

Summary
Oligodendrocytes (OLs), the myelin‐forming cells of the central nervous system, form a functional unit with axons and play a crucial role in axonal integrity. An episode of hypoxia–ischemia causes rapid and severe damage to these particularly vulnerable cells via multiple pathways such as overactivation of glutamate and ATP receptors, oxidative stress, and disruption of mitochondrial function. The cardinal effect of OL pathology is demyelination and dysmyelination, and this has profound effects on axonal function, transport, structure, metabolism, and survival. The OL is a primary target of ischemia in adult‐onset stroke and especially in periventricular leukomalacia and should be considered as a primary therapeutic target in these conditions. More emphasis is needed on therapeutic strategies that target OLs, myelin, and their receptors, as these have the potential to significantly attenuate white matter injury and to establish functional recovery of white matter after stroke. In this review, we will summarize recent progress on the role of OLs in white matter ischemic injury and the current and emerging principles that form the basis for protective strategies against OL death.
Keywords: Excitotoxicity, Hypoxia–ischemia, Oligodendrocyte, Oxidative stress, White matter
Introduction
After an episode of cerebral hypoxia–ischemia (HI), early events include energy crisis, cell depolarization from the breakdown of transmembrane gradients, cytotoxic edema, reactive oxygen species (ROS) production, and endothelial dysfunction 1. These events prompt a complex cascade resulting in neuronal and glial damage and death. OLs, the myelin‐forming cells of the CNS, are acutely damaged by short periods of HI. Cell swelling occurs as early as 30 min after arterial occlusion, and large numbers of OLs die within 3 h 2. It has been reported 3 that 30 min of oxygen–glucose deprivation (OGD) results in the death of 90% of OLs within 9 h. OL pathology results in demyelination and dysmyelination which have profound consequences for axonal function, transport, structure, metabolism, and survival 4, 5, 6. The most devastating effects of HI on these cells occur in premature infants of <32 weeks' gestation, which show pathological symptoms of chronic myelination disturbance, leading to periventricular white matter injury 7. The white matter of these infants is immature and poorly vascularized and contains oligodendrocyte progenitors (pre‐OL) which are sensitive to ischemia and infection.
Research in neurological disorders is progressively embracing the concept of the neurovascular unit, which emphasizes that a successful neurorestorative therapy cannot exclusively target neurons, but must also encompass glial and endothelial cells 8. Thus, therapeutic strategies that target OLs, myelin, and their receptors have the potential to significantly attenuate white matter injury in HI. This review highlights the mechanisms of OL injury and death in HI at all stages of development and focuses on the oligoprotective and oligorestorative therapies that have been investigated thus far.
Intrinsic Susceptibility of Oligodendrocytes to Hypoxic–Ischemic Damage
OLs display a number of features that render them more vulnerable to HI than other CNS glial cells, and in certain brain regions and stages of development, more vulnerable than neurons 9 (Figure 1).
Figure 1.
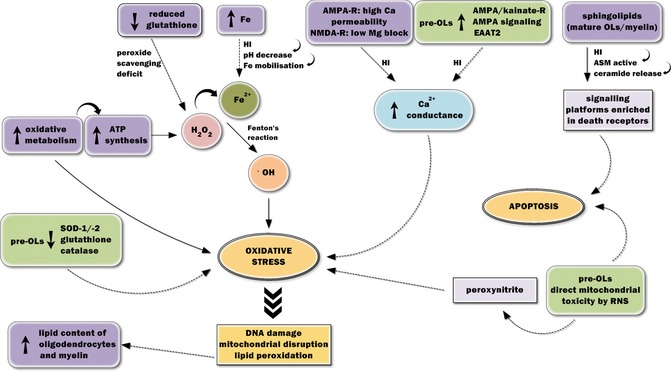
Features of oligodendrocytes (OLs) and pre‐OLs which render them vulnerable to hypoxia–ischemia (HI). The amplified vulnerability of OLs to HI derives from their high iron content, low reduced glutathione content, high rate of oxidative metabolism, high lipid and sphingolipid content, and high permeability of glutamate receptors. Pre‐OLs are even more vulnerable than their mature counterparts due to low levels of antioxidant enzymes, upregulation of AMPA/kainate receptors and enhanced AMPA/kainate signaling, increased expression of the glutamate transporter EAAT2, and a susceptibility to direct mitochondrial toxicity by reactive nitrogen species. In the event of HI, these properties lead to higher levels of oxidative stress and apoptosis, hence, severe damage, and death to cells of this lineage.
Of all the cell types in the brain, OLs contain the highest levels of immobilized, protein‐bound iron, which is a basic requirement for their function and oxidative metabolism, and for the synthesis of myelin components 10, 11. Apart from its important functional role, ferrous iron (Fe2+) can be a potent cytotoxin by catalyzing the conversion of hydrogen peroxide to hydroxyl radicals (OH), via the Fenton reaction 12, 13. In cerebral ischemia, an energy crisis leads to lactic acidosis, which results in mobilization of protein‐bound iron stores. This increases the levels of free cytosolic Fe2+ that participates in the Fenton reaction to bring about oxidative stress 14, 15. This effect is further amplified in OLs by their low content of reduced glutathione (GSH) 16, 17 which is an electron donor for the function of glutathione peroxidase, which in turn, scavenges peroxides. OLs contain less than half of the glutathione content of astrocytes and <15% of the glutathione peroxidase activity, which leads to a peroxide‐scavenging deficit 17. OLs also have the highest rate of oxidative metabolism by volume and can support a myelin membrane up to 100 times the weight of their cell bodies 4, 11, 18. This high metabolic activity generates more ROS 18 and requires a correspondingly high consumption of oxygen and ATP, the synthesis of which generates hydrogen peroxide as a by‐product 19, 20, 21.
The subunit composition of glutamate receptors in OLs continues to predispose them to injury during HI. Their AMPA receptors are especially permeable to Ca2+ 22, 23, and their NMDA receptors are only weakly blocked by Mg2+, enabling them to generate a substantial current even at resting membrane potential 22, 24, 25.
Sphingolipids, constituents of the myelin membrane, may also increase the susceptibility of OLs to damage under pathological conditions 4, 26. The simplest sphingolipid, ceramide, can activate the major pathways that govern cell death 27 and kill cells by limiting access to extracellular nutrients 28. Many apoptotic stimuli activate acid sphingomyelinase, an enzyme that mediates ceramide release from biological membranes 29, 30. Ceramide‐enriched signaling platforms that contain death receptors are formed in the plasmalemma, and these transmit apoptotic signals into the cell 29, 31. Ceramide released intracellularly also acts as a second messenger, leading to caspase‐mediated OL apoptosis within hours 26, 32, 33.
Even more susceptible to injury than mature OLs are the O4+/O1− late OL progenitors, which comprises about 90% of all OLs during the high‐risk period for periventricular leukomalacia (PVL) 7, 34. This vulnerability is a consequence of:
Amplified oxidative damage that results from a developmental deficit in superoxide dismutases (SOD‐1 and ‐2) and a hydrogen peroxide‐scavenging deficit 35, 36, 37 combined with active iron acquisition 11.
Higher vulnerability to reactive nitrogen species attack by direct mitochondrial toxicity with translocation of apoptosis‐inducing factor 38 and formation of peroxynitrite 39, 40.
Significant developmental upregulation of non‐NMDA glutamate receptors 41, 42 accompanied by enhanced AMPA‐mediated calcium signaling 43, which increases excitotoxicity. Furthermore, pre‐OLs also exhibit a transiently increased expression of the glutamate transporter (GluT) EAAT2, which may become a source of glutamate under pathological conditions 44.
Mechanisms of Oligodendrocyte Damage in HI
Neurotransmitter‐Mediated Toxicity
OLs express neurotransmitter receptors that allow for axon‐to‐OL signaling and mediate their own development and function. The major excitatory neurotransmitters involved are glutamate and ATP 45, 46. These bind to their respective receptors on the OL plasmalemma and result in an influx of ions, most notably Ca2+, which acts as a chemical signal under physiological conditions, triggering OL differentiation and myelination 47.
OLs are extremely sensitive to disruptions in intracellular calcium homeostasis 25. In HI, energy crisis and metabolic stress lead to prolonged overstimulation of neurotransmitter receptors, resulting in a cytosolic Ca2+ surge which is worsened by the activation of voltage‐gated calcium channels (VGCC) and the reversal of the Na+/Ca2+ exchanger (NCX) 48; (Figure 2). This Ca2+ is sequestered by mitochondria and leads to mitochondrial bioenergetic dysfunction, which is characterized by impaired oxidative phosphorylation, ROS generation, the release of apoptogenic proteins, such as cytochrome C, and cell death by apoptosis or necrosis 49.
Figure 2.
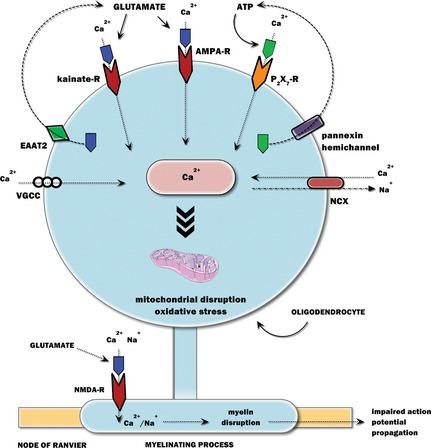
The major pathways governing neurotransmitter‐mediated oligodendrocyte death in hypoxia–ischemia (HI). The glutamate surge that occurs during HI leads to the overactivation of AMPA/kainate receptors on oligodendrocyte somata and NMDA receptors on myelinating processes. ATP is also released in HI, partly from the oligodendrocyte itself via pannexin hemichannels, leading to the overactivation of purinergic P2X7 receptors and enhanced Ca2+ signaling. The Ca2+ surge leads to the activation of voltage‐gated calcium channels (VGCC) and reversal of Na+/Ca2+ exchanger (NCX), further increasing the intracellular Ca2+. The glutamate transporter EAAT2 also starts to operate in reverse, contributing to the surge in extracellular glutamate. The excess cytosolic Ca2+ is sequestered in mitochondria where it leads to mitochondrial disruption and oxidative stress and eventual death to oligodendrocytes.
Glutamate‐Mediated Toxicity
Glutamate excitotoxicity is one of the major contributors toward ischemic injury in the CNS 50. OLs are sensitive to glutamate‐induced cell death 48 with an EC50 of 200 μM for a 24‐h exposure period 51. The glutamate signaling is governed by ionotropic and metabotropic glutamate receptors (iGluRs and mGluRs, respectively) and GluTs 52, 53. OLs express three main types of iGluRs: the AMPA and kainate receptors, predominantly located on their cell body, and NMDA receptors, clustered on their myelinating processes 25, 54. Pre‐OLs strongly express all three groups of mGluRs, but these are downregulated in mature OLs 55. GluTs are responsible for the uptake of glutamate from the extracellular space and maintenance of low extracellular glutamate levels (1–2 μM). However, under conditions of energy failure that result from HI, GluTs on OLs, astrocytes and microglia operate in reverse, with release of glutamate into the extracellular space 48, 56.
OLs have for a very long time, been known to be vulnerable to AMPA/kainate receptor‐mediated excitotoxicity 57, 58. The AMPA receptors on mature, myelinating OLs contain subunits GluR3 and GluR4, but not GluR1 59, and although mRNA for GluR2 is present, immunoprecipitation experiments indicate that GluR2 does not assemble with the other subunits 22, 60, which renders them highly permeable to Ca2+ when activated 23, 61. In fact, Ca2+ influx via these receptors alone is enough to induce death of OLs by excitotoxicity in culture, 61 and blockade of AMPA/kainate receptors alone prevents death of OLs by OGD during cerebral ischemic injury 3. Prolonged activation of these receptors leads to caspase‐dependent and caspase‐independent death pathways 62, and this toxicity is dose‐dependent 58. Recent work suggests the involvement of disturbed Zn2+ homeostasis in AMPA‐induced excitotoxicity. It has been demonstrated 63 that activation of OL AMPA receptors leads to mobilization of intracellular Zn2+ and a surge in cytosolic Zn2+, which contributes toward ROS production and mitochondrial depolarization, by a mechanism which is altogether separate from the Fenton reaction.
Oligodendrocytes express mRNA for the kainate subunits GluR6, GluR7, KA‐1, and KA‐2, but not for GluR5 22. Nontoxic concentrations of glutamate can sensitize these cells to complement attack, inducing OL death, in a process that is mediated exclusively by activation of kainate receptors. This complement‐induced death of OLs occurs via formation of the membrane attack complex, which increases membrane conductance and leads to a Ca2+ surge 64.
Excitotoxic OL death was previously thought to be exclusively mediated by AMPA and kainate glutamate receptors. Three recent reports 24, 54, 65 have manifestly altered this view by showing in vivo that OLs and myelin possess NMDA receptors and that these are involved in ischemic injury. White matter OLs at all stages of development contain NR1, NR2C, and NR3A NMDA receptor subunits, allowing for inward currents upon binding of glutamate 66. OL NMDA receptors are enriched with NR2C and NR3A subunits, which are blocked weakly by extracellular Mg2+ and allow for the generation of a current even at the cell's resting membrane potential 65, 66, 67. As NMDA receptors are clustered on the myelinating processes of OLs, receptor activation leads to a drastic increase in ion concentration because of the small intracellular volume 25, 54, 68 with disruption of myelin structure and action potential propagation 69.
In pre‐OLs, glutamate toxicity also occurs via a non‐receptor‐mediated mechanism, referred to as oxytosis, or oxidative glutamate toxicity 70. This involves system , a plasmalemmal antiport protein that transports cystine into the cytosol in exchange for glutamate to the extracellular space, in a 1:1 ratio 51. Once in the cytosol, the cystine is converted to cysteine, which is used in the production of glutathione 71. High extracellular glutamate concentrations can reverse the direction of this transport, promoting the efflux of cystine with consequent depletion of intracellular glutathione, and enhancement of oxidative stress 51, 72. Although this phenomenon is not exclusive to pre‐OLs, they exhibit enhanced, maturation‐dependent vulnerability because of low levels of glutathione peroxidase and SODs, especially SOD‐2 35, 36, 37.
ATP‐Mediated Toxicity
ATP activates ionotropic P2X and metabotropic P2Y purinoreceptors, both of which are expressed by OLs 73. P2X receptors consist of P2X1‐7 subunits that are most permeable to Ca2+ ions 74, 75. During ischemia, ATP‐mediated toxicity to OLs occurs mainly via P2X7 receptor subtypes, the sustained activation of which induces cell death, myelin damage, and white matter injury 53, 76, 77.
During situations of metabolic stress, such as cerebral ischemia, anoxic depolarization causes ATP to be released from glial cells, leading to a surge in the extracellular ATP concentration 78. It has been suggested 76 that OLs may release ATP during ischemia via pannexin hemichannels, resulting in depolarization of mitochondria and release of ROS. ATP released from dying cells can continue to aggravate P2X7‐mediated injury 6. Functional P2Y and P2X receptors are also expressed by pre‐OLs 79, the latter of which exhibit postischemic downregulation 80.
ATP‐mediated toxicity leads to apoptosis or necrosis of OLs, the mode of cell death being determined by the intensity of the Ca2+ surge, which, in turn, depends on the intensity of the ischemic insult 6. Prolonged stimulation of P2X7 receptors also leads to several enzyme and secondary messenger cascades, with release of cytokines such as interleukin‐1β and activation of mitogen‐activated protein kinase (MAPK) and nuclear factor‐κB, among others 75, 81.
Mitochondrial Disruption and Oxidative Stress
Oxidative damage is a cardinal consequence of neurotransmitter‐mediated toxicity. HI rapidly causes oxidative stress in OLs, which is characterized by enhanced production of the superoxide radical (), lipid peroxidation, and reduction of Fe3+ to the oxidant Fe2+ 82. The exposure of OLs to systems which generate free radicals, or free radical donors, such as and NO, leads to their rapid necrosis or apoptosis 5, 83.
The drastic rise in cytoplasmic Ca2+ that occurs during HI has profound consequences for mitochondria, which sequester this cation in large amounts and generate ROS at levels dependent on Ca2+ uptake 84. The oxidative stress that ensues activates several signaling pathways that modulate the functions of enzymes and transcription factors. These signals cause changes in gene expression that influence the cell's survivability 85. and NO radicals are particularly toxic to mitochondria as they interact with and block several key proteins of the respiratory chain 18. These radicals also lead to a diffusion‐limited generation of peroxynitrite, which causes death of OLs by lipid peroxidation, release of Zn2+, activation of extracellular signal‐regulated kinases and of 12‐lipoxygenase, and formation of additional ROS 86.
Auxiliary Mechanisms
Kinins are peptides produced at sites of tissue injury or inflammation 87. They activate specific B1 or B2 receptors, which mediate a number of signaling transduction mechanisms 88. In the CNS, kinins act as neuromediators 89. They also promote the synthesis of other pro‐inflammatory mediators, including cytotoxins and prostanoids, which lead to tissue damage and blood–brain barrier breakdown 88, 90. Functional kinin receptors are expressed by OLs, and their activation leads to a cytosolic Ca2+ surge, inflammation, and turnover of phosphoinositide 1, 91. Following ischemia, expression of B1 and B2 kinin receptors is upregulated, and the concentrations of bradykinin and kallidin also increase and result in damage and death of neural and glial tissue. Because of this, B1R and B2R receptor antagonists may be useable as neuroprotective and glioprotective agents during stroke, especially because they target multiple mechanisms that are involved in different stages of brain pathology 1.
The activation of dopamine D2 and D3 receptors 92, GABAA receptors 22 and adenosine A2A receptors 93 has also been implicated in ischemic damage of OLs. Moreover, A1 adenosine receptors are found on pre‐OLs, and their activation in HI inhibits maturation of these cells 94, with consequent shortage of myelinating OLs.
The Role of Neighboring Glia
Neighboring glia cause bystander damage to OLs in HI. Glutamate activates AMPA/kainate receptors in both resting and activated microglia at the site of injury and thereby enhances production and release of the cytokine, tumor necrosis factor‐α 95. This can kill OLs by apoptosis and by potentiation of interferon γ toxicity and is more toxic to pre‐OLs than to mature OLs 96, 97. Reactive microglia also release interleukin‐1β, glutamate 98, and reactive oxygen and nitrogen species, such as peroxynitrite 40, which further inhibit glutamate uptake and amplify excitotoxic damage 99.
Activation of microglia is a major source of damage to pre‐OLs in PVL, especially as the number of microglia in cerebral white matter peaks during the period of highest vulnerability to PVL 100. Reactive astrocytes, microglia, and macrophages also damage pre‐OLs in PVL, by the release of interferon γ 101, which leads to an increase in inducible nitric oxide synthase (iNOS) that becomes upregulated during HI 102. iNOS generates NO, which injures pre‐OLs by peroxynitrite formation and nitrosative damage. Antimicroglial agents, such as minocycline and melatonin, provide promising routes to the attenuation of pre‐OL damage and demyelination in PVL 97.
Recovery from Trauma and Role of Adult Oligodendrocyte Progenitor Cells (OPCs)
An important task of the adult CNS after an episode of HI is the replacement of affected OLs and the remyelination of affected axons, to restore saltatory conduction, improving motor function 103. In vivo rodent models of stroke have demonstrated that a few days following an insult, OLs surrounding the infarct tend to increase in number 104. Axons that have been demyelinated as a result of trauma or disease can be remyelinated by immature cells that “respond to demyelination by differentiating into myelinating OLs” 105. These cells, now referred to as adult OPCs, form part of a larger subtype of glial cells, NG2+ glia, which express the NG2 proteoglycan and platelet‐derived growth factor‐alpha (PDGF‐α) receptors 106. Also known as polydendrocytes, these cells are closely intermingled with other glial cells in the CNS, but nonetheless represent a distinct cell population 107.
Adult OPCs are not pre‐OLs but mature cells which develop after birth. They become activated during axonal inflammation and/or demyelination and develop into mature, myelinating OLs 108. Many chemical signals appear to be responsible for their activation, including axonal signals released on demyelination, growth factors and cytokines from other activated glial cells, as well as other injury‐induced stimuli, such as ATP and glutamate surges 4. It is of interest that, although TNF‐α causes death of OL by apoptosis 96, lack of TNF‐α leads to a delay in remyelination and a reduction in the population of proliferating adult NG2+ OPCs, which is followed by a decrease in the number of myelinating OLs. Apparently, the binding of this cytokine to TNF receptor 2 (TNFR2) is critical for the regeneration of OLs after trauma 109.
Recently, several therapies have been evaluated to target the protection or multiplication of these progenitors and allow for replacement of OLs and remyelination. Sun et al. 110 report that the synthetic cannabinoid agonist WIN55, 212‐2, has been shown to reduce injury to NG2+ glia cells and to promote their multiplication in the stroke penumbra. Adenosine was found to accelerate the maturation of OPCs in culture 111 and erythropoietin to stimulate oligodendrogenesis and maturation in vivo 112. The transplantation of predifferentiated human embryonic stem cells, which develop into myelinating OPCs, has also been proposed 103.
In PVL, pre‐OLs and immature OLs also exhibit a defensive reaction in response to HI. These cells typically take the form of an enlarged soma with elaborate, thickened processes that are not typical of OLs at this stage of development and with a concentrated distribution around areas of injury 34. HI also promotes accelerated maturation of pre‐OLs to immature OLs, which are less vulnerable to ischemia 7.
Protective Strategies for Oligodendrocyte Injury in HI
Numerous neuroprotective agents have been developed and tested for their ability to block specific cell damaging pathways in the ischemic cascade. Although many of these gave promising results in animal models, clinical trials have been, for the most part, disappointing, because of a lack of efficacy and/or clinical safety concerns. This failure may be explained, in part, by the histological and morphological differences between human and rodent brains 113. Ginsberg 114 also suggests that many agents may have been taken to clinical trials without sufficient preclinical evidence of efficacy. More rigorous experimentation is necessary to elucidate efficacious and clinically safe neuroprotective and glioprotective agents, with a focus on targeting multiple biochemical cascades and CNS cell types, and combinatorial therapies. A summary of the agents that have been deemed most promising in conferring protection to OLs is provided in Table 1.
Table 1.
Therapeutic candidates for oligoprotection in hypoxia–ischemia
| Mechanism | Oligoprotective agent | Oligodendrocyte maturation stage | Experimental model | References |
|---|---|---|---|---|
| AMPA antagonist | NBQX | Mature | Brain slices (mouse) | 3 |
| Pre‐OLs | In vivo(rat) | 42 | ||
| Topiramate | Pre‐OLs | In vivo(rat) | 117 | |
| SPD502 | Mature | In vivo (rat) | 118 | |
| GYKI52466 | Mature | Brain slices (mouse) | 3 | |
| CNQX | Mature | Optic nerve oligodendrocyte culture | 58 | |
| NMDA‐antagonist | D‐AP5 | Mature | Live adult rat optic nerve | 24 |
| Pre‐OLs, immature, mature | Brain slices (rat) | 65 | ||
| MK801 | Mature | Live rat optic nerve | 24 | |
| Pre‐OLs, immature, mature | Brain slices (rat) | 65 | ||
| Memantine | Mature | Brain slices (rat) | 69 | |
| Pre‐ols | In vivo (rat) | 120 | ||
| 7‐CKA | Mature | Live adult rat optic nerve | 24 | |
| Reverse glutamate transport inhibitor | Dihydrokainic acid | Immature | Cultured rat OLs | 119 |
| P2X7 antagonist | BBG | Mature | Rat optic nerve oligodendrocyte culture + isolated optic nerve | 53 |
| Oxidized ATP | ||||
| P2X antagonist | PPADS | |||
| ATP degrader | Apyrase | 76 | ||
| Pannexin hemichannel blocker | Mefloquine | |||
| Adenosine receptor antagonist | SCH58261 | Mature | In vivo (rat) | 93 |
| Caffeine | Pre‐OLs | In vivo (mouse) | 94 | |
| Antioxidant/radical scavenger | Mangiferin | Mature | Optic nerve oligodendrocyte culture | 84 |
| Morin | ||||
| N‐acetyl cysteine | Pre‐OLs | Rat oligodendrocyte progenitor cultures | 124 | |
| Edaravone | Mature | In vivo (rat) | 127 | |
| Mature | Clinical trial | 128 | ||
| α‐phenyl‐tert‐butyl‐nitrone | Mature | In vivo (rat) | 126 | |
| Vitamin K | Pre‐OLs | Cultured rat OLs | 129 | |
| Ebselen | Mature | In vivo (rat) | 130 | |
| Mature | Clinical trial | 133 | ||
| Erythropoietin | Pre‐OLs | In vivo (sheep) | 135 | |
| Melatonin | Pre‐OLs | In vivo (rat) | 136, 137 | |
| Estradiol | Mature | In vivo (mouse) | 138 | |
| Iron chelator | Deferoxamine | Mature | Cultured rat OLs | 134 |
| Antiapoptotic agent | IGF‐1 | Pre‐OLs | In vivo (rat) | 97, 140 |
| Pre‐OLs | In vivo (lamb) | 97, 141 | ||
| Mature | In vivo (mouse) | 142 | ||
| CNTF | Pre‐OLs | In vivo (mouse) | 97, 143 | |
| Estradiol | Pre‐OLs | Cultured rat OL + in vivo (rat) | 97, 144 | |
| Antimicroglial agent | Minocycline | Pre‐OLs | In vivo (rat) | 97, 145 |
| Cannabinoid agonist | WIN55, 212‐2 | OPCs | In vivo (rat) | 110 |
BBG, brilliant blue‐G; OLs, oligodendrocytes; OPC, oligodendrocyte progenitor cell.
Protection Against Neurotransmitter‐Mediated Injury
Excitotoxic OL, pre‐OL, and neuronal injury can be attenuated by administration of the AMPA antagonist NBQX, which preserves white matter structure and improves motor deficits 3, 42, 115, although this compound may not be clinically safe 116. Topiramate, a clinically safe anticonvulsant, protects pre‐OLs against HI when administered postinsult, as does NBQX 117. SPD 502, a competitive AMPA antagonist, protects both gray and white matter, including OLs, when administered intravenously 15 min before the insult, and for 4 h after the insult 118. Other AMPA antagonists that have been shown to protect OLs against excitotoxic damage include GYKI 52466 3 and CNQX 58. Dihydrokainic acid, an inhibitor of glutamate release via reverse transport, significantly protected immature OLs from ischemic injury in culture 119.
NMDA receptors are excellent targets for antagonists because they contain several sites at which ligands can bind in a subunit‐selective manner, such as glutamate‐binding sites, ion‐channel pores, and allosteric sites on the N‐terminal domain. NMDA receptor antagonists that target NR3A and NR2C subunits have the potential of acting as major therapeutic targets for white matter preservation in stroke 67. The NMDA antagonists, D‐AP5, and MK801 protect OLs and myelin from excitotoxic death, but are not clinically safe 24, 65, 72. Memantine, a clinically safe, uncompetitive NMDA receptor blocker is also effective against injury in both OLs and pre‐OLs 72, 120, and 7‐CKA protects OLs and myelin during chemical ischemia in vitro 24. Of interest is that blockade of NMDA receptors or removal of extracellular Ca2+ worsens, rather than improves, functional recovery in aging animals 121, which emphasizes the importance of age‐specific stroke treatment.
Another possible therapeutic route is the upregulation of GluTs, as these allow for ischemic tolerance subsequent to ischemic preconditioning. EAAT2 promoters, such as valproic acid, can protect glia against ischemia by enhanced removal of glutamate from the extracellular space 122, 123, 124.
Ischemia‐induced mitochondrial depolarization and oxidative stress are partially reversed by P2X7 receptor antagonists, by the ATP‐degrading enzyme apyrase, and by pannexin hemichannel blockers such as mefloquine. P2X7 receptor antagonists do not interfere with normal physiological function because of their selective activation 76, 77. The P2X7 antagonists Brilliant Blue‐G (BBG), oxidized ATP (oATP), and the nonselective P2X antagonist PPADS prevent ATP‐mediated OL toxicity 53. The calmodulin antagonist calmidazolium has been shown to inhibit P2X7‐receptor evoked glutamate release and may therefore have potential in oligoprotection during ischemia 125. The administration of the selective adenosine A2A receptor antagonist SCH58261 also protects OLs against cerebral ischemia by reducing the activation of the MAPK, JNK 93. Caffeine, an adenosine receptor antagonist, was found to be protective in PVL as it promotes the maturation of pre‐OLs after HI 94.
Protection Against Oxidative Stress
Antioxidants are potent therapeutic candidates for oxidative damage to OLs in cerebral ischemia. Mangiferin and morin, two natural antioxidant polyphenols, protect OLs from excitotoxic insult by free radical scavenging and cytosolic Ca2+ handling 84. N‐acetyl cysteine also attenuates AMPA/kainate OL cytotoxicity by increasing intracellular glutathione levels 124. Pretreatment with the spin‐trap agent α‐phenyl‐tertbutyl‐nitrone (PBN) reduced the number of damaged OLs by 55%, 40 min after the insult 126. The radical scavenger edaravone protects all components of the neurovascular unit against oxidative stress 8, 127, 128, while Vitamin K prevents oxidative damage to pre‐OLs and neurons during HI, with clinical safety 129. 12‐lipoxygenase inhibitors may also be of protective value to OLs at all stages of development, as 12‐lipoxygenase is a potent generator of ROS 97.
Ebselen, a mimic of glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase, exerts potent antioxidant effects on OLs and neurons 130, 131, 132. When administered intravenously, 2 h after stroke onset, it can salvage damaged tissue without major side effects 130. In a clinical trial, ebselen demonstrated a significant improvement in stroke patients who started ebselen treatment within 24 h of onset of the insult 133. The iron chelator deferoxamine protects OLs from cytotoxic effects induced by H2O2 and suppresses free radical formation 134. In clinical trials for PVL, erythropoietin, an antiinflammatory, antiapoptotic, antioxidant, and neurotrophic agent was found to reduce injury and preserve myelination in infants with moderate damage, without significant adverse effects 135. Melatonin, a free radical scavenger and up‐regulator of SOD, catalase, and glutathione peroxidase, has been found to promote pre‐OL maturation after perinatal brain damage 136 and decreases white matter inflammation, promoting myelination after neonatal stroke 137. The administration of the hormone 17β‐estradiol was recently shown to attenuate OL loss in the corpus callosum of male mice, and results in decreased demyelination and microglial activation 138, by a quinol‐based cyclic antioxidant mechanism 139.
The ability to visualize OLs in living brain through cell type‐selective transfer of genes encoding fluorescent proteins 146 provides new opportunities to understand cell–cell interactions of recovery in diseases of the myelinating unit.
Conclusion
Largely ignored for many years, the importance of OLs in the pathophysiology of a variety of neurological disorders has become evident. We now know that OLs are major targets of cerebral ischemia, both in the case of adult‐onset stroke and especially in PVL, which means that treatment strategies that exclusively target neuronal recovery cannot be optimally successful. This has led and should continue to lead researchers to make new links and explore new pathways of investigation, with the objective of treating cerebral ischemia in a more comprehensive manner.
New, groundbreaking research on oligodendrocyte pathophysiology in ischemia is constantly being made available. A notable example is the relatively recent discovery of functional NMDA receptors on OLs, antagonists of which are now being considered a possibly valid and valuable therapeutic route. Further work should continue to elucidate the exact underlying mechanisms of oligodendrocyte pathophysiology and to shed light on therapies that simultaneously target multiple mechanisms of injury and multiple components of the neurovascular unit. Therefore, it is hoped that future investigations should continue to work toward generating animal models of white matter stroke along with well‐designed clinical trials to extrapolate the findings on experimental animals to human neurological disease. These therapies are expected to be more innovative, more extensive, and more clinically viable.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
This study was supported in part by University of Malta research funding, coordinator M. Valentino. The authors thank EU COST Action CM1103 “Structure‐based drug design for diagnosis and treatment of neurological diseases: dissecting and modulating complex function in the monoaminergic systems of the brain.”
References
- 1. Albert‐Weißenberger C, Sirén AL, Kleinschnitz C. Ischemic stroke and traumatic brain injury: The role of the Kallikrein‐Kinin System. Prog Neurobiol 2013;101–102:65–82. [DOI] [PubMed] [Google Scholar]
- 2. Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996;27:1641–1647. [DOI] [PubMed] [Google Scholar]
- 3. Tekkök SB, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci 2001;21:4237–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem 2008;107:1–19. [DOI] [PubMed] [Google Scholar]
- 5. Merrill JE, Scolding NJ. Mechanisms of damage to myelin and oligodendrocytes and their relevance to disease. Neuropathol Appl Neurobiol 1999;25:435–458. [DOI] [PubMed] [Google Scholar]
- 6. Matute C, Ransom BR. Roles of white matter in central nervous system pathophysiologies. ASN Neuro 2012;4:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Back SA, Han BH, Luo NL, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia–ischemia. J Neurosci 2002;22:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee BJ, Egi Y, van Leyen K, Lo EH, Arai K. Edaravone, a free radical scavenger, protects components of the neurovascular unit against oxidative stress in vitro. Brain Res 2010;1307:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petito CK, Olarte JP, Roberts B, Nowak TS, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol 1998;57:231–238. [DOI] [PubMed] [Google Scholar]
- 10. Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: The role of iron. Glia 2009;57:467–478. [DOI] [PubMed] [Google Scholar]
- 11. Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia 1996;17:83–93. [DOI] [PubMed] [Google Scholar]
- 12. Winterbourn C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol Lett 1995;82–83:969–974. [DOI] [PubMed] [Google Scholar]
- 13. Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci 2002;22:5848–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oubidar M, Boquillon M, Marie C, Schreiber L, Bralet L. Ischemia‐induced brain iron delocalization: Effect of iron chelators. Free Radic Biol Med 1994;16:861–867. [DOI] [PubMed] [Google Scholar]
- 15. Selim MH, Ratan RR. The role of iron neurotoxicity in ischemic stroke. Ageing Res Rev 2004;3:345–353. [DOI] [PubMed] [Google Scholar]
- 16. Thorburne SK, Juurlink BH. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem 1996;67:1014–1022. [DOI] [PubMed] [Google Scholar]
- 17. Juurlink BH, Thorburne SK, Hertz L. Peroxide‐scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia 1998;22:371–378. [DOI] [PubMed] [Google Scholar]
- 18. Bradl M, Lassmann H. Oligodendrocytes: Biology and pathology. Acta Neuropathol 2010;119:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richter‐Landsberg C, Vollgraf U. Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp Cell Res 1998;244:218–229. [DOI] [PubMed] [Google Scholar]
- 20. Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen‐activated protein kinases in an oligodendrocyte cell line. J Neurochem 1999;72:112–119. [DOI] [PubMed] [Google Scholar]
- 21. Laszkiewicz I, Mouzannar R, Wiggins RC, Konat GW. Delayed oligodendrocyte degeneration induced by brief exposure to hydrogen peroxide. J Neurosci Res 1999;55:303–310. [DOI] [PubMed] [Google Scholar]
- 22. Káradóttir R, Attwell D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 2007;145:1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci 1994;17:31–108. [DOI] [PubMed] [Google Scholar]
- 24. Micu I, Jiang Q, Coderre E, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2005;439:988–992. [DOI] [PubMed] [Google Scholar]
- 25. Butt AM. Neurotransmitter‐mediated calcium signalling in oligodendrocyte physiology and pathology. Glia 2006;54:666–675. [DOI] [PubMed] [Google Scholar]
- 26. Casaccia‐Bonnefil P, Aibel L, Chao MV. Central glial and neuronal populations display differential sensitivity to ceramide‐dependent cell death. J Neurosci Res 1996;43:382–389. [DOI] [PubMed] [Google Scholar]
- 27. Morales A, Lee H, Goni FM, Kolesnic R, Fernandez‐Checa JC. Sphingolipids and cell death. Apoptosis 2007;12:923–939. [DOI] [PubMed] [Google Scholar]
- 28. Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci USA 2008;105:17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gulbins E. Regulation of death receptor signaling and apoptosis by ceramide. Pharmacol Res 2003;47:393–399. [DOI] [PubMed] [Google Scholar]
- 30. Carpinteiro A, Dumitru C, Schenck M, Gulbins E. Ceramide‐induced cell death in malignant cells. Cancer Lett 2008;264:1–10. [DOI] [PubMed] [Google Scholar]
- 31. Gulbins E, Grassmé H. Ceramide and cell death receptor clustering. Biochim Biophys Acta 2002;1585:139–145. [DOI] [PubMed] [Google Scholar]
- 32. Larocca JN, Farooq M, Norton WT. Induction of oligodendrocyte apoptosis by C2‐ceramide. Neurochem Res 1997;22:529–534. [DOI] [PubMed] [Google Scholar]
- 33. Craighead M, Pole J, Waters C. Caspases mediate C2‐ceramide‐induced apoptosis of the human oligodendroglial cell line, MO3. 13. Neurosci Lett 2000;278:125–128. [DOI] [PubMed] [Google Scholar]
- 34. Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 2001;21:1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Folkerth RD, Haynes RL, Borenstein NS, et al. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J Neuropathol Exp Neurol 2004;63:990–999. [DOI] [PubMed] [Google Scholar]
- 36. Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase‐catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci 2004;24:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baud O, Haynes RF, Wang H, et al. Developmental up‐regulation of MnSOD in rat oligodendrocytes confers protection against oxidative injury. Eur J Neurosci 2004;20:29–40. [DOI] [PubMed] [Google Scholar]
- 38. Baud O, Li J, Zhang Y, Neve RL, Volpe JJ, Rosenberg PA. Nitric oxide‐induced cell death in developing oligodendrocytes is associated with mitochondrial dysfunction and apoptosis‐inducing factor translocation. Eur J Neurosci 2004;20:1713–1726. [DOI] [PubMed] [Google Scholar]
- 39. Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg P. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 2003;62:441–450. [DOI] [PubMed] [Google Scholar]
- 40. Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci USA 2005;102:9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosenberg PA, Dai W, Gan XD, et al. Mature myelin basic protein‐expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res 2003;71:237–245. [DOI] [PubMed] [Google Scholar]
- 42. Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci 2000;20:9235–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Itoh T, Beesley J, Itoh A, et al. AMPA glutamate receptor‐mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J Neurochem 2002;81:390–402. [DOI] [PubMed] [Google Scholar]
- 44. Desilva TM, Kinney HC, Borenstein NS, et al. The glutamate transporter EAAT2 is transiently expressed in developing human cerebral white matter. J Comp Neurol 2007;501:879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chiu SY, Kriegler S. Neurotransmitter‐mediated signaling between axons and glial cells. Glia 1994;11:191–200. [DOI] [PubMed] [Google Scholar]
- 46. Fields RD, Stevens‐Graham B. New insights into neuron‐glia communication. Science 2002;298:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci 1996;19:346–352. [DOI] [PubMed] [Google Scholar]
- 48. Matute C, Domercq M, Sánchez‐Gómez MV. Glutamate‐mediated glial injury: Mechanisms and clinical importance. Glia 2006;53:212–224. [DOI] [PubMed] [Google Scholar]
- 49. Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004;36:257–264. [DOI] [PubMed] [Google Scholar]
- 50. Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature 1999;399:7–14. [DOI] [PubMed] [Google Scholar]
- 51. Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: Pharmacology, mechanisms, and prevention. J Neurosci 1993;13:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matute C. Glutamate and ATP signalling in white matter pathology. J Anat 2011;219:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matute C, Torre I, Perez‐Cerda F, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 2007;27:9525–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salter MG, Fern R. NMDA receptors are expressed in oligodendrocytes and mediate injury. Nature 2005;438:1167–1171. [DOI] [PubMed] [Google Scholar]
- 55. Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci USA 2004;101:7751–7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rothstein JD, Dykes‐Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996;16:675–686. [DOI] [PubMed] [Google Scholar]
- 57. Mcdonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor‐mediated excitotoxicity. Nat Med 1998;4:291–297. [DOI] [PubMed] [Google Scholar]
- 58. Sánchez‐Gómez MV, Matute C. AMPA and kainate receptors each mediate excitotoxicity in oligodendroglial cultures. Neurobiol Dis 1999;6:475–485. [DOI] [PubMed] [Google Scholar]
- 59. Li S, Stys PK. Mechanisms of ionotropic glutamate receptor‐mediated excitotoxicity in isolated spinal cord white matter. J Neurosci 2000;20:1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wosik K, Ruffini F, Almazan G. OLivier A, Nalbantoglu J, Antel JP. Resistance of human adult oligodendrocytes to AMPA/kainate receptor‐mediated glutamate injury. Brain 2004;127:2636–2648. [DOI] [PubMed] [Google Scholar]
- 61. Alberdi E, Sánchez‐Gómez MV, Marino A, Matute C. Ca2+ Influx through AMPA or kainate receptors alone is sufficient to initiate excitotoxicity in cultured oligodendrocytes. Neurobiol Dis 2002;9:234–243. [DOI] [PubMed] [Google Scholar]
- 62. Sanchez‐Gomez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. Caspase‐ dependent and caspase‐independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci 2003;23:9519–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mato S, Sánchez‐Gómez MV, Bernal‐Chico A, Matute C. Cytosolic zinc accumulation contributes to excitotoxic oligodendroglial death. Glia 2013;61:750–764. [DOI] [PubMed] [Google Scholar]
- 64. Alberdi E, Sánchez‐Gómez MV, Torre I, et al. Activation of kainate receptors sensitizes oligodendrocytes to complement attack. J Neurosci 2006;26:3220–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005;438:1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burzomato V, Frugier G, Pérez‐Otaño I, Kittler JT, Attwell D. The receptor subunits generating NMDA receptor mediated currents in oligodendrocytes. J Physiol 2010;588:3403–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paoletti P, Neyton J. NMDA receptor subunits: Function and pharmacology. Curr Opin Pharmacol 2007;7:39–47. [DOI] [PubMed] [Google Scholar]
- 68. Benarroch EE. Oligodendrocytes: Susceptibility to injury and involvement in neurologic disease. Neurology 2009;72:1779–1785. [DOI] [PubMed] [Google Scholar]
- 69. Bakiri Y, Hamilton NB, Káradóttir R, Attwell D. Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia 2008;56:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A. Mechanisms of oxidative glutamate toxicity: The glutamate/cystine antiporter system xc as a neuroprotective drug target. CNS Neurol Disord Drug Targets 2010;9:373–382. [DOI] [PubMed] [Google Scholar]
- 71. Lo M, Wang YZ, Gout PW. The x(c)‐ cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J Cell Physiol 2008;215:593–602. [DOI] [PubMed] [Google Scholar]
- 72. Conrad M, Sato H. The oxidative stress‐inducible cystine/glutamate antiporter, system xc −: Cystine supplier and beyond. Amino Acids 2012;42:231–246. [DOI] [PubMed] [Google Scholar]
- 73. James G, Butt AM. P2X and P2Y purinoreceptors mediate ATP‐evoked calcium signalling in optic nerve glia in situ. Cell Calcium 2001;30:251–259. [DOI] [PubMed] [Google Scholar]
- 74. Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: An overview. Trends Neurosci 2009;32:19–29. [DOI] [PubMed] [Google Scholar]
- 75. North RA. Molecular physiology of P2X receptors. Physiol Rev 2002;82:1013–1067. [DOI] [PubMed] [Google Scholar]
- 76. Domercq M, Perez‐Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 2010;58:730–740. [DOI] [PubMed] [Google Scholar]
- 77. Matute C. P2X7 receptors in oligodendrocytes: A novel target for neuroprotection. Mol Neurobiol 2008;38:123–128. [DOI] [PubMed] [Google Scholar]
- 78. Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Pharmacol 2009;7:160–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Agresti C, Meomartini ME, Amadio S, et al. Metabotropic P2 receptor activation regulates oligodendrocyte progenitor migration and development. Glia 2005;50:132–144. [DOI] [PubMed] [Google Scholar]
- 80. Wang LY, Cai WQ, Chen PH, Deng QY, Zhao CM. Downregulation of P2X7 receptor expression in rat oligodendrocyte precursor cells after hypoxia ischemia. Glia 2009;57:307–319. [DOI] [PubMed] [Google Scholar]
- 81. Suprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol 2009;71:333–359. [DOI] [PubMed] [Google Scholar]
- 82. Shereen A, Nemkul N, Yang D, et al. Ex vivo diffusion tensor imaging and neuropathological correlation in a murine model of hypoxia–ischemia‐induced thrombotic stroke. J Cereb Blood Flow Metab 2011;31:1155–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mronga T, Stahnke T, Goldbaum O, Richter‐Landsberg C. Mitochondrial pathway is involved in hydrogen‐peroxide‐induced apoptotic cell death of oligodendrocytes. Glia 2004;46:446–455. [DOI] [PubMed] [Google Scholar]
- 84. Ibarretxe G, Sánchez‐Gómez MV, Campos‐Esparza MR, Alberdi E, Matute C. Differential oxidative stress in oligodendrocytes and neurons after excitotoxic insults and protection by natural polyphenols. Glia 2006;53:201–211. [DOI] [PubMed] [Google Scholar]
- 85. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: Signaling for suicide and survival. J Cell Physiol 2002;192:1–15. [DOI] [PubMed] [Google Scholar]
- 86. Zhang Y, Wang H, Li J, et al. Intracellular zinc release and ERK phosphorylation are required upstream of 12‐lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J Biol Chem 2006;281:9460–9470. [DOI] [PubMed] [Google Scholar]
- 87. Rodi D, Couture R, Ongali B, Simonato M. Targeting kinin receptors for the treatment of neurological diseases. Curr Pharm Des 2005;11:1313–1326. [DOI] [PubMed] [Google Scholar]
- 88. Walker K, Perkins M, Dray A. Kinins and kinin receptors in the nervous system. Neurochem Int 1995;26:1–16. [DOI] [PubMed] [Google Scholar]
- 89. Raidoo DM, Bhoola KD. Kinin receptors on human neurones. J neuroimmunol 1997;77:39–44. [DOI] [PubMed] [Google Scholar]
- 90. Wagner S, Kalb P, Lukosava M, Hilgenfeldt U, Schwaninger M. Activation of the tissue kallikrein–kinin system in stroke. J Neurol Sci 2002;202:75–76. [DOI] [PubMed] [Google Scholar]
- 91. Noda M, Kariura Y, Amano T, et al. Kinin receptors in cultured rat microglia. Neurochem Int 2004;45:437–442. [DOI] [PubMed] [Google Scholar]
- 92. Rosin C, Colombo S, Calver AA, Bates TE, Skaper SD. Dopamine D2 and D3 receptor agonists limit oligodendrocyte injury caused by glutamate oxidative stress and oxygen/glucose deprivation. Glia 2005;52:336–343. [DOI] [PubMed] [Google Scholar]
- 93. Melani A, Cipriani S, Vannucchi MG, et al. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain 2009;132:1480–1495. [DOI] [PubMed] [Google Scholar]
- 94. Back SA, Craig A, Ling Luo N, et al. Protective effects of caffeine on chronic hypoxia‐induced perinatal white matter injury. Ann Neurol 2006;60:696–705. [DOI] [PubMed] [Google Scholar]
- 95. Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA–kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci 2000;20:251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Buntinx M, Moreels M, Vandenabeele F, et al. Cytokine‐induced cell death in human oligodendroglial cell lines: I. Synergistic effects of IFN‐γ and TNF‐α on apoptosis. J Neurosci Res 2004;76:834–845. [DOI] [PubMed] [Google Scholar]
- 97. Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int J Dev Neurosci 2011;29:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin‐1β promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol 2003;53:588–595. [DOI] [PubMed] [Google Scholar]
- 99. Kaur C, Ling EA. Periventricular white matter damage in the hypoxic neonatal brain: Role of microglial cells. Prog Neurobiol 2009;87:264–280. [DOI] [PubMed] [Google Scholar]
- 100. Billiards SS, Haynes RL, Folkerth RD, et al. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol 2006;497:199–208. [DOI] [PubMed] [Google Scholar]
- 101. Folkerth RD, Keefe RJ, Haynes RL, Trachtenberg FL, Volpe JJ, Kinney HC. Interferon‐γ expression in periventricular leukomalacia in the human brain. Brain Pathol 2004;14:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Haynes RL, Folkerth RD, Trachtenberg FL, Volpe JJ, Kinney HC. Nitrosative stress and inducible nitric oxide synthase expression in periventricular leukomalacia. Acta Neuropathol 2009;118:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sharp J, Keirstead HS. Therapeutic applications of oligodendrocyte precursors derived from human embryonic stem cells. Curr Opin Biotechnol 2007;18:434–440. [DOI] [PubMed] [Google Scholar]
- 104. Mandai K, Matsumoto M, Kitagawa K, et al. Ischemic damage and subsequent proliferation of oligodendrocytes in focal cerebral ischemia. Neuroscience 1997;77:849–861. [PubMed] [Google Scholar]
- 105. Gensert JM, Goldman JE. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 1997;19:197–203. [DOI] [PubMed] [Google Scholar]
- 106. Nishiyama A, Chang A, Trapp BD. NG2+ glial cells: A novel glial cell population in the adult brain. J Neuropathol Exp Neurol 1999;58:1113–1124. [DOI] [PubMed] [Google Scholar]
- 107. Di Bello IC, Dawson MR, Levine JM, Reynolds R. Generation of oligodendroglial progenitors in acute inflammatory demyelinating lesions of the rat brain stem is associated with demyelination rather than inflammation. J Neurocytol 1999;28:365–381. [DOI] [PubMed] [Google Scholar]
- 108. Greenwood K, Butt AM. Evidence that perinatal and adult NG2‐glia are not conventional oligodendrocyte progenitors and do not depend on axons for their survival. Mol Cell Neurosci 2003;23:544–558. [DOI] [PubMed] [Google Scholar]
- 109. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JPY. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001;4:1116–1122. [DOI] [PubMed] [Google Scholar]
- 110. Sun J, Fang YQ, Ren H, et al. WIN55, 212‐2 protects oligodendrocyte precursor cells in stroke penumbra following permanent focal cerebral ischemia in rats. Acta Pharmacol Sin 2013;34:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: A neuron‐glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 2002;36:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Iwai M, Stetler RA, Xing J, et al. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010;41:1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dronne MA, Grenier E, Chapuisat G, Hommel M, Boissel JP. A modelling approach to explore some hypotheses of the failure of neuroprotective trials in ischemic stroke patients. Prog Biophys Mol Biol 2008;97:60–78. [DOI] [PubMed] [Google Scholar]
- 114. Ginsberg MD. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology 2008;55:363–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kanellopoulos GK, Xu XM, Hsu CY, Lu X, Sundt TM, Kouchoukos NT. White matter injury in spinal cord ischemia protection by AMPA/kainate glutamate receptor antagonism. Stroke 2000;31:1945–1952. [DOI] [PubMed] [Google Scholar]
- 116. Martinez‐Vila E, Sieira PI. Current status and perspectives of neuroprotection in ischemic stroke treatment. Cerebrovasc Dis 2001;11:60–70. [DOI] [PubMed] [Google Scholar]
- 117. Follett PL, Deng W, Dai W, et al. Glutamate receptor‐mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J Neurosci 2004;24:4412–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. McCracken E, Fowler JH, Dewar D, Morrison S, McCulloch J. Grey matter and white matter ischemic damage is reduced by the competitive AMPA receptor antagonist, SPD 502. J Cereb Blood Flow Metab 2002;22:1090–1097. [DOI] [PubMed] [Google Scholar]
- 119. Fern R, Möller T. Rapid ischemic cell death in immature oligodendrocytes: A fatal glutamate release feedback loop. J Neurosci 2000;20:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Manning SM, Talos DM, Zhou C, et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci 2008;28:6670–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Baltan S. Ischemic injury to white matter: An age‐dependent process. Neuroscientist 2009;15:126–133. [DOI] [PubMed] [Google Scholar]
- 122. Zhang M, Li WB, Geng JX, et al. The upregulation of glial glutamate transporter‐1 participates in the induction of brain ischemic tolerance in rats. J Cereb Blood Flow Metab 2007;27:1352–1368. [DOI] [PubMed] [Google Scholar]
- 123. Romera C, Hurtado O, Mallolas J, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARγ target gene involved in neuroprotection. J Cereb Blood Flow Metab 2007;27:1327–1338. [DOI] [PubMed] [Google Scholar]
- 124. Liu HN, Giasson BI, Mushynski WE, Almazan G. AMPA receptor‐mediated toxicity in oligodendrocyte progenitors involves free radical generation and activation of JNK, calpain and caspase 3. J Neurochem 2002;82:398–409. [DOI] [PubMed] [Google Scholar]
- 125. Cervetto C, Mazzotta MC, Frattaroli D, et al. Calmidazolium selectively inhibits exocytotic glutamate release evoked by P2X7 receptor activation. Neurochem Int 2012;60:768–772. [DOI] [PubMed] [Google Scholar]
- 126. Irving EA, Yatsushiro K, McCulloch J, Dewar D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: Involvement of free radicals. J Cereb Blood Flow Metab 1997;17:612–622. [DOI] [PubMed] [Google Scholar]
- 127. Kubo K, Nakao S, Jomura S, et al. Edaravone, a free radical scavenger, mitigates both gray and white matter damages after global cerebral ischemia in rats. Brain Res 2009. Jul;1279:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nakase T, Yoshioka S, Suzuki A. Free radical scavenger, edaravone, reduces the lesion size of lacunar infarction in human brain ischemic stroke. BMC Neurol 2011;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li J, Lin JC, Wang H, et al. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci 2003;23:5816–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Imai H, Masayasu H, Dewar D, Graham DI, Macrae IM. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke 2001;32:2149–2154. [DOI] [PubMed] [Google Scholar]
- 131. Porciúncula LO, Rocha JB, Cimarosti H, et al. Neuroprotective effect of ebselen on rat hippocampal slices submitted to oxygen–glucose deprivation: Correlation with immunocontent of inducible nitric oxide synthase. Neurosci Lett 2003;346:101–104. [DOI] [PubMed] [Google Scholar]
- 132. Namura S, Nagata I, Takami S, Masayasu H, Kikuchi H. Ebselen reduces cytochrome c release from mitochondria and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke 2001;32:1906–1911. [DOI] [PubMed] [Google Scholar]
- 133. Yamaguchi T, Sano K, Takakura K, et al. Ebselen in acute ischemic stroke a placebo‐controlled, double‐blind clinical trial. Stroke 1998;29:12–17. [DOI] [PubMed] [Google Scholar]
- 134. Vollgraf U, Wegner M, Richter‐Landsberg C. Activation of AP‐1 and NF‐kappaB transcription factors is involved in hydrogen peroxide‐induced apoptotic cell death of oligodendrocytes. J Neurochem 1999;73:2501–2509. [DOI] [PubMed] [Google Scholar]
- 135. Rees S, Harding R, Walker D. The biological basis of injury and neuroprotection in the fetal and neonatal brain. Int J Dev Neurosci 2011;29:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Olivier P, Fontaine RH, Loron G, et al. Melatonin promotes oligodendroglial maturation of injured white matter in neonatal rats. PLoS ONE 2009;4:e7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Villapol S, Fau S, Renolleau S, Biran V, Charriaut‐Marlangue C, Baud O. Melatonin promotes myelination by decreasing white matter inflammation after neonatal stroke. Pediatr Res 2011;69:51–55. [DOI] [PubMed] [Google Scholar]
- 138. Taylor LC, Puranam K, Gilmore W, Ting JP, Matsushima GK. 17β‐estradiol protects male mice from cuprizone‐induced demyelination and oligodendrocyte loss. Neurobiol Dis 2010;39:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Prokai L, Prokai‐Tatrai K, Perjesi P, et al. Quinol‐based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci USA 2003;100:11741–11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lin SY, Fan LW, Pang Y, Rhodes PG, Mitchell HJ, Cai ZW. IGF‐1 protects oligodendrocyte progenitor cells and improves neurological functions following cerebral hypoxia‐ischemia in the neonatal rat. Brain Res 2005;1063:15–26. [DOI] [PubMed] [Google Scholar]
- 141. Cao Y, Gunn AJ, Bennet L, et al. Insulin‐like growth factor (IGF)‐1 suppresses oligodendrocyte caspase‐3 activation and increases glial proliferation after ischemia in near‐term fetal sheep. J Cereb Blood Flow Metab 2003;23:739–747. [DOI] [PubMed] [Google Scholar]
- 142. Mason JL, Ye P, Suzuki K, D'ercole AJ, Matsushima GK. Insulin‐like growth factor‐1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci 2000;20:5703–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Linker RA, Maurer M, Gaupp S, et al. CNTF is a major protective factor in demyelinating CNS disease: A neurotrophic cytokine as modulator in neuroinflammation. Nat Med 2002;8:620–624. [DOI] [PubMed] [Google Scholar]
- 144. Gerstner B, Lee J, DeSilva TM, Jensen FE, Volpe JJ, Rosenberg PA. 17β–Estradiol protects against hypoxic/ischemic white matter damage in the neonatal rat brain. J Neurosci Res 2009;87:2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lechpammer M, Manning SM, Samonte F, et al. Minocycline treatment following hypoxic‐ischemic injury attenuates white matter injury in a rodent model of periventricular leukomalacia. Neuropathol Appl Neurobiol 2008;34:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ness J, Valentino M, McIver SR, Goldberg MP. Identification of oligodendrocytes in experimental disease models. Glia 2005;50:321–328. [DOI] [PubMed] [Google Scholar]