
Taken together, findings of this strongly suggest that: (1) MELAS exhibited increasing COX expression that seemed to have a preservative respiratory function, but had higher Pi/PCr during exercise and delayed ATP/PCr recovery during recovery compared to CPEO, indicating even worse energy metabolism and mitochondrial dysfunction, (2) this conflict may be partially explained by excessive COX, suggesting that COX might lose the normal function and involved in MELAS pathology, and (3) noninvasive 31P‐MRS is a promising tool for detecting energy metabolism inefficiency and mitochondrial function impairment in skeletal muscle of mitochondrial encephalomyopathy.
Disclosures
None.
Conflict of Interest
We declare no conflict of interest.
Supporting information
Table S1. Mitochondrial disease criteria (MDC, simplified version for beside use)*, 2006.
Acknowledgments
We thank Brad Peterson for revising the paper. This work was supported by the National Nature Science Foundation of China (81300977, 81300925, 81230026,81171085, 81200876) and the bureau of health (LJ201101) of Jiangsu Province of China.
The first two authors contributed equally to this work.
References
- 1. Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci 2008;1142:133–158. [DOI] [PubMed] [Google Scholar]
- 2. Naini A, Kaufmann P, shanske S, et al. Hypocitrullinemia in patients with MELAS: An insight into the “MELAS paradox”. J Neurol Sci 2005;229–230:187–193. [DOI] [PubMed] [Google Scholar]
- 3. Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion 2008;8:396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsujikawa T, Yoneda M, Shimizu Y, et al. Pathophysio‐logic evaluation of MELAS strokes by serially quantified MRS and CASL perfusion images. Brain Dev 2010;32:143–149. [DOI] [PubMed] [Google Scholar]
- 5. Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006;67:1823–1826. [DOI] [PubMed] [Google Scholar]
- 6. Niu FN, Chang LL, Meng FQ, et al. Evaluation of a mitochondrial disease criteria scoring system on mitochondrial encephalomyopathy in Chinese patients. Int J Neurosci 2013;123:93–98. [DOI] [PubMed] [Google Scholar]
- 7. Layec G, Bringard A, Vilmen C, et al. Accurate work‐rate measurements during in vivo MRS studies of exercising human quadriceps. MAGMA 2008;21:227–235. [DOI] [PubMed] [Google Scholar]
- 8. Layec G, Malucelli E, Le Fur Y, et al. Effects of exercise‐induced intracellular acidosis on the phosphocreatine recovery kinetics: A (31) P MRS study in three muscle groups in humans. NMR Biomed 2013;26:1403–1411. [DOI] [PubMed] [Google Scholar]
- 9. Zhang B, Li M, Sun ZZ, et al. Evaluation of functional MRI markers in mild cognitive impairment. J Clin Neurosci 2009;16:635–641. [DOI] [PubMed] [Google Scholar]
- 10. Wu FY, Tu HJ, Qin B, et al. Value of dynamic (31)P magnetic resonance spectroscopy technique in in vivo assessment of the skeletal muscle mitochondrial function in type 2 diabetes. Chin Med J (Engl) 2012;125:281–286. [PubMed] [Google Scholar]
- 11. Park JH, Niermann KJ, Ryder NM, et al. Muscle abnormalities in juvenile dermatomyositis patients: P‐31 magnetic resonance spectroscopy studies. Arthritis Rheum 2000;43:2359–2367. [DOI] [PubMed] [Google Scholar]
- 12. Sarnat HB, Marin‐Garcia J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci 2005;32:152–166. [DOI] [PubMed] [Google Scholar]
- 13. Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol 2007;83:84–92. [DOI] [PubMed] [Google Scholar]
- 14. Aure K, Fayet G, Leroy JP, et al. Apoptosis in mitochondrial myopathies is linked to mitochondrial proliferation. Brain 2006;129:1249–1259. [DOI] [PubMed] [Google Scholar]
- 15. DiMauro S, Tanji K, Schon EA. The many clinical faces of cytochrome c oxidase deficiency. Adv Exp Med Biol 2012;748:341–357. [DOI] [PubMed] [Google Scholar]
- 16. van Elderen SG, Doornbos J, van Essen EH, et al. Phosphorus‐31 magnetic resonance spectroscopy of skeletal muscle in maternally inherited diabetes and deafness A3243G mitochondrial mutation carriers. J Magn Reson Imaging 2009;29:127–131. [DOI] [PubMed] [Google Scholar]
- 17. El‐Hattab AW, Emrick LT, Craigen WJ, et al. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab 2012;107:247–252. [DOI] [PubMed] [Google Scholar]
- 18. Moller HE, Kurlemann G, Putzler M, et al. Magnetic resonance spectroscopy in patients with MELAS. J Neurol Sci 2005;229–230:131–139. [DOI] [PubMed] [Google Scholar]
- 19. Kang HC, Lee YM, Kim HD. Mitochondrial disease and epilepsy. Brain Dev 2013;35:757–761. [DOI] [PubMed] [Google Scholar]
- 20. Ito H, Mori K, Kagami S. Neuroimaging of stroke‐like episodes in MELAS. Brain Dev 2011;33:283–288. [DOI] [PubMed] [Google Scholar]
- 21. Wu JS, Buettner C, Smithline H, et al. Evaluation of skeletal muscle during calf exercise by 31‐phosphorus magnetic resonance spectroscopy in patients on statin medications. Muscle Nerve 2011;43:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bendahan D, Mattei JP, Guis S, et al. Non‐invasive investigation of muscle function using 31P magnetic resonance spectroscopy and 1H MR imaging. Rev Neurol (Paris) 2006;162:467–484. [DOI] [PubMed] [Google Scholar]
- 23. Lanza IR, Bhagra S, Nair KS, et al. Measurement of human skeletal muscle oxidative capacity by 31P‐MR spectroscopy: A cross‐validation with in vitro measurements. J Magn Reson Imaging 2011;34:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mitochondrial disease criteria (MDC, simplified version for beside use)*, 2006.