

Summary
Aim
Visfatin, a novel adipokine, is predominantly produced by visceral adipose tissue and exists in intracellular and extracellular compartments. The intracellular form of visfatin is proved to be nicotinamide phosphoribosyltransferase (NAMPT) and exhibits neuroprotection through maintaining intracellular NAD + pool. However, whether extracellular form of visfatin has NAMPT activity and the effect of extracellular visfatin in cerebral ischemia are unknown.
Methods and Results
Plasma concentrations of visfatin, NAD +, and ATP were increased in mice upon cerebral ischemia. Cultured glia, but not neuron, was able to secrete visfatin. Oxygen‐glucose deprivation (OGD) stress increased the secretion of visfatin from glia. Extracellular recombinant mouse wild‐type visfatin, but not mouse H247A‐mutant enzymatic‐dead visfatin, had NAMPT enzymatic function in vitro. Treatment of wild‐type visfatin, but not H247A‐mutant enzymatic‐dead visfatin, significantly attenuated detrimental effect of OGD on the cell viability and apoptosis in both cultured mouse neuron and glia. Treatment of neutralizing antibody, abolished the protective effect of extracellular visfatin on cell viability, but failed to block the antiapoptotic effect of extracellular visfatin. At last, we observed that plasma visfatin concentrations decreased in 6‐month‐old but not 3‐month‐old SHR‐SP compared with that in age‐matched Wistar‐Kyoto rats. Inhibition of NAMPT enzymatic function of visfatin (by FK866) accelerated the occurrence of stroke in SHR‐SP.
Conclusions
Extracellular visfatin has NAMPT enzymatic activity and maybe be neuroprotective just as intracellular visfatin in cerebral ischemic injury.
Keywords: Cerebral ischemia, Extracellular, Neuroprotection, Nicotinamide phosphoribosyltransferase, Visfatin
Introduction
Ischemic stroke is a leading cause of morbidity and mortality worldwide that occurs mainly due to a cerebral artery occlusion by a thrombus 1. Few therapeutic advances appear to be of value for ischemic stroke in the clinic yet; recombinant tissue plasminogen activator is still the only drug approved for treatment in acute ischemic stroke 1. Upon ischemic stress, endogenous defense responses against ischemia‐induced cellular injury are evoked in neuronal and adjacent cells. These protective responses include activation of superoxide dismutase/glutathione peroxidase 2, microRNAs 3, hypoxia‐inducible factor‐1 4, endothelial progenitor cells 5 and so on. Elucidation of the defense mechanisms against stroke injury may be a key to the development of new therapies for stroke prevention and treatment.
Visfatin is a recently discovered adipokine with important roles in cellular biological functions 6. A large number of works have documented that visfatin is a multifaceted molecule existing in both intracellular and extracellular compartments. On the one hand, intracellular visfatin is able to convert nicotinamide into nicotinamide mononucleotide (NMN) as the rate‐limiting enzyme for mammalian NAD+ biosynthesis, which is required for lymphocyte development 7, myeloid differentiation 8, metabolic regulation 9, and cardiovascular functions 10. On the other hand, the extracellular visfatin, which was originally isolated as a presumptive cytokine named pre‐B‐cell colony‐enhancing factor that enhances the maturation of B‐cell precursors 11, is released from adipocytes and exerts multiple effects both in vitro and in vivo 6. Interestingly, this protein lacks a signal sequence for secretion and the presence of visfatin in extracellular space might be due to either cell lysis or cell death 12. And the putative receptor of extracellular visfatin is still undiscovered.
Our group and another group independently reported that intracellular visfatin protects against cerebral ischemic injury 13, 14, 15, 16. Both the results from us and them demonstrate that the NAD+ biosynthesis activity is critical for the neuroprotection of intracellular visfatin 15, 16, 17, 18. However, the effects of extracellular visfatin on ischemic neuronal injury are unknown yet. It was reported that plasma concentration of visfatin (extracellular) was higher in patients with ischemic stroke compared with healthy individuals 19. We hypothesized that the extracellular visfatin may play a role in cerebral ischemia‐induced neuronal injury, just as intracellular visfatin.
Materials and methods
Animals and Agents
Male 8‐week‐old C57L/BJ6 mice, stroke‐prone spontaneously hypertensive rats (SHR‐SP) and Wistar‐Kyoto (WKY) rats were provided by the Animal Center of our university. Animals were housed in a facility with controlled temperature (23 ± 1°C) with free access to tap water and chow 20. All procedures were performed in compliance with our institutional guidelines for animal care and the Guide for Care and Directive 2010/63/EU 21.
Specific chemical inhibitor of visfatin FK866 was kindly provided by TopoTarget A/S (formerly Apoxis SA, Lausanne, Switzerland). CCK‐8 kit was purchased from Dojndo Lab (Tokyo, Japan). Enzyme immunoassay (EIA) for visfatin was purchased from Phoenix Pharmaceuticals (Belmont, CA, USA). Luminescent ATP Detection Assay Kit was purchased from Promega (Madison, WI, USA). Anti‐Tuj1 and anti‐GFAP were purchased from Millipore (Billerica, MA, USA). DAPI, TUNEL assay and Neurobasal medium were purchased from Invitrogen (Carlsbad, CA, USA).
Determination of Plasma Visfatin and NAD+ Levels
Plasma visfatin levels were determined by visfatin (C‐terminal) EIA kit as described previously 22. The EIA kit has an intraassay CV% of < 5%, an interassay CV% of < 12% and a sensitivity of 0.1 ng/mL. Plasma NAD+ levels were determined using a commercial assay as described previously 23 using a microplate luminometer 24.
Bioluminescent Determination of ATP Concentrations
The plasma ATP levels were determined following the manufacturer's instructions as described previously 25. The plasma (25 μL) was pipetted into a tube containing 25 μL of stabilizing solution containing 118 mmol NaCl, 5 mmol KCl, 40 mmol tricine buffer, 4.15 mmol EDTA, 5 nmol NBTI and 100 μmol IBMX, pH adjusted to 7.4 with 2 M KOH. Then, 50 μL of luciferase reagent was added. The emitted light was linearly related to the ATP concentration and measured using a microplate luminometer 26.
Purification of Recombinant Mouse Wild‐type and H247A‐mutant Enzymatic‐dead Visfatin
The expression and purification of mouse visfatin was described previously 27. Briefly, the plasmid containing his‐tagged wild‐type (WT) mouse visfatin (gift from Dr. Imai S, Washington University School of Medicine) 27 and H247A‐mutant enzymatic‐dead mouse visfatin (gift from Dr. Cynthia Wolberger, Johns Hopkins University School of Medicine) 28 were expressed in BL21‐CondonPlus(DE3)‐RIL cells (Stratagen, San Diego, CA, USA) 29 at 28°C in 2 × YT medium containing 100 μg/mL kanamycin and 37 μg/mL chloramphenicol and then purified with nickel‐nitrilotriacetic acid resin (Qiagen, Valencia, CA, USA). The purity of the protein was verified more than 95% by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Coomassie staining 30.
Enzymatic Activity of Extracellular Visfatin
To determine the enzymatic activity of extracellular visfatin, the concentration of NMN, an enzymatic product of visfatin, was measured using a method reported previously 27. First, 10 μL of 20% acetophenone in dimethyl sulfoxide (DMSO) and 10 μL of 2 M KOH were added into 25 μL of samples. Then, the mixture was incubated in an ice bath for 2 min before the addition of 45 μL of 88% formic acid. After incubation at 37°C for 10 min, the solution was transferred into a flat‐bottom 96‐well black plate (Greiner, Frickenhausen, Germany). The fluorescence was measured using a Tecan Infinity M200 plate reader (Tecan Group, Durham, NC, USA), setting the excitation and emission wave‐lengths to 382 and 445 nm, respectively 31.
Primary Neuron and Glia Culture
Primary mouse neuronal cells were prepared from the cerebral cortex of neonatal animals within 12 h after birth as described previously 13, 17. For primary neuron culture, cortices were isolated, dissociated in sterile PBS and triturated into single‐cell suspensions. Cells were plated in 12‐well plate and cultured in DMEM containing 10% FBS overnight. On the second day, the cultures were replenished with Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen). Glial growth was suppressed by addition of 5‐fluoro‐2‐deoxyuridine and uridine (10 μM), yielding cultured cells with >90% neurons.
For primary mix glia culture, the mice were decapitated, and the cortices were isolated and dissociated in sterile PBS. Glia isolated from the cerebral cortex were triturated into single‐cell suspensions, plated in 12‐well and cultured in DMEM containing 10% FBS for 2 weeks. The mixed glia cultures were maintained in a humidified atmosphere containing 5% CO2 at 37°C and the medium was changed every 3 days. Experiments were performed when the mixed glia had been cultured for 12–14 days.
Conditioned Medium of Neurons and Glia
Neurons and glia were cultured in medium for 48 h. The medium was collected into 15‐mL tubes and concentrated around 10 times using Microcon YM‐10 with 10 kDa molecular‐weight centrifugal filters (Millipore). Then, visfatin in the medium of neurons and glia was detected by immunoblotting and enzyme immunoassay, respectively.
Oxygen‐Glucose Deprivation Model
Oxygen‐glucose deprivation (OGD) model (in vitro) was prepared in cultured neurons as described previously 32, 33. Primary mouse cortical neurons or glia were isolated from the cerebral cortex of neonatal mice within 6 h. To establish OGD conditions, the cultured neurons or glia (Day 7–14) were washed three times and incubated with glucose‐free Earle's balanced salt solution (EBSS) and placed for different times within a hypoxic chamber (Forma Scientific, Marietta, OH, USA) that was continuously flushed with 95% N2 and 5% CO2 at 37°C to obtain <0.5% O2. Control neurons or glia were placed in EBSS containing glucose (25 mM) and incubated under normal culture conditions for the same period (2 h for neuron and 8 h for glia) 34.
Treatments
Primary neurons and glia were cultured in 12‐well plates for cell viability or glass bottom dishes (Corning Costar Corp., Cambridge, MA, USA) for TUNEL assay. Cells were treated by recombinant visfatin (300 ng/mL) or H247A mutant visfatin (300 ng/mL) and then subjected to GD treatment (4 h for neurons and 8 h for glia). Then, cell viability or TUNEL assays were performed. For neutralizing antibody assay, the antibody (Santa Cruz Biotechnology, CA, USA; clone number: H‐300; catalogue: sc‐67020) against the amino acids 1‐300 mapping at the N‐terminus of visfatin was used. Antibody (1:300) plus recombinant visfatin (300 ng/mL) or antibody (300 μL/well) plus H247A mutant visfatin (300 ng/mL) were added into primary neurons and glia cultures. Cells were then subjected to OGD treatment.
Cell Viability Assay
Cell viability analysis was performed as described previously 22, 35. Cell viability was evaluated by a nonradioactive cell counting kit (CCK‐8) assay (Dojndo Laboratories) according to the manufacturer's instruction.
Immunofluorescence and TUNEL Staining
Immunofluorescence and TUNEL staining were performed as described previously 13, 17. Cultured cells were placed on confocal dish (Corning) and were fixed in 4% paraformaldehyde, blocked by 8% normal donkey serum, and incubated in specific primary antibodies (mouse anti‐Tuj‐1, 1:1000; mouse anti‐GFAP, 1:200). Then, the cells were incubated with corresponding Alexa 488‐conjugated secondary antibodies 36, 37 or immunofluorescent TUNEL reaction mixture for 1 h in box 38, 39. DAPI was used to stain nuclei 24, 40.
Immunoblotting
Immunoblotting for the secretary proteins in conditional medium was performed as described previously 30, 41. Samples were separated by 10% SDS‐PAGE, transferred to nitrocellulose membranes, and probed by rabbit polyclonal antibody against visfatin followed by secondary antibody.
Drug Treatment in SHR‐SP
Stroke‐prone spontaneously hypertensive rats were randomly divided into two groups. One group was given FK866 (1 mg/kg/day) 13, 15 via drinking water at the age of 6 months for lifelong treatment to determine the effect of visfatin inhibition on stroke and survival. The other group was given normal water control. Every rat enrolled in the result was necropsied to confirm stroke as described in our previous reports 42, 43.
Statistical Analysis
Data was expressed as mean ± SEM. Statistical differences were evaluated with t‐test or ANOVA method. P < 0.05 was regarded as statistically significant.
Results
Cerebral Ischemic Stress Increases Plasma Concentrations of Visfatin, NAD+ and ATP in vivo
We compared the level of plasma visfatin between pMCAO and sham‐operated mice. Plasma visfatin concentration in pMCAO mice was higher than those in sham‐operated mouse (26.8 ± 1.7 vs. 22.3 ± 1.2 ng/mL, P < 0.05, Figure 1A). Level of plasma NAD+, the enzymatic product of extracellular visfatin, was also higher in pMCAO mice compared with that in sham‐operated mouse (7.7 ± 2.3 vs. 5.8 ± 2.2 μM, Figure 1B). ATP is an essential activator of visfatin NAMPT enzymatic activity 28, 44. Bioluminescent assay showed that the mouse plasma ATP concentration was about 18.2 ± 3.7 nM (Figure 1C), which is similar to human plasma ATP concentration (~20–50 nM) reported recently 25, 45. Moreover, the plasma ATP concentration in pMCAO mice was significantly higher than that in sham‐operated mice (31.5 ± 4.2 vs. 18 ± 4 nM, P < 0.01, Figure 1C). These results suggest that plasma concentrations of visfatin, NAD+, and ATP were increased upon cerebral ischemic stress.
Figure 1.
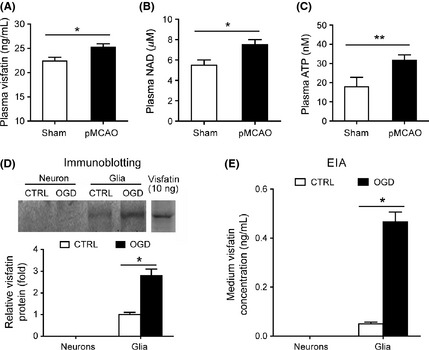
Mouse plasma concentrations of visfatin, NAD+, and ATP and secretion of visfatin from cultured glia but not neuron. (A) Plasma visfatin levels in sham‐operated and pMCAO mice. *P < 0.05 versus sham, n = 6. (B) Plasma NAD+ levels in sham‐operated and pMCAO mice. *P < 0.05 versus sham, n = 6. (C) Plasma ATP levels in sham‐operated and pMCAO mice. **P < 0.01 versus sham, n = 6. Sham, sham‐operated; pMCAO, permanent middle cerebral artery occlusion. (D) Conditional mediums of neuron and glia were collected, and the secreted visfatin was detected by immunoblotting. *P < 0.05 versus control, n = 4. Recombinant wild‐type mouse visfatin protein (10 ng) was also loaded as a positive control. (E) Conditional mediums of neuron and glia were collected, and the secreted visfatin was detected by EIA assay. *P < 0.05 versus control, n = 4.
Cerebral Ischemic Stress Enhances Secretion of Visfatin from Glia but not Neuron in vitro
As shown in Figure 1D, immunoblotting assay detected the released visfatin from cultured glia but not from neurons. Interestingly, the secretion of visfatin from glia was increased by OGD treatment (Figure 1D). EIA assay confirmed these results (Figure 1E). These results indicate that the neural cells may be influenced by local autocrined or paracrined visfatin under ischemic condition.
Extracellular Visfatin has NAMPT Enzymatic Activity
Based on the above‐mentioned results, we assume that the increased exogenous visfatin, which exists in circulating blood and local compartments, may affect neural survival under cerebral ischemia condition through regulating NAD+ level‐like intracellular visfatin. One key point in this notion is that whether the extracellular visfatin has NAMPT enzymatic activity. A previous work claimed that there was no or little ATP, an essential activator for the NAMPT enzymatic activity of visfatin, in extracellular spaces and thus extracellular visfatin cannot be enzymatic 44. We determined NAMPT enzymatic activity of extracellular visfatin using a fluorometric approach by converting nicotinamide mononucleotide (NMN), the direct enzymatic product of NAMPT, to a fluorescent derivative according to our previous work 27. The concentrations of mouse plasma NMN were 120 ± 22.4 nM (Figure 2A). As NMN is directly derived from NAMPT, these results imply that the extracellular blood visfatin may have NAMPT enzymatic activity.
Figure 2.
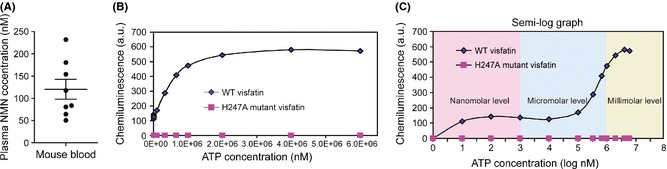
Determination of NAMPT enzymatic activity of extracellular visfatin. (A) Plasma NMN concentrations in mouse plasma. (B–C) Parabolic curve (B) and semi‐log graph (C) of NAMPT enzymatic activity of extracellular wild‐type (WT) and H247A‐mutant visfatin under different concentrations of ATP.
Next, we tested the influence of ATP concentrations on NAMPT enzymatic activity of extracellular visfatin using two kinds of exogenous visfatin protein: recombinant mouse wild‐type (WT) visfatin and H247A‐mutant enzymatic‐dead visfatin. As the ATP concentrations went up, the NMN levels went up: a typical parabolic curve was observed in WT visfatin group (Figure 2B). According to this curve, we obtained a semi‐log curve (Figure 2B). The fluorescent density kept at a low level when ATP concentrations were 10–105 nM (Figure 2C). When the ATP concentration was above 105 nM, the fluorescent density increased swiftly (Figure 2C). In line with the previous report 28, H247A‐mutant visfatin had no NAMPT enzymatic activity (Figure 2C). These results further support that extracellular visfatin has NAMPT enzymatic activity, although the NAMPT enzymatic activity of extracellular visfatin maintained at a relatively low level when the ATP concentration was under 105 nM.
Extracellular Visfatin is Neuroprotective in OGD Model
Our previous works demonstrated that intracellular visfatin is neuroprotective in cerebral ischemia 13, 17. We subjected primary cultured mouse neurons to OGD stress. OGD significantly increased the number of apoptotic TUNEL‐positive neurons (Figure 3A and B), and reduced the number of Tuj‐1‐positive neurons (Figure 3A and C). These effects were partly attenuated by exogenous recombinant WT visfatin treatment (Figure 3A–C). Exogenous visfatin treatment also partly blocked the harmful effect of OGD on neuron viability (Figure 3D). However, when recombinant H247A‐mutant visfatin was administrated, the neuroprotective effect was not observed (Figure 3A–D). We next studied the influence of exogenous visfatin treatment on glia in OGD model. Similarly, OGD significantly increased the number of apoptotic TUNEL‐positive glia (Figure 4A and B), reduced the number of GFAP‐1‐positive glia (Figure 4A and C) and decreased glial cell viability (Figure 4D), which were partly prevented by exogenous recombinant WT visfatin treatment but not recombinant H247A‐mutant visfatin (Figure 4A–D).
Figure 3.
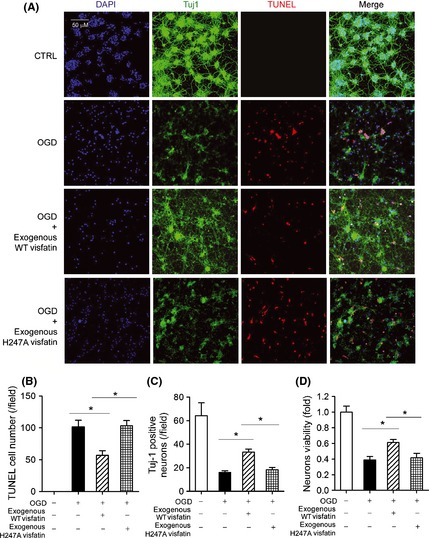
Effects of exogenous recombinant wild‐type (WT) visfatin (300 ng/mL) and H247A‐mutant visfatin (300 ng/mL) on neuron in OGD model. (A) Representative images of mediums of neuron in OGD model (2 h). Tuj‐1 (green) was a marker of neuron axon. TUNEL (red) was used to detect apoptosis. DAPI was used to stain nucleus. (B) Quantitative analysis of TUNEL‐positive apoptotic neurons. *P < 0.05, n = 6. (C) Quantitative analysis of Tuj‐1‐positive survival neurons. *P < 0.05, n = 6. (D) Cell viability assay on neurons. *P < 0.05, n = 6.
Figure 4.
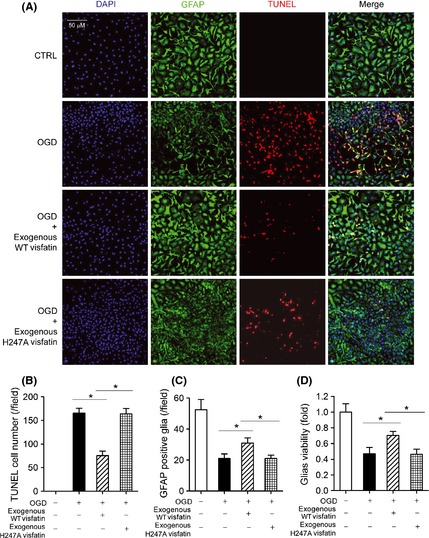
Effects of exogenous recombinant wild‐type (WT) visfatin (300 ng/mL) and H247A‐mutant visfatin (300 ng/mL) on glia in OGD model. (A) Representative images of mediums of glia in OGD model (8 h). GFAP (green) was a marker of glia. TUNEL (red) was used to detect apoptosis. DAPI was used to stain nucleus. (B) Quantitative analysis of TUNEL‐positive apoptotic glia. *P < 0.05, n = 6. (C) Quantitative analysis of GFAP‐positive survival glia. *P < 0.05, n = 6. (D) Cell viability assay on glia. *P < 0.05, n = 6.
Influence of Neutralizing Antibody on the Neuroprotective Effect of Extracellular Visfatin
We used neutralizing antibody of visfatin to determine whether it could block the effect of extracellular visfatin in cultured neural cells. As shown in Figure 5A, neutralizing antibody abolished the protective effect of extracellular visfatin on cell viability. However, TUNEL assay showed that it failed to block the antiapoptotic effect of extracellular visfatin (Figure 5B and C).
Figure 5.
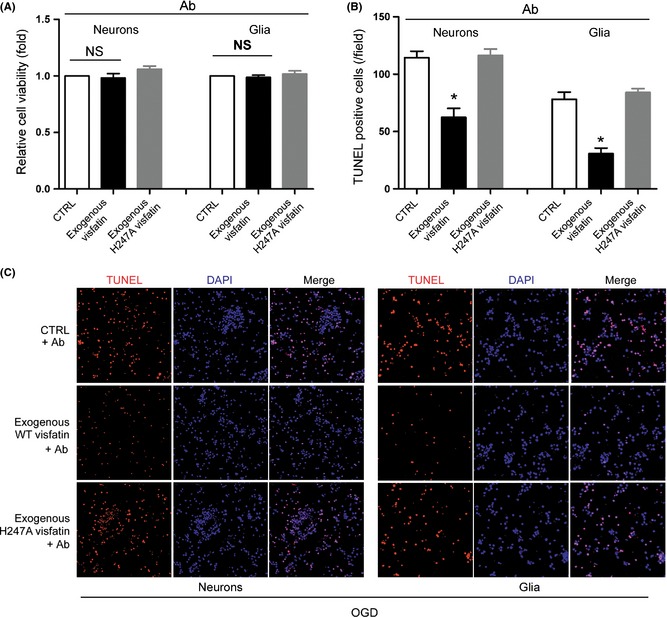
Influence of neutralizing antibody on the neuroprotective effect of extracellular visfatin. (A) Effect of neutralizing antibody on the protective effect of extracellular visfatin in cell viability. (B) and (C) Quantitative analysis (B) and representative images (C) of the effect of neutralizing antibody on the protective effect of extracellular visfatin in apoptosis. *P < 0.05 versus CTRL, n = 3. Ab, antibody (neutralizing). NS, no significance.
Decrease of Plasma Visfatin in SHR‐SP
We also measured the plasma visfatin levels in SHR‐SP, a generally accepted animal model of stroke. SHR‐SP die from stroke, ischemic stroke in a majority and hemorrhagic stroke in a minority, with an average lifespan of 45 weeks 42. The susceptibility to stroke in the SHR‐SP emerges at around 6 months old 46. Interestingly, we found there was no difference of plasma visfatin levels between 3‐month‐old SHR‐SP and control WKY animal (Figure 6A). However, the plasma visfatin levels in 6‐month‐old SHR‐SP were significantly lower than those in 6‐month‐old WKY rats (Figure 6A). To determine whether or not modulation of plasma visfatin levels influences the occurrence of stroke, SHR‐ SP rats were treated with the visfatin inhibitor FK866 from the age of 6 months on. Among the 25 rats studied, 20 animals that died from stroke were confirmed by displaying neurological symptoms of stroke and/or brain pathological examination (Figure 6B) as described in our previous reports 42, 43. FK866 significantly accelerated stroke occurrence and death in SHR‐SP (Figure 6C).
Figure 6.
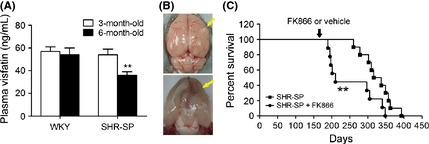
Decrease of plasma visfatin levels in SHR‐SP. (A) Comparison of plasma visfatin levels between SHR‐SP and control WKY rats (6‐month‐old and 3‐month‐old). **P < 0.01 versus WKY, n = 6. (B) Representative images for brain lesions (yellow arrow) of SHR‐SP with neurological signs. (C) Effect of visfatin inhibition (from 6‐month‐old) by FK866 on stroke occurrence and death in SHR‐SP, n = 20.
Discussion
In this study, we demonstrated that extracellular blood visfatin is enzymatic and protective for neuronal cells upon OGD stress like intracellular visfatin. Since the discovery of visfatin existence within and outside cell, how extracellular visfatin exerts its functions was studied intensively. A large number of works found that the extracellular visfatin has evident effects on many biological functions of cells. Extracellular visfatin was initially found to mimic the effects of insulin via binding insulin receptor 47. Although this function of visfatin was not confirmed by later work 9, extracellular visfatin was still reported to regulate insulin secretion in β cells as a systemic NAD+ biosynthetic enzyme 9. Extracellular visfatin activated migration, invasion, and tube formation in human umbilical vein endothelial cells through activating extracellular signal‐regulated kinase 1/2 (ERK1/2) signaling 48. Moreover, extracellular visfatin inhibited insulin‐like growth factor‐1 function in articular chondrocytes by activating the ERK1/2 pathway 49, and induced human endothelial vascular endothelial growth factor and matrix prometalloproteinase‐2/9 production via activating mitogen‐activated protein kinase and Akt kinase signaling pathways 50. It is widely believed that the diverse biological functions of extracellular visfatin are attributed to its NAMPT activity 51. Nicotinamide, a substrate for NAD+ synthesis, postpones stroke in stroke‐prone spontaneously hypertensive rats 52. However, Hara et al.44 recently showed that NMN, the enzymatic product of NAMPT, as well as ATP, the essential factor for NAMPT activity, were not observed in mouse plasma. Moreover, they found that only when ATP was present at millimolar levels, NAMPT efficiently catalyzed the NMN formation 44. They concluded that extracellular blood visfatin does not have NAMPT activity. As a result, how the extracellular visfatin functions remains to be a matter of debate.
ATP is an energy‐bearing molecule found in all living cells 53. In the present study, we measured the plasma levels of ATP using luciferin/luciferase assay and NMN based on a method generated by our laboratory previously 27. Our results are apparently in conflict with the results of Hara et al.44. We found that mouse plasma ATP concentration was 18 ± 4 nM and NMN was 120 ± 22.4 nM. In fact, ATP can be detected in human blood. Human venous plasma ATP concentrations were previously reported to be generally in the 1 μM range 54, 55, 56. Recently, Gorman and his colleague reported that human blood ATP concentration was about ~28 nM with a new method 25. Our result in mouse plasma (18 ± 4 nM) was rather close to this data. Importantly, we also demonstrated that extracellular visfatin had NAMPT enzymatic activity in this concentration range (Figure 3B and C). Additionally, we detected NAMPT product NMN (~120 nM) in mouse plasma (Figure 3A). These indicate that extracellular visfatin has enzymatic activity, and thereby support the concept of “NAMPT‐mediated systemic NAD+ biosynthesis” 9.
Another important finding of our study is that visfatin is a neural cells‐derived factor and extracellular visfatin is neuroprotective upon ischemic stimuli. Genetic deletion of visfatin is lethal in mice, indicating the importance of visfatin for life 9. Intracellular visfatin positively regulates the activity of SIRT1, a putative longevity protein 57, and exerts antiapoptotic and antiaging action in many cell types 57. In ischemic condition, intracellular visfatin has been shown to promote cell survival in cardiomyocytes 58 and neuronal cells 13, 14, 17. In this study, we, for the first time, demonstrate that visfatin can be released from glia (Figure 2), suggesting that visfatin is a neural cells‐derived factor. Moreover, we showed that extracellular recombinant WT visfatin, but not recombinant H247‐mutant enzymatic‐dead visfatin, protected against OGD‐induced cell injury in both neuron and glia. Thus, the NAMPT enzymatic activity is required for the neuroprotection of extracellular visfatin. When this manuscript was prepared, a recent report showed that cerebral ischemic injury in cultured neural cells was exacerbated by extracellular visfatin 59. In that paper, extracellular visfatin did not affect the cell viability of cultured neuron cells under normal or OGD conditions. However, in neuron‐glial mixed culture, either wild type or H247A mutant visfatin increased the percentage of necrotic cells and of necrotic neurons, which was prevented by TNF‐α neutralizing antibody 59. Our results are apparently in contrast with that study. The OGD time is the major difference between our work and that study 59. In our work, the OGD treatment on neurons was conducted for 4 h, whereas OGD was conducted only one hour in that study. The cell viability of neurons was only decreased about 15% by one hour OGD 59. In contrast, 4 h of OGD led to a more significant decrease of cell viability of neurons (~45%). This inconsistency may explain the discrepancy between the two works partly.
A previous report showed that blood visfatin concentration increased during ischemic stroke in human 19, as well as in mouse reported in this study. And, the secretion of visfatin from glia increased about 3~5 times under OGD stress (Figure 2). These results imply that extracellular visfatin may exhibit neuroprotective effect on ischemic neurons via two possible sources: endocrine (blood) and paracrine (glia). Interestingly, plasma visfatin concentrations decreased in 6‐month‐old but not 3‐month‐old SHR‐SP, and the susceptibility to stroke in the SHR‐SP emerges at around 6‐months old 46. This supports the notion that the decrease of plasma visfatin level in 6‐month‐old SHR‐SP may be associated with the onset of susceptibility to stroke in SHR‐SP.
There are some limitations in our study. Because we previously demonstrated that NMN, the enzymatic product of visfatin, was able to protect against ischemic injury in MCAO model 13, we thus did not show the neuroprotective effect of extracellular visfatin protein in this animal model. Alternatively, we tested the neuroprotective effect of extracellular visfatin in cultured cell model (neurons and glial cells), and used FK866 in SHR‐SP, a spontaneous stroke animal model. FK866, a chemical inhibitor of visfatin, is able to block NAMPT enzymatic activity of both extracellular and intracellular visfatin. To specifically block the extracellular visfatin, the best tool appears to use visfatin neutralizing antibody. However, application of visfatin neutralizing antibody only blocked the neuroprotective effect of extracellular visfatin on cell viability (CCK‐8 assay) but failed to affect the apoptosis (TUNEL assay). We did not know why there was a discrepancy between cell viability and apoptosis data. In addition, application of visfatin neutralizing antibody in SHR‐SP for long‐term observation (more than 3 months) is hard to achieve.
In conclusion, our results provide direct evidence that plasma extracellular visfatin has NAMPT enzymatic activity. Moreover, we found that glia, but not neuron, is able to secrete visfatin, which is further enhanced under ischemic stress. Administration of WT visfatin with NAMPT enzymatic activity, but not H247A‐mutant enzymatic‐dead visfatin, protects neuron and glia against ischemic stress in OGD model. These findings highlight the neuroprotective role of extracellular visfatin in ischemic stroke.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors wish to thank TopoTarget Switzerland SA. for providing FK866 for this study. This work was supported by grants from the National Natural Science Foundation of China (81100866 to P.W. and 81373414 to C.‐Y.M.), the National Basic Research Program of China (2009CB521902 to C.‐Y.M.), and the Shanghai Project (12ZZ078 to P.W. and 10GG19 to C.‐Y.M.)
The first two authors contributed equally to this work.
References
- 1. Langhorne P, Bernhardt J, Kwakkel G. Stroke rehabilitation. Lancet 2011;377:1693–1702. [DOI] [PubMed] [Google Scholar]
- 2. Reddy MK, Labhasetwar V. Nanoparticle‐mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia‐reperfusion injury. FASEB J 2009;23:1384–1395. [DOI] [PubMed] [Google Scholar]
- 3. Sun Y, Gui H, Li Q, et al. MicroRNA‐124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther 2013;19:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang P, Yang FJ, Du H, et al. Involvement of leptin receptor long isoform (LepRb)‐STAT3 signaling pathway in brain fat mass‐ and obesity‐associated (FTO) downregulation during energy restriction. Mol Med 2011;17:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao YH, Yuan B, Chen J, et al. Endothelial progenitor cells: Therapeutic perspective for ischemic stroke. CNS Neurosci Ther 2013;19:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dahl TB, Holm S, Aukrust P, Halvorsen B. Visfatin/NAMPT: A multifaceted molecule with diverse roles in physiology and pathophysiology. Annu Rev Nutr 2012;32:229–243. [DOI] [PubMed] [Google Scholar]
- 7. Rongvaux A, Galli M, Denanglaire S, et al. Nicotinamide phosphoribosyl transferase/pre‐B cell colony‐enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol 2008;181:4685–4695. [DOI] [PubMed] [Google Scholar]
- 8. Skokowa J, Lan D, Thakur BK, et al. NAMPT is essential for the G‐CSF‐induced myeloid differentiation via a NAD(+)‐sirtuin‐1‐dependent pathway. Nat Med 2009;15:151–158. [DOI] [PubMed] [Google Scholar]
- 9. Revollo JR, Korner A, Mills KF, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 2007;6:363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adya R, Tan B, Chen J, Randeva HS. Visfatin and endothelial angiogenesis. Cardiovasc Res 2012;96:223–226. [Google Scholar]
- 11. Samal B, Sun Y, Stearns G, et al. Cloning and characterization of the cDNA encoding a novel human pre‐B‐cell colony‐enhancing factor. Mol Cell Biol 1994;14:1431–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanaka M, Nozaki M, Fukuhara A, et al. Visfatin is released from 3T3‐L1 adipocytes via a non‐classical pathway. Biochem Biophys Res Commun 2007;359:194–201. [DOI] [PubMed] [Google Scholar]
- 13. Wang P, Xu TY, Guan YF, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1‐dependent adenosine monophosphate‐activated kinase pathway. Ann Neurol 2011;69:360–374. [DOI] [PubMed] [Google Scholar]
- 14. Zhang W, Xie Y, Wang T, et al. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab 2010;30:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang P, Miao CY. Autophagy in the disorders of central nervous system: Vital and/or fatal? CNS Neurosci Ther 2012;18:955–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wei K, Wang P, Miao CY. A double‐edged sword with therapeutic potential: An updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther 2012;18:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang P, Guan YF, Du H, et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012;8:77–87. [DOI] [PubMed] [Google Scholar]
- 18. Bi J, Li H, Ye SQ, Ding S. Pre‐B‐cell colony‐enhancing factor exerts a neuronal protection through its enzymatic activity and the reduction of mitochondrial dysfunction in in vitro ischemic models. J Neurochem 2012;120:334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu LF, Yang SS, Wang CP, et al. Elevated visfatin/pre‐B‐cell colony‐enhancing factor plasma concentration in ischemic stroke. J Stroke Cerebrovasc Dis 2009;18:354–359. [DOI] [PubMed] [Google Scholar]
- 20. Douglas G, Bendall JK, Crabtree MJ, et al. Endothelial‐specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(‐)/(‐) mice. Cardiovasc Res 2012;94:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruiz‐Meana M, Martinson EA, Garcia‐Dorado D, Piper HM. Animal ethics in cardiovascular research. Cardiovasc Res 2012;93:1–3. [DOI] [PubMed] [Google Scholar]
- 22. Wang P, Xu TY, Guan YF, et al. Perivascular adipose tissue‐derived visfatin is a vascular smooth muscle cell growth factor: Role of nicotinamide mononucleotide. Cardiovasc Res 2009;81:370–380. [DOI] [PubMed] [Google Scholar]
- 23. Chuang YL, Hsu CY. Changes in mitochondrial energy utilization in young and old worker honeybees (Apis mellifera). Age (Dordr) 2013;35:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang B, Zhang B, Liang P, et al. Nucleolin protects the heart from ischaemia‐reperfusion injury by up‐regulating heat shock protein 32. Cardiovasc Res 2013;99:92–101. [DOI] [PubMed] [Google Scholar]
- 25. Gorman MW, Feigl EO, Buffington CW. Human plasma ATP concentration. Clin Chem 2007;53:318–325. [DOI] [PubMed] [Google Scholar]
- 26. Rosc‐Schluter BI, Hauselmann SP, Lorenz V, et al. NOX2‐derived reactive oxygen species are crucial for CD29‐induced pro‐survival signalling in cardiomyocytes. Cardiovasc Res 2012;93:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang RY, Qin Y, Lv XQ, et al. A fluorometric assay for high‐throughput screening targeting nicotinamide phosphoribosyltransferase. Anal Biochem 2011;412:18–25. [DOI] [PubMed] [Google Scholar]
- 28. Wang T, Zhang X, Bheda P, et al. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat Struct Mol Biol 2006;13:661–662. [DOI] [PubMed] [Google Scholar]
- 29. Kronstein R, Seebach J, Grossklaus S, et al. Caveolin‐1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 2012;93:130–140. [DOI] [PubMed] [Google Scholar]
- 30. Thiagarajan PS, Yakubenko VP, Elsori DH, et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin‐1. Cardiovasc Res 2013;99:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cai WF, Pritchard T, Florea S, et al. Ablation of junctin or triadin is associated with increased cardiac injury following ischaemia/reperfusion. Cardiovasc Res 2012;94:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fan YY, Zhang XN, He P, et al. Transient lack of glucose but not O2 is involved in ischemic postconditioning‐induced neuroprotection. CNS Neurosci Ther 2013;19:30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Du CP, Tan R, Hou XY. Fyn kinases play a critical role in neuronal apoptosis induced by oxygen and glucose deprivation or amyloid‐beta peptide treatment. CNS Neurosci Ther 2012;18:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao J, Li YX, Hao YJ, et al. Effects of oxysophoridine on rat hippocampal neurons sustained oxygen‐glucose deprivation and reperfusion. CNS Neurosci Ther 2013;19:138–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun Q, Kawamura T, Masutani K, et al. Oral intake of hydrogen‐rich water inhibits intimal hyperplasia in arteri‐alized vein grafts in rats. Cardiovasc Res 2012;94:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim HJ, Myers R, Sihn CR, Rafizadeh S, Zhang XD. Slc26a6 functions as an electrogenic Cl‐/HCO3‐ exchanger in cardiac myocytes. Cardiovasc Res 2013;100:383–391. [DOI] [PubMed] [Google Scholar]
- 37. Ruggiero A, Chen SN, Lombardi R, Rodriguez G, Marian AJ. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc Res 2013;97:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Appukuttan A, Kasseckert SA, Kumar S, Reusch HP, Ladilov Y. Oxysterol‐induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc Res 2013;99:734–742. [DOI] [PubMed] [Google Scholar]
- 39. Kim SW, Kim HW, Huang W, et al. Cardiac stem cells with electrical stimulation improve ischaemic heart function through regulation of connective tissue growth factor and miR‐378. Cardiovasc Res 2013;100:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prigent‐Tessier A, Quirie A, Maguin‐Gate K, et al. Physical training and hypertension have opposite effects on endothelial brain‐derived neurotrophic factor expression. Cardiovasc Res 2013;100:374–382. [DOI] [PubMed] [Google Scholar]
- 41. Klawitter J, Agardi E, Corby K, et al. Association of DJ‐1/PTEN/AKT‐ and ASK1/p38‐mediated cell signalling with ischaemic cardiomyopathy. Cardiovasc Res 2013;97:66–76. [DOI] [PubMed] [Google Scholar]
- 42. Zhang W, Liu AJ, Yi‐Ming W, et al. Pressor and non‐pressor effects of sodium loading on stroke in stroke‐prone spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 2008;35:83–88. [DOI] [PubMed] [Google Scholar]
- 43. Liu AJ, Ma XJ, Shen FM, et al. Arterial baroreflex: A novel target for preventing stroke in rat hypertension. Stroke 2007;38:1916–1923. [DOI] [PubMed] [Google Scholar]
- 44. Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS ONE 2011;6:e22781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Farias M 3rd, Gorman MW, Savage MV, Feigl EO. Plasma ATP during exercise: Possible role in regulation of coronary blood flow. Am J Physiol Heart Circ Physiol 2005;288:H1586–H1590. [DOI] [PubMed] [Google Scholar]
- 46. Wang P, Tian WW, Song J, Guan YF, Miao CY. Deficiency of NG2+ cells contributes to the susceptibility of stroke‐prone spontaneously hypertensive rats. CNS Neurosci Ther 2011;17:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005;307:426–430. [DOI] [PubMed] [Google Scholar]
- 48. Kim SR, Bae SK, Choi KS, et al. Visfatin promotes angiogenesis by activation of extracellular signal‐regulated kinase 1/2. Biochem Biophys Res Commun 2007;357:150–156. [DOI] [PubMed] [Google Scholar]
- 49. Yammani RR, Loeser RF. Extracellular nicotinamide phosphoribosyltransferase (NAMPT/visfatin) inhibits insulin‐like growth factor‐1 signaling and proteoglycan synthesis in human articular chondrocytes. Arthritis Res Ther 2012;14:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP‐2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin‐induced angiogenesis. Cardiovasc Res 2008;78:356–365. [DOI] [PubMed] [Google Scholar]
- 51. Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol Metab 2009;20:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo JM, Dong WZ, Liu AJ, Cheng MH, Su DF. Nicotinamide postpones stroke in stroke‐prone spontaneously hypertensive rats. CNS Neurosci Ther 2012;18:267–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res 2012;95:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harkness RA, Coade SB, Webster AD. ATP, ADP and AMP in plasma from peripheral venous blood. Clin Chim Acta 1984;143:91–98. [DOI] [PubMed] [Google Scholar]
- 55. Born GV, Kratzer MA. Source and concentration of extracellular adenosine triphosphate during haemostasis in rats, rabbits and man. J Physiol 1984;354:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lader AS, Prat AG, Jackson GR Jr, et al. Increased circulating levels of plasma ATP in cystic fibrosis patients. Clin Physiol 2000;20:348–353. [DOI] [PubMed] [Google Scholar]
- 57. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004;279:50754–50763. [DOI] [PubMed] [Google Scholar]
- 58. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 2009;105:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhao B, Zhang M, Han X, et al. Cerebral ischemia is exacerbated by extracellular nicotinamidephosphoribosyltransferase via a non‐enzymatic mechanism. PLoS ONE 2013;8:e85403. [DOI] [PMC free article] [PubMed] [Google Scholar]