

Summary
Aims
Fibrillar aggregates of β‐amyloid protein (Aβ) are the main constituent of senile plaques and considered to be one of the causative events in the pathogenesis of Alzheimer's disease (AD). Compounds that could inhibit Aβ fibrils formation, disaggregate preformed Aβ fibrils as well as reduce their associated neurotoxicity might have therapeutic values for treating AD. In this study, the inhibitory effects of bis (heptyl)‐cognitin (B7C), a multifunctional dimer derived from tacrine, on aggregation and neurotoxicity of Aβ1‐40 were evaluated both in vitro and in vivo.
Methods
Thioflavin T fluorescence assay was carried out to evaluate Aβ aggregation, MTT and Hoechst‐staining assays were performed to investigate Aβ‐associated neurotoxicity. Fluorescent probe DCFH‐DA was used to estimate the accumulation of intracellular reactive oxygen stress (ROS). Morris water maze was applied to determine learning and memory deficits induced by intracerebroventricular infusion of Aβ in rats.
Results
B7C (0.1–10 μM), but not tacrine, effectively inhibited Aβ fibrils formation and disaggregated preformed Aβ fibrils following co‐incubation of B7C and Aβ monomers or preformed fibrils, respectively. In addition, B7C markedly reduced Aβ fibrils‐associated neurotoxicity in SH‐SY5Y cell line, as evidenced by the increase in cell survival, the decrease in Hoechst‐stained nuclei and in intracellular ROS. Most encouragingly, B7C (0.1 and 0.2 mg/kg), 10 times more potently than tacrine (1 and 2 mg/kg), inhibited memory impairments after intracerebroventricular infusion of Aβ in rats, as evidenced by the decrease in escape latency and the increase in the spatial bias in Morris water maze test along with upregulation of choline acetyltransferase activity and downregulation of acetylcholinesterase activity.
Conclusion
These findings provide not only novel molecular insight into the potential application of B7C in treating AD, but also an effective approach for screening anti‐AD agents.
Keywords: Alzheimer's disease, Aβ fibrils, Bis (heptyl)‐cognitin, Morris water maze, Reactive oxygen stress, Thioflavin T
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that predominantly affects the elderly and is associated with impaired cognition and memory 1. By far, no effective cure is available for treating AD. Currently used anti‐AD drugs, including acetylcholinesterase (AChE) inhibitors (donepezil, rivastigmine and galantamine) and N‐methyl‐D‐aspartate (NMDA) receptor antagonist (memantine), offer only symptomatic and limited benefits without modifying disease progression. Thus, there is an urgent need to develop more effective therapeutic approaches that could slowdown or halt the pathological processes of AD.
Pathologically, AD is characterized largely by insoluble neurofibrillary tangles and senile plaques composed of fibrillar β‐amyloid (Aβ) 2. Aβ peptide, derived by proteolytic cleavage of β‐amyloid precursor protein (APP), has a high propensity to rapidly aggregate into highly toxic oligomers and fibrils 3. Considerable evidence indicates that the abnormal aggregation of Aβ is one of the causative factors in AD pathology and may accelerate the disease progress 4, 5, 6. Several possible mechanisms have been suggested to account for the neurotoxicity of oligomers and fibrils, for instance, induction of oxidative stress 7, direct or indirect trigger of apoptosis 8, 9, 10, unbalance in Ca2+ homeostasis 11, and reduction in acetylcholine concentration by activating acetylcholinesterase 12 or inhibiting choline acetyltransferase (ChAT) activities 13. Therefore, an approach aimed at inhibiting Aβ fibrils formation, disaggregating preformed fibrils as well as blocking Aβ fibrils‐associated neurotoxicity may represent an attractive therapeutic strategy with disease‐modifying potential for AD treatment.
Bis (heptyl)‐cognitin (B7C, Figure 1) is a multifunctional dimer modified from tacrine by us and has been identified as a promising anti‐AD agent on the basis of its inhibitory properties on AChE (IC50, 1.6 nM, 149 times more potent than tacrine) 14, neuronal nitric oxide synthase (nNOS, IC50, 2.9 μM) 15 and NMDA receptor (IC50, 0.19 μM) 16. In addition, B7C has been demonstrated to protect neurons in several in vivo models associated with AD, for instance, in middle cerebral artery occlusion‐ and scopolamine‐induced brain damage 17, 18, suggesting that B7C may cross the blood–brain barrier and have the potential to be developed as a drug for central nervous system disorders. In this study, we extended our efforts in evaluating the inhibitory effects of B7C on Aβ fibrillization and Aβ fibrils‐associated neurotoxicity in vitro and in vivo. It was found that B7C effectively inhibited Aβ fibrils formation and disaggregated preformed Aβ fibrils, blocked Aβ‐induced neuronal apoptosis and intracellular accumulation of reactive oxygen stress (ROS) in SH‐SY5Y cells. Most importantly, B7C potently reversed cognition and memory impairments in rats insulted by Aβ.
Figure 1.
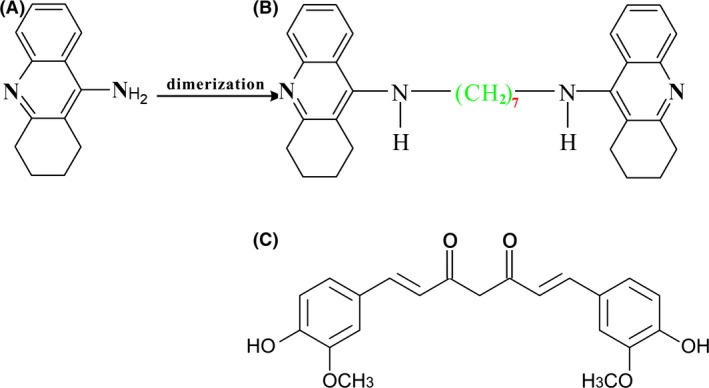
Dimeric bis (heptyl)‐cognitin (B) was synthesized from tacrine (A). Curcumin (C) with symmetrical structure was originally extracted from the rhizome of the plant Curcuma longa.
Materials and Method
Chemicals and Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin, and other regents for cell culture were purchased from Invitrogen (Carlsbad, CA, USA). Hexafluoroisopropanol (HFIP), Hoechst 33342, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT), tacrine, Thioflavin T (ThT), 2, 7‐dichlorofluorescin diacetate (DCFH‐DA), choline bromide, and acetylcholine iodide were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Purified synthetic Aβ1‐40 peptide was from GL Biochem (Shanghai, China). [14C] Acetyl coenzyme A was from Perkin Elmer (Boston, MA, USA).
Aβ1‐40 Preparation
Aβ1‐40 preparation was performed as we previously described 19. Briefly, Aβ1‐40 peptide was dissolved in HFIP, sonicated for 10 min in water bath, aliquoted in microcentrifuge tubes, and stored at −80°C. Immediately before use, the HFIP‐treated monomeric Aβ1‐40 was dried in the fume hood and dissolved in Milli‐Q water to 1 mM.
ThT Fluorescence Assay
ThT assay for quantifying Aβ fibrils formation was carried out as we previously described 10, 19. For the study of inhibition on Aβ fibrils formation, 20 μM monomeric Aβ1‐40 was mixed with various compounds and 5 μM ThT, and incubated for 6 days at 37°C. For the study of disaggregation of preformed fibrils, monomeric Aβ1‐40 was incubated for 3 days at 37°C to generate fibrils. Preformed Aβ fibrils were then mixed with compounds and 5 μM ThT and incubated for 3 days at 37°C. The fluorescence intensity of Aβ or compounds‐modified Aβ was measured at 440 nm (excitation) and 485 nm (emission) using a 1‐cm light‐path quartz cuvette.
SH‐SY5Y Cell Cultures
SH‐SY5Y cells (ATCC No. CRL‐2266) were cultured in DMEM containing 10% FBS and 100 units/mL penicillin/streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
MTT Assay
MTT assay for evaluating cell viability was performed as we previously reported 19, 20. Briefly, 24 h after plating, SH‐SY5Y cells were incubated for 48 h in FBS‐free medium containing Aβ or compounds‐modified Aβ. Thereafter, MTT solution was added to each well and the amount of resulted formazan was measured by observing absorbance at 570 nm.
Hoechst Staining Assay
Hoechst 33342 staining assay for detecting apoptotic nuclei was performed as we previously reported 21. After treatment, cells were washed twice with phosphate buffer saline (PBS), fixed for 10 min in 4% paraformaldehyde, and then stained for 5 min with 5 μg/mL Hoechst 33342. Images were acquired under a fluorescence microscope (Nikon Instruments Inc., Melville, NY, USA). Approximately 400 cells in 4 randomly chosen visual fields were counted.
Intracellular ROS Assay
Intracellular accumulation of ROS was evaluated using fluorescent probe DCFH‐DA. Briefly, after drug treatment, cells were incubated for 30 min at 37°C with DCFH‐DA (10 μM) in FBS‐free medium, washed twice with PBS, and then observed under the Nikon fluorescence microscope.
Animals
All the animal experiments were conducted in accordance with the standard guidelines for the care and use of laboratory animals. A total of 60 male Sprague Dawley rats, aged 7 weeks and weighing 220–280 g, were obtained from Animal Care Facility of the Hong Kong University of Science and Technology. They were housed in an animal room that was maintained at a temperature of 22 ± 2°C with a 12‐h light–dark cycle.
Stereotaxic Surgery and Drug Treatment
Rats were anesthetized with chloral hydrate (350 mg/kg, i.p.), and their head were then fixed into a stereotaxic apparatus. A guide cannulae was implanted in the left cerebral ventricle (A 1.4, L 0.9, V 4.0). Aβ1‐40 (800 pmol, i.c.v.) was slowly infused on Days 5, 8, and 11 after surgery in a total volume of 10 μL at the rate of 2 μL/min. From Day 5, rats (n = 10 for each group) were administered with saline (vehicle), B7C (0.1, 0.2 mg/kg, i.p.), or tacrine (1, 2 mg/kg, i.p.) once per day for 12 consecutive days.
Morris Water Maze (MWM) Test
The MWM test for evaluating learning performance was carried out as we previously reported 19. MWM equipment was composed of a circular water tank (150 cm in diameter and 60 cm in height) that was filled with water (23°C) to a depth of 30 cm. The tank was divided into four quadrants (north, south, west and east) that were used as start points. An escape platform (10 cm in diameter) was placed in the center of one randomly chosen quadrant (southeast for this study), with 1.5 cm below the surface of water.
Spatial learning performance was assessed 1 day after the last injection of Aβ1‐40. Rats were trained for 4 consecutive days to find the submerged platform. To evaluate long‐term memory retention, on Day 16 after the last day of training, probe trials were performed by removing the platform and allowing the rats to swim for 60 s to search for it. A video camera located above the center of the tank was used to record the time taken to reach the submerged platform (escape latency) and the percentage of total distance swum in the target quadrant for each rat (spatial bias).
Assay of ChAT and AChE Activities
After the removal of rat brain, the frontal cortex and hippocampus were dissected out, quickly frozen in liquid nitrogen. Following homogenization in 0.1 M phosphate buffer, ChAT activity in both brain regions was examined by observing the rate of formation of acetylcholine from acetyl‐CoA, using a radiochemical method 22. Briefly, the rat brain homogenates (5%, w/v) were mixed in a vial with 0.2 mM [14C] acetyl coenzyme A, 300 mM NaCl, 50 mM sodium phosphate buffer (pH 7.4), 8 mM choline bromide, and 0.1 mM physostigmine. The mixture was incubated at 37°C for 30 min and then stopped by the addition of 10 mM sodium phosphate buffer (pH 7.4). Thereafter, 2 mL of acetonitrile containing 10 mg Kalignost and 10 mL toluene scintillation mixture were added to the vial. The newly synthesized [14C] acetylcholine was extracted and quantified by liquid scintillation counting.
AChE activity was quantified using a colorimetric method 23. Briefly, a mixture of 2 mL in volume which contained 0.1 mL acetylcholine iodide (12 mM), 1.8 mL sodium phosphate buffer (0.1 mM, pH 7.4), and 0.1 mL homogenate was incubated at 37°C for 5 min. The reaction was stopped by the addition of 1 mL of 3% (w/v) sodium lauryl sulfate. About 1 mL of 0.2% (w/v) 5,5′‐dithiobis (2‐nitrobenzoin) acid was then added into the mixture to form a yellow complex. The color production was then examined spectrophotometrically at 420 nm.
Statistical Analysis
All the result data were presented as means ± SEM. Statistical analysis for single comparison was performed by Student's t‐test. When necessary, multiple comparisons were performed using one‐way ANOVA followed by an LSD post hoc analysis using SPSS software. P < 0.05 or less was considered to be statistically significant.
Results
B7C Markedly Inhibits Aβ Fibrils Formation and Disaggregates Preformed Aβ Fibrils in vitro
The fibrillar aggregates of Aβ are the major component of senile plaques that may result in neurodegeneration. With the use of ThT fluorescence assay, we tested the inhibitory effects of various compounds on Aβ1‐40 fibrils formation by co‐incubating them with monomeric Aβ1‐40 for 6 days at 37°C. Compared to Aβ1‐40 sample, the samples co‐incubated with B7C (0.1–10 μM) showed less ThT fluorescence increase (maximum of 50% reduction at 10 μM), an observation with similar potency and efficacy to those with curcumin that has been identified as an inhibitor of Aβ fibrils (Figure 2A) 24, 25.
Figure 2.
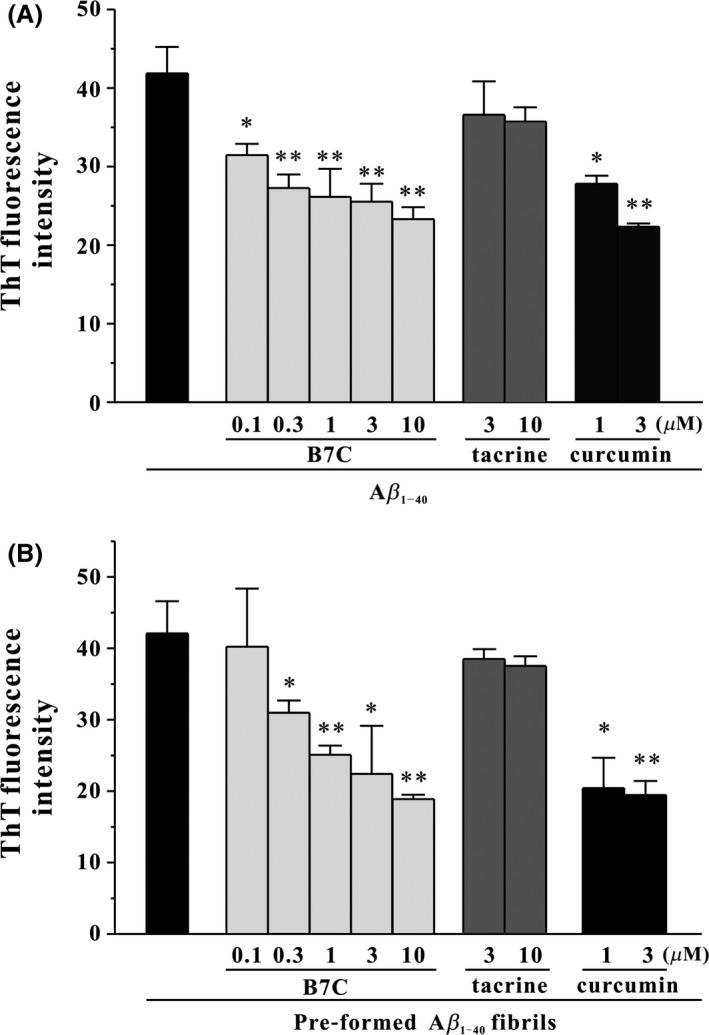
B7C effectively inhibits Aβ1‐40 fibrils formation and disaggregates preformed Aβ1‐40 fibrils in a dose‐dependent manner. (A) HFIP‐reconstituted Aβ monomers (20 μM) were incubated at 37°C for 6 days with or without B7C (0.1–10 μM), tacrine (3–10 μM), or curcumin (1–3 μM). The extent of Aβ aggregation was evaluated using ThT fluorescence assay. *P < 0.05, **P < 0.01, compared to Aβ group. (B) The preformed Aβ fibrils were allowed for 3 days and then incubated at 37°C for another 3 days with compounds described in (A). *P < 0.05, **P < 0.01, compared to Aβ group.
Next, we investigated whether these compounds could further disaggregate preformed Aβ1‐40 fibrils. Aβ1‐40 fibrils that were generated by 3‐days incubation of monomeric Aβ1‐40 at 37°C were incubated with tested compounds for another 3 days. It was shown that the samples co‐incubated with B7C or curcumin decreased ThT fluorescence by almost 50% (Figure 2B), suggesting that B7C was able to destabilize preformed Aβ1‐40 fibrils. However, tacrine, the monomer of B7C, did not show any effects on Aβ1‐40 fibrils formation and disaggregation of preformed Aβ1‐40 fibrils.
B7C Efficiently Blocks Aβ‐Induced Toxicity in SH‐SY5Y Cells
To judge whether compounds‐modified Aβ aggregates were less toxic than Aβ fibrils, we co‐incubated compounds with monomeric Aβ or preformed Aβ fibrils and added these Aβ samples to cultured SH‐SY5Y cells. As shown in Figure 3, compared to the group of Aβ fibrils, an approximate 20% increase in cell viability was observed in that of B7C or curcumin‐modified Aβ aggregates in both two cases. Meanwhile, tacrine‐modified Aβ aggregates did not enhance cell viability.
Figure 3.
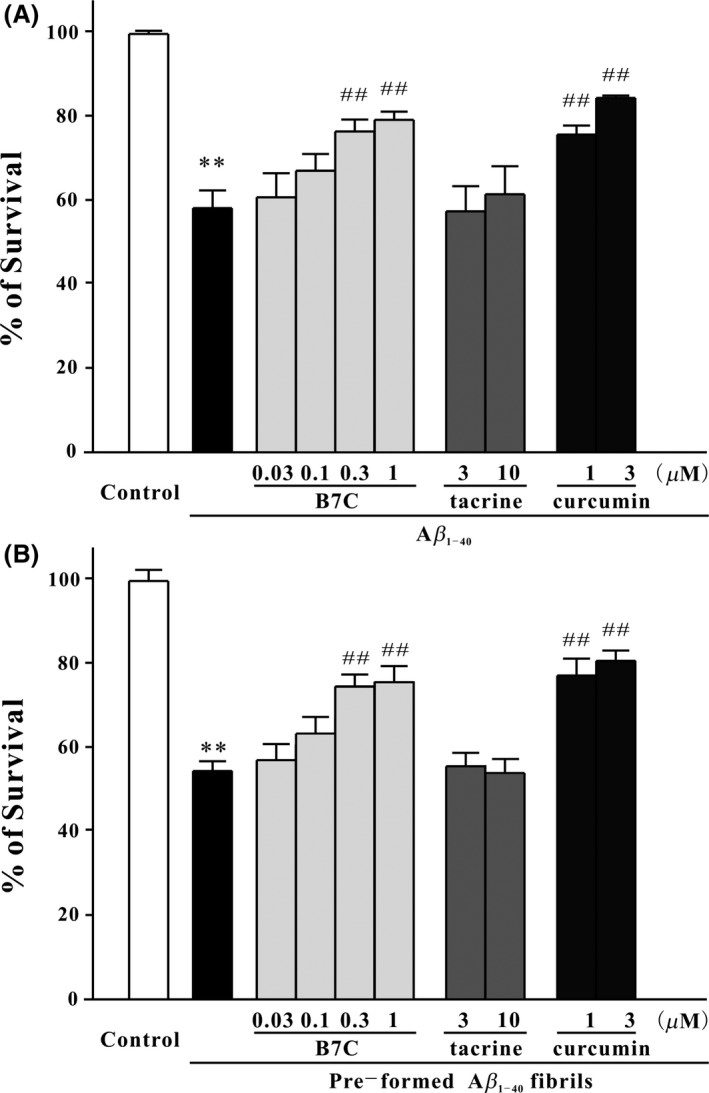
B7C markedly blocks Aβ1‐40‐induced neuronal death in SH‐SY5Y cells. (A) Aβ monomers (20 μM) were incubated at 37°C for 6 days with or without B7C (0.1–10 μM), tacrine (3–10 μM), or curcumin (1–3 μM) and then added into SH‐SY5Y cells. (B) The preformed Aβ fibrils were allowed for 3 days, then incubated at 37°C for another 3 days with compounds described in (A), and finally added into cells. Forty‐eight h after treatment, MTT assay was used to measure cell viability. **P < 0.01, compared to control group. ## P < 0.01, compared to Aβ group.
Furthermore, to confirm the neuroprotective effects produced by B7C, Hoechst‐staining assay was performed. As demonstrated in Figure 4, there was a significant decrease in the number of apoptotic nuclei stained by Hoechst 33342 in B7C‐modified Aβ aggregates group, in comparison with the Aβ fibrils group.
Figure 4.
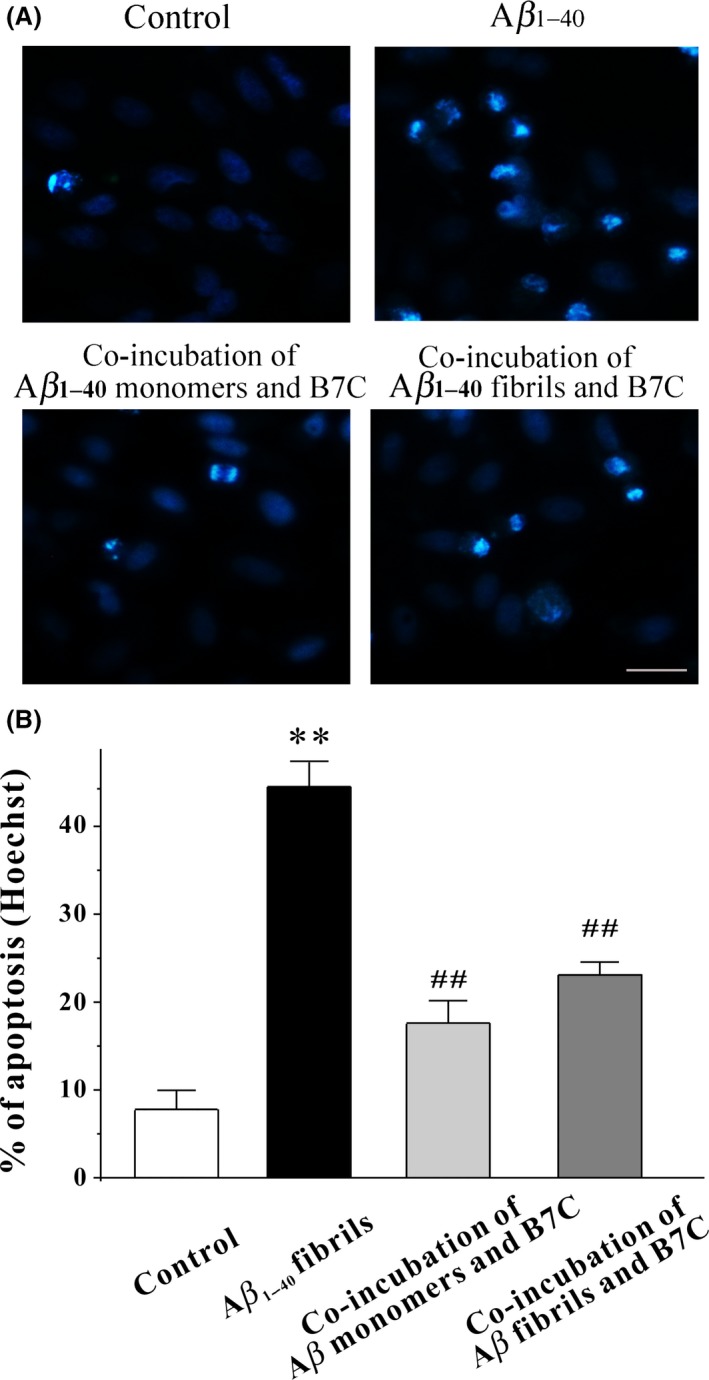
B7C efficiently inhibits Aβ1‐40‐induced apoptosis in SH‐SY5Y cells. (A) B7C (0.1–10 μM) was incubated with Aβ monomers for 6 days or preformed Aβ fibrils for 3 days and then added into SH‐SY5Y cells. Forty‐eight h after treatment, cell morphology was observed by the Nikon fluorescence microscopy. Scale bar = 50 μm. (B) Apoptosis was examined by counting the condensed nuclear stained by Hoechst 33342 from 4 randomly chosen fields. **P < 0.01, compared to control group. ## P < 0.01, compared to Aβ group.
B7C Substantially Decreases Aβ‐Induced Intracellular Accumulation of ROS in SH‐SY5Y Cells
Oxidative stress caused by elevated intracellular ROS generation is believed to an important mechanism underlying Aβ neurotoxicity. We therefore tested whether B7C‐modified Aβ aggregates were less toxic in neurons in terms of inhibition of intracellular accumulation of ROS. As shown in Figure 5, Aβ insult elicited a 2.5‐fold increase of DCF fluorescence intensity compared with the untreated control group, which was substantially inhibited by B7C‐modified Aβ aggregates.
Figure 5.
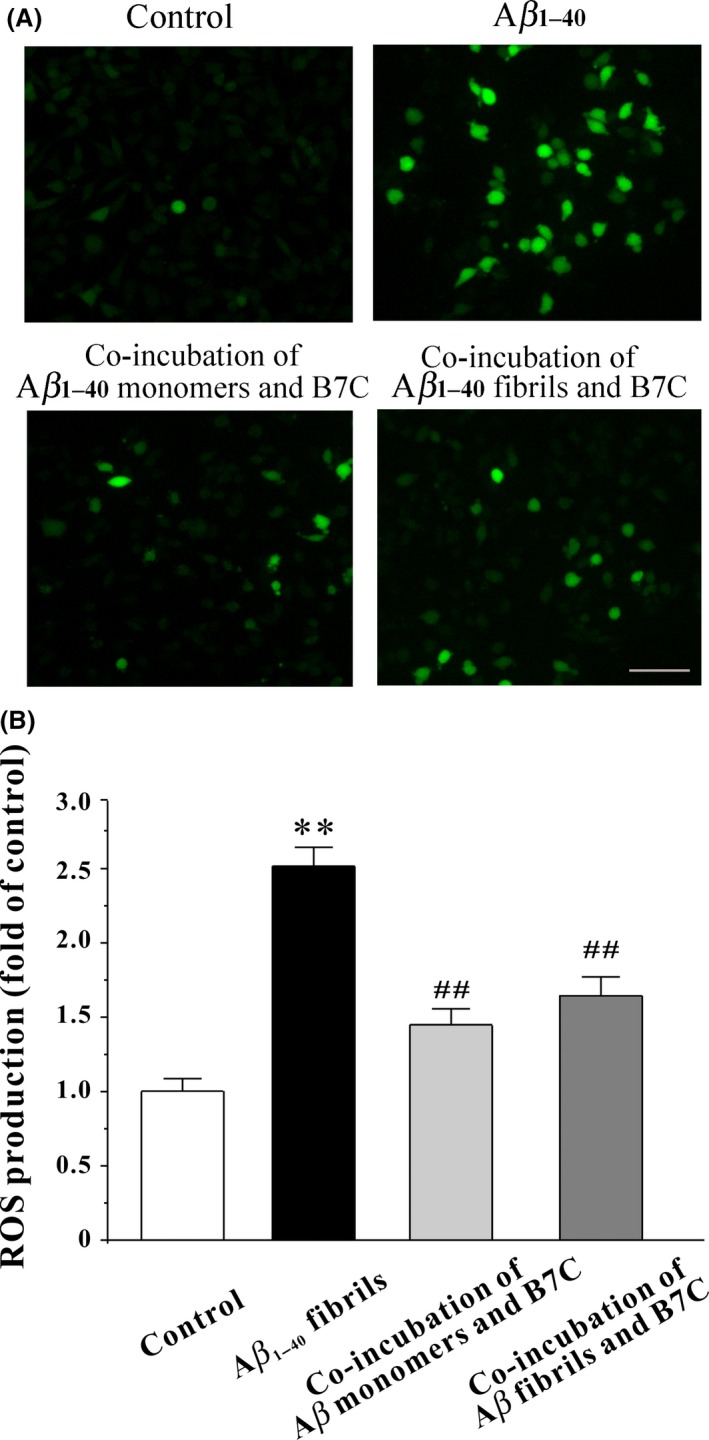
B7C substantially decreases Aβ1‐40‐induced intracellular accumulation of ROS in SH‐SY5Y cells. (A) B7C (0.1–10 μM) was incubated with Aβ monomers for 6 days or preformed Aβ fibrils for 3 days and then added into SH‐SY5Y cells. Forty‐eight h after treatment, cells were incubated for 30 min at 37°C with DCFH‐DA (10 μM) in FBS‐free medium, washed twice with PBS, and then observed under the Nikon fluorescence microscope. Scale bar = 100 μm. (B) The DCF fluorescence intensity values were analyzed from 4 randomly chosen fields. **P < 0.01, compared to control group. ## P < 0.01, compared to Aβ group.
B7C Potently Reverses Aβ‐Induced Learning and Memory Impairment in Rats
The experimental protocol is shown in Figure 6A. The learning and memory performance of rats were tested using MWM. All groups of rats showed a progressive decrease in escape latency during consecutive 4 days of training (Day 12–Day 15). The escape latency of Aβ1‐40‐treated group was higher than that of vehicle, suggesting that Aβ1‐40‐treated rats took longer time to find the hidden platform. While, the escape latency of rats treated by B7C (0.1–0.2 mg/kg) or tacrine (2 mg/kg) was significantly lower than that by Aβ1‐40 (Figures 6B, C, and D).
Figure 6.
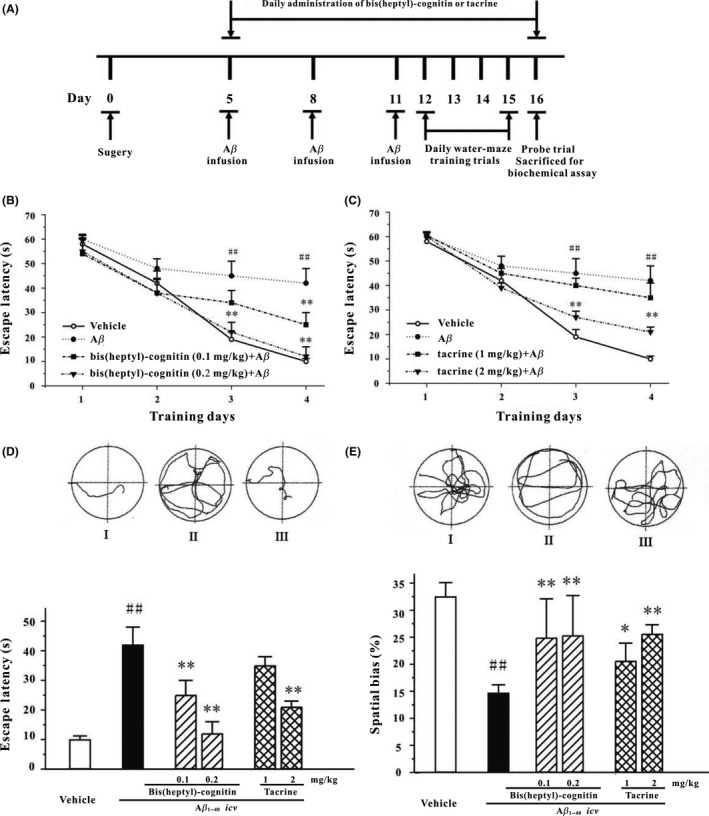
B7C potently reverses the memory deficits induced by icv infusion of Aβ1–40 in rats. (A) The schedules of animal experiments. (B, C) Mean latency to escape from the water onto the hidden platform. Each rat was subjected to two trials per day for 4 consecutive days. (D) Upper: typical swimming‐tracking path of vehicle control (I), groups of rats treated with Aβ1–40 (II), and groups of rats treated with 0.2 mg/kg B7C plus Aβ1–40 (III), on the fourth training day in Morris water maze. Lower: mean latency to escape from the water onto the hidden platform in fourth training day. (E) Upper: typical swimming‐tracking path of vehicle control (I), groups of rats treated with Aβ1–40 (II), and groups of rats treated with 0.2 mg/kg B7C plus Aβ1–40 (III), on the fifth probe trial day in Morris water maze. Lower: the swum distance in the target quadrant (southeast, in which the platform had been placed during the training phase) in the probe trial (swimming 60 s without platform). Data represent means ± SEM. ## P < 0.01 vs. vehicle group; **P < 0.01 vs. Aβ1–40‐treated group.
Moreover, the representative navigation paths at the end of the MWM training (Day 15) showed that a spatial learning acquisition impairment was observed in rats treated by Aβ1‐40, which was substantially reversed by treatment with B7C and Aβ1‐40 (Figure 6D). On the day of probe trial (Day 16), rats treated with B7C or tacrine swam longer in the probe quadrant (southeast) than rats in vehicle group (Figure 6E). In terms of escape latency and spatial bias, B7C was 10 times more potent than tacrine in reversing memory impairments induced by Aβ1‐40.
B7C Effectively Modulates Aβ‐Induced Dysfunction of Activities of AChE and ChAT
Inhibition of ChAT and activation of AChE activities are two important factors responsible for cholinergic dysfunction induced by Aβ. As shown in Table. 1, a significant decrease in ChAT activity was observed in both brain cortex and hippocampus of rats insulted by Aβ1‐40. In addition, Aβ1‐40 also elicited a marked increase in AChE activity in cortex but not hippocampus. B7C treatment reversed Aβ1‐40‐induced decrease of ChAT (in cortex and hippocampus) and increase of AChE (cortex) more potently than tacrine did.
Table 1.
B7C reverses the dysfunction of ChAT and AChE activities induced by icv Aβ1–40 in rats
| Aβ1‐40 | ||||||
|---|---|---|---|---|---|---|
| B7C (mg/kg) | Tacrine (mg/kg) | |||||
| Vehicle | 0.1 | 0.2 | 1 | 2 | ||
| ChAT (% of control) | ||||||
| Cortex | 100 ± 8.1 | 78.9 ± 9.0# | 97.0 ± 7.2** | 92.0 ± 8.2* | 84.4 ± 4.3 | 88.2 ± 7.9 |
| Hippocampus | 100 ± 8.9 | 80.1 ± 10.5## | 96.9 ± 6.6* | 97.5 ± 13.2* | 90.5 ± 4.2* | 90.3 ± 4.1* |
| AChE (% of control) | ||||||
| Cortex | 100 ± 13.6 | 122.1 ± 6.9# | 117.0 ± 15.5 | 94.0 ± 6.6** | 103.2 ± 4.4* | 100. ± 4.7* |
| Hippocampus | 100 ± 8.0 | 104.1 ± 10.1 | 101.6 ± 8.0 | 102.1 ± 28.3 | 101.5 ± 8.5 | 100.7 ± 9.6 |
Rats were killed 2 h after the fifth performance of water maze. Saline (Vehicle), B7C or tacrine was administered once per day for 12 consecutive days. Data are expressed as means ± SEM (n = 7). # P < 0.05 and ## P < 0.01 vs. vehicle group; *P < 0.05 and **P < 0.01 vs. Aβ1–40‐treated group.
Discussion
The aggregation of Aβ peptide into Aβ oligomers and fibrils which are associated with neurotoxicity in vitro and in vivo is considered to be one of key causative events in the pathogenesis of AD. Therefore, inhibition of Aβ aggregation and destabilization of preformed mature Aβ oligomers or fibrils would be a potential therapeutic strategy for the prevention and treatment of AD. Very recently, we have shown that the dimeric compound B7C prevented Aβ oligomers‐induced inhibition of long‐term potentiation and protected against Aβ oligomers‐induced synaptic and memory impairments via altering Aβ assembly 26. In the current study, we further demonstrated that B7C efficiently inhibited monomeric Aβ into fibrils and disaggregated preformed Aβ fibrils, as well as blocked Aβ‐associated neurotoxicity in cellular and animal models of AD, offering novel molecular insights into the potential application of B7C for the treatment of AD.
Aβ monomers derived from APP could be prone to spontaneously self‐aggregate into multiple physical forms, including oligomeric, protofibrillar, and fibrillar aggregates 27. These Aβ aggregates differing in their aggregation state are believed to be causative neurotoxic factors in the development of AD. Considerable evidence has demonstrated that intracellular accumulation of Aβ fibrils is involved in various pathogenic mechanisms, such as oxidative stress, synaptotoxicity, mitochondrial toxicity, and neuronal apoptosis, which leads to neurodegeneration and drive disease progression, indicating that Aβ fibrillization may be a therapeutic target for treating AD 28, 29, 30.
A sensitive method for measuring the amount of Aβ fibrils was introduced in the 1990s by Wood and his colleagues that involved monitoring ThT fluorescence 31. An enhanced ThT fluorescence will be observed when ThT binds to Aβ fibrils, possibly through the interaction with β‐pleated sheet structure and intercalation into growing fibrils 32. In this work, the ThT fluorescence results suggested that dimeric B7C efficiently inhibited Aβ fibrils formation and disaggregation of mature preformed Aβ fibrils (Figure 2), with a potency and efficacy similar to curcumin that also has a symmetrical structure (Figure 1C), while tacrine, the monomer of B7C, did not show any effects on Aβ aggregation (Figure 2). A growing body of evidence indicates that symmetrical structures are shared by various compounds that have been identified as inhibitors of Aβ aggregation. As explained by earlier studies 33, the symmetrical structures may be suitable for specific binding of Aβ, inhibition of Aβ aggregation as well as destabilization of β‐sheet‐rich conformation of Aβ fibrils. Therefore, there is a high likelihood that B7C might interact with Aβ through its symmetrical entities at both ends to inhibit Aβ fibrillization. Molecular dynamics simulation technique is being undertaken in our group to explore the exact binding sites of B7C and Aβ.
An efficient inhibitor of Aβ should interfere with Aβ aggregation as well as block Aβ‐associated neurotoxicity in vitro and in vivo. When we co‐incubated B7C with Aβ monomers or preformed mature Aβ fibrils, it was found that B7C‐modified Aβ aggregates were less toxic to neurons in comparison with Aβ fibrils, as evidenced by the increase in cell viability and decrease in the percentage of Hoechst‐stained apoptotic nuclei (Figures 3 and 4), suggesting that the neuroprotection of B7C is highly likely due to its ability to inhibit monomeric Aβ aggregation into neurotoxic fibrils or disaggregate Aβ fibrils into less or nontoxic Aβ species. Moreover, based on the fact that oxidative stress is an important mechanism that underlies Aβ‐induced neurotoxicity 34, 35, 36, we further tested the inhibitory effects of B7C on intracellular accumulation of ROS. Similarly, there was a significant decrease in DCF fluorescence intensity caused by B7C‐modified Aβ aggregates (Figure 5), again indicating that B7C might interact with Aβ species to interfere with Aβ aggregation and disassembly of Aβ fibrils.
After intracerebroventricular injection of Aβ into the brain of rodent animals, this peptide assembles into insoluble fibrils that are resistant to degeneration and associated with memory deficits 37. Several possible mechanisms, including activation of AChE and inhibition of ChAT activities 13, overstimulation of NMDA receptors 38 as well as trigger of oxidative stress and apoptosis 7, 35, have been demonstrated to be involved in Aβ‐mediated neurotoxicity. Herein, we further evaluated the neuroprotection of B7C and tacrine in a rat Alzheimer's model consisting of intracerebroventricular injection of Aβ1‐40, and assessed the learning performance of rats using water maze test, evidenced by the increase in spatial bias and decrease in escape latency (Figure 6), along with the upregulation of ChAT and downregulation of AChE activities (Table. 1), B7C was able to reverse the memory impairments induced by Aβ1‐40, possibly through its interference with Aβ aggregation. Based on the fact that Aβ could disrupt the transmission of neurotransmitter in synaptic clefts, including cholinergic and/or glutamatergic systems 12, 38, compounds that could modulate these two neurotransmitter systems may confer neuroprotection against Aβ insults. Therefore, tacrine, which was capable of inhibiting AChE but lack of anti‐Aβ fibrillization ability, was also shown to be effective in our model, although being 10 times less potent than B7C. Admittedly, as B7C is a superior AChE inhibitor and NMDA receptor antagonist, we could not rule out the possibility that inhibiting AChE and NMDA receptor may contribute to the in vivo neuroprotection of B7C. Further studies, such as Thioflavin S technique that stains plaques in brain, will be carried out using AChE inhibitors and NMDA receptor antagonist in combination or alone as a reference.
In conclusion, we provided evidence that B7C could inhibit Aβ fibrils formation and disaggregated the preformed Aβ fibrils. Moreover, B7C‐modified Aβ aggregates were less toxic to neurons, as evidenced by the increase in cell viability, the decrease in the number of apoptotic nuclei, and decrease in intracellular accumulation of ROS. Most encouragingly, B7C showed a high potency (10 times) in reversing Aβ‐induced memory deficits than its monomeric form tacrine. As there is increasing lines of evidence indicating that multifunctional compounds may exhibit greater therapeutic efficacy for treating AD by concurrently and synergistically acting multiple targets, the findings revealed herein and published previously may lead us to conjecture that B7C, which confers anti‐Aβ aggregation, anti‐AChE, and anti‐NMDA receptor activities, may provide promising therapeutic value against AD and/or other related neurodegenerative disorders.
Conflict of Interest
The authors declared no conflict of interest.
Acknowledgment
This work was supported by grants from the Research Grants Council of Hong Kong (561011, 15101014), The Hong Kong Polytechnic University (G‐SB10 and G‐UC15), the National Natural Science Foundation of China (81202510) the Science and Technology Development Fund of Macao SAR (Ref. No. 134/2014/A3), and the Research Committee, University of Macau (Ref. No. MYRG2015‐00214‐ICMS‐QRCM3, MYRG2015‐00172‐ICMS‐QRCM).
References
- 1. Strittmatter WJ. Medicine. Old drug, new hope for Alzheimer's disease. Science 2012;335:1447–1448. [DOI] [PubMed] [Google Scholar]
- 2. Choi SH, Kim YH, Hebisch M, et al. A three‐dimensional human neural cell culture model of Alzheimer's disease. Nature 2014;515:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmit JD, Ghosh K, Dill K. What drives amyloid molecules to assemble into oligomers and fibrils? Biophys J 2011;100:450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bieschke J, Russ J, Friedrich RP, et al. EGCG remodels mature alpha‐synuclein and amyloid‐beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A 2010;107:7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharoar MG, Thapa A, Shahnawaz M, et al. Kaempferol‐3‐O‐rhamnoside abrogates amyloid beta toxicity by modulating monomers and remodeling oligomers and fibrils to non‐toxic aggregates. J Biomed Sci 2012;19:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiao SS, Yao XQ, Liu YH, et al. Edaravone alleviates Alzheimer's disease‐type pathologies and cognitive deficits. Proc Natl Acad Sci U S A 2015;112:5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan KH, Lam KS, Cheng OY, et al. Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid‐beta neurotoxicity. PLoS ONE 2012;7:e52354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu MS, Suen KC, Kwok NS, So KF, Hugon J, Chang RC. Beta‐amyloid peptides induces neuronal apoptosis via a mechanism independent of unfolded protein responses. Apoptosis 2006;11:687–700. [DOI] [PubMed] [Google Scholar]
- 9. Nelson TJ, Alkon DL. Protection against beta‐amyloid‐induced apoptosis by peptides interacting with beta‐amyloid. J Biol Chem 2007;282:31238–31249. [DOI] [PubMed] [Google Scholar]
- 10. Du WJ, Guo JJ, Gao MT, et al. Brazilin inhibits amyloid beta‐protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity. Sci Rep 2015;5:7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fonseca AC, Moreira PI, Oliveira CR, Cardoso SM, Pinton P, Pereira CF. Amyloid‐Beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol Neurobiol 2014;51:610–622. [DOI] [PubMed] [Google Scholar]
- 12. Hu W, Gray NW, Brimijoin S. Amyloid‐beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J Neurochem 2003;86:470–478. [DOI] [PubMed] [Google Scholar]
- 13. Nunes‐Tavares N, Santos LE, Stutz B, et al. Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid‐beta peptide oligomers. J Biol Chem 2012;287:19377–19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H, Carlier PR, Ho WL, et al. Effects of bis(7)‐tacrine, a novel anti‐Alzheimer's agent, on rat brain AChE. NeuroReport 1999;10:789–793. [DOI] [PubMed] [Google Scholar]
- 15. Li W, Xue J, Niu C, et al. Synergistic neuroprotection by bis(7)‐tacrine via concurrent blockade of N‐methyl‐D‐aspartate receptors and neuronal nitric‐oxide synthase. Mol Pharmacol 2007;71:1258–1267. [DOI] [PubMed] [Google Scholar]
- 16. Luo J, Li W, Zhao Y, et al. Pathologically activated neuroprotection via uncompetitive blockade of N‐methyl‐D‐aspartate receptors with fast off‐rate by novel multifunctional dimer bis(propyl)‐cognitin. J Biol Chem 2010;285:19947–19958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han RW, Zhang RS, Chang M, et al. Reversal of scopolamine‐induced spatial and recognition memory deficits in mice by novel multifunctional dimers bis‐cognitins. Brain Res 2012;1470:59–68. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y, Li W, Chow PC, et al. Bis(7)‐tacrine, a promising anti‐Alzheimer's dimer, affords dose‐ and time‐dependent neuroprotection against transient focal cerebral ischemia. Neurosci Lett 2008;439:160–164. [DOI] [PubMed] [Google Scholar]
- 19. Hu S, Wang R, Cui W, et al. Inhibiting beta‐amyloid‐associated Alzheimer's pathogenesis in vitro and in vivo by a multifunctional dimeric Bis(12)‐hupyridone derived from Its natural analogue. J Mol Neurosci 2015;55:1014–1021. [DOI] [PubMed] [Google Scholar]
- 20. Hu SQ, Cui W, Xu DP, et al. Substantial neuroprotection against K+ deprivation‐induced apoptosis in primary cerebellar granule neurons by novel dimer bis(propyl)‐cognitin via the activation of VEGFR‐2 signaling pathway. CNS Neurosci Ther 2013;19:764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu S, Han R, Mak S, Han Y. Protection against 1‐methyl‐4‐phenylpyridinium ion (MPP+)‐induced apoptosis by water extract of ginseng (Panax ginseng C.A. Meyer) in SH‐SY5Y cells. J Ethnopharmacol 2011;135:34–42. [DOI] [PubMed] [Google Scholar]
- 22. Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem 1975;24:407–409. [DOI] [PubMed] [Google Scholar]
- 23. Yu H, Li WM, Kan KK, et al. The physicochemical properties and the in vivo AChE inhibition of two potential anti‐Alzheimer agents, bis(12)‐hupyridone and bis(7)‐tacrine. J Pharm Biomed Anal 2008;46:75–81. [DOI] [PubMed] [Google Scholar]
- 24. Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti‐amyloidogenic effects for Alzheimer's beta‐amyloid fibrils in vitro. J Neurosci Res 2004;75:742–750. [DOI] [PubMed] [Google Scholar]
- 25. Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 2005;280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 26. Chang L, Cui W, Yang Y, et al. Protection against beta‐amyloid‐induced synaptic and memory impairments via altering beta‐amyloid assembly by bis(heptyl)‐cognitin. Sci Rep 2015;5:10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010;362:329–344. [DOI] [PubMed] [Google Scholar]
- 28. Chang PT, Talekar RS, Kung FL, et al. A newly designed molecule J2326 for Alzheimer's disease disaggregates amyloid fibrils and induces neurite outgrowth. Neuropharmacology 2015;92:146–157. [DOI] [PubMed] [Google Scholar]
- 29. Minarini A, Milelli A, Tumiatti V, et al. Cystamine‐tacrine dimer: A new multi‐target‐directed ligand as potential therapeutic agent for Alzheimer's disease treatment. Neuropharmacology 2012;62:997–1003. [DOI] [PubMed] [Google Scholar]
- 30. Gao X, Zheng CY, Yang L, Tang XC, Zhang HY. Huperzine A protects isolated rat brain mitochondria against beta‐amyloid peptide. Free Radic Biol Med 2009;46:1454–1462. [DOI] [PubMed] [Google Scholar]
- 31. Wood SJ, MacKenzie L, Maleeff B, Hurle MR, Wetzel R. Selective inhibition of Abeta fibril formation. J Biol Chem 1996;271:4086–4092. [DOI] [PubMed] [Google Scholar]
- 32. Schenk D, Basi GS, Pangalos MN. Treatment strategies targeting amyloid beta‐protein. Cold Spring Harb Perspect Med 2012;2:a006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ono K, Hamaguchi T, Naiki H, Yamada M. Anti‐amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer's disease. Biochim Biophys Acta 2006;1762:575–586. [DOI] [PubMed] [Google Scholar]
- 34. Kim EA, Cho CH, Kim DW, Choi SY, Huh JW, Cho SW. Antioxidative effects of ethyl 2‐(3‐(benzo[d]thiazol‐2‐yl)ureido)acetate against amyloid beta‐induced oxidative cell death via NF‐kappaB, GSK‐3beta and beta‐catenin signaling pathways in cultured cortical neurons. Free Radic Res 2015;49:411–421. [DOI] [PubMed] [Google Scholar]
- 35. Rege SD, Geetha T, Broderick TL, Babu JR. Resveratrol protects beta amyloid‐induced oxidative damage and memory associated proteins in H19‐7 hippocampal neuronal cells. Curr Alzheimer Res 2015;12:147–156. [DOI] [PubMed] [Google Scholar]
- 36. Gray NE, Sampath H, Zweig JA, Quinn JF, Soumyanath A. Centella asiatica attenuates amyloid‐beta‐induced oxidative stress and mitochondrial dysfunction. J Alzheimers Dis 2015;45:933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rambaran RN, Serpell LC. Amyloid fibrils: Abnormal protein assembly. Prion 2008;2:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Talantova M, Sanz‐Blasco S, Zhang X, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA 2013;110:E2518–E2527. [DOI] [PMC free article] [PubMed] [Google Scholar]