

Summary
Background
Demyelination is one of the most important pathological factors of spinal cord injury. Oligodendrocyte apoptosis is involved in triggering demyelination. However, fewer reports on pathological changes and mechanism of demyelination have been presented from compressed spinal cord injury (CSCI). The relative effect of oligodendrocyte apoptosis on CSCI‐induced demyelination and the mechanism of apoptosis remain unclear.
Aims
In this study, a custom‐designed model of CSCI was used to determine whether or not demyelination and oligodendrocyte apoptosis occur after CSCI. The pathological changes in axonal myelinated fibers were investigated by osmic acid staining and transmission electron microscopy. Myelin basic protein (MBP), which is used in myelin formation in the central nervous system, was detected by immunofluorescence and Western blot assays. Oligodendrocyte apoptosis was revealed by in situ terminal‐deoxytransferase‐mediated dUTP nick‐end labeling. To analyze the mechanism of oligodendrocyte apoptosis, we detected caspase‐12 [a representative of endoplasmic reticulum (ER) stress], cytochrome c (an apoptotic factor and hallmark of mitochondria), and inhibitor of DNA binding 2 (Id2, an oligodendrocyte lineage gene) by immunofluorescence and Western blot assays.
Results
The custom‐designed model of CSCI was successfully established. The rats were spastic, paralyzed, and incontinent. The Basso, Beattie, and Bresnahan (BBB) locomotor rating scale scores were decreased as time passed. The compressed spinal cord slices were ischemic. Myelin sheaths became swollen and degenerative; these sheaths were broken down as time passed after CSCI. MBP expression was downregulated after CSCI and consistent with the degree of demyelination. Oligodendrocyte apoptosis occurred at 1 day after CSCI and increased as caspase‐12 expression was enhanced and cytochrome c was released. Id2 was distributed widely in the white matter. Id2 expression increased with time after CSCI.
Conclusion
Demyelination occurred after CSCI and might be partly caused by oligodendrocyte apoptosis, which was positively correlated with ER–mitochondria interactions and enhanced Id2 expression after CSCI in rats.
Keywords: Compressed spinal cord injury, Demyelination, Oligodendrocyte apoptosis
Introduction
Compressed spinal cord injury (CSCI) has become a global issue because of vertebral fracture, spinal tuberculosis, ligamenta flava rigidification, tumor in marrow, epidural hematoma, ossification of the posterior longitudinal ligament, and disk protrusion 1, 2, 3, 4, 5. CSCI impairs motor and sensory functions insidiously and progressively. For these reasons, CSCI is considered as a serious disorder at personal and societal levels 2.
Demyelination is one of the most important pathological changes manifested in CSCI, and this condition has been reported since the beginning of the twentieth century 6, 7, 8, 9, 10, but individuals have not considered demyelination in CSCI for a long time. Demyelination after CSCI significantly contributes to neurological deficits, including disruptions in motor coordination, hind limb paralysis, spasticity, and incontinence 8, 9.
Until now, studies on demyelination have partially explained the mechanisms based on three general models of experimental central nervous system (CNS) demyelination: toxin‐induced model 11, 12, 13; virus‐induced model 14, 15; and autoimmune‐induced model (experimental autoimmune encephalomyelitis, EAE) 16, 17, 18. Fewer reports on demyelination incurred from CSCI have been published than numerous demyelination models induced by toxins, viruses, and EAE. Studies on the pathological mechanisms of demyelination related to CSCI have been impeded in the past by the lack of good in vivo models. In toxin‐, virus‐induced, and EAE models, diseases are caused by different mechanisms and have different patterns of clinical neurological deficits, pathologies, and repair mechanisms 19.
Oligodendrocytes are myelinating cells of the vertebrate CNS 20.These cells synthesize a myelin basic protein (MBP) on free polysomes in the cytoplasm 21, 22. MBP, the second most abundant protein in the CNS, presents a major dense line of myelin 23, 24, 25 and is essential for myelin formation 21. Apoptosis of oligodendrocytes can result in the breakdown of MBP and contribute to posttraumatic demyelination. MBP breakdown is a characteristic of demyelination. In general, apoptosis may be induced by toxins, protein synthesis inhibitors, several parasites, and pathogenic yeasts 26, 27, 28. During apoptosis, endoplasmic reticulum (ER)–mitochondria interactions are involved 29. Apoptosis can be further induced by the inhibitor of DNA binding 2 (Id2) of interleukin‐3 (IL‐3)‐dependent 32D.3 myeloid progenitors and U2OS osteosarcoma cells 30. Id2 is one of the oligodendrocyte lineage genes belonging to the family of helix‐loop‐helix (HLH) transcription factors that regulate MBP negatively in normal animals 31, 32, 33. However, studies about the relative effect of oligodendrocyte apoptosis on CSCI‐induced demyelination are scarce. Furthermore, the mechanisms of oligodendrocyte apoptosis remain unclear. This study aimed to investigate whether or not demyelination occurs and identify the pathological changes in demyelination after CSCI. We also identified the relative effect of oligodendrocyte apoptosis on demyelination, the possible changes in Id2 expression, and ER–mitochondria interactions involved in oligodendrocyte apoptosis following CSCI.
Materials and methods
A total of 108 Sprague‐Dawley rats weighing 250 g to 320 g were provided by the Experimental Animal Center of Chongqing Medical University. Approval and consent for our study were obtained from our institution's Animal Care and Ethics Committee and formulated by the Ministry of Science and Technology of the People's Republic of China. The rats were housed in a temperature‐controlled room (24°C) with a 12:12 dark/light cycle. These rats were given food and water ad libitum. They were randomly divided into three groups: normal (n = 24), sham (n = 24), and CSCI (n = 60). We investigated the relative mechanisms only at acute stages because demyelinated axons do not persist for extended periods of time in animal models 34. Hence, the rats were sacrificed at intervals of 1, 3, and 7 days.
Surgical Procedure
Compressed Spinal Cord Injury Group
The rats were fasted 24 h prior to surgery, anesthetized by intraperitoneal injection with 3.5% chloral hydrate, and subjected to continuous oxygen inhalation. The rats were then fixed prone on a surgical table. A limited laminectomy was performed at L1. The anterior and the posterior articular processes were excised without damaging the spinal cord. A small rectangular stainless steel board (3 mm × 2 mm) was placed in the center of the spinal cord surface at L1. A custom‐made swallow tail‐like stainless steel fixation was positioned between T12 and L2. The center of the stainless steel fixation was inserted using a small screw (2 mm in diameter and 2 mm long with a flat and smooth screw tip) slowly and vertically until the front end of the screw reached the small rectangular stainless steel board to establish a rat model showing a compressed spinal cord. The incision was then gradually sutured in layers. The rats were allowed to recover from anesthesia and housed as described previously. The screw was maintained on the dura mater for 1, 3, and 7 days.
Sham Group
The rats were subjected to laminectomy without compression.
Normal Group
The rats were not subjected to any experimental procedure.
Neurological Function Assessment
All of the rats were evaluated based on the Basso, Beattie, and Bresnahan (BBB) locomotor rating scale in an open field by two independent observers in a double‐blind manner according to the indications in the previously published articles 35. A total of 21 scores from 0 (complete paralysis) to 21 (normal gait) were recorded at 1, 3, and 7 days.
Evaluation of Infarcted Zone by TTC Staining
At 1, 3, and 7 days, the rats were deeply anesthetized and sacrificed after CSCI. The spinal cord tissues were removed rapidly and sliced coronally at 2‐mm intervals. All of the slices were incubated in 2% 2,3,5‐triphenyltetrazolium chloride (TTC; TaKaRa, China) at 37°C for 20 min and fixed in 4% paraformaldehyde solution for 24 h. The undamaged spinal cord tissue appeared bright red; the infarcted zones appeared white or pale.
Tissue Processing for Osmic Acid Staining, Ultrastructure Observation, TUNEL Staining, and Immunofluorescence
At 1, 3, and 7 days after CSCI, the rats from different groups were anesthetized. Physiological saline (250 mL; 4°C) and 4% paraformaldehyde in 0.1 mol/L phosphate‐buffered saline (PBS 500 mL; 4°C; pH 7.4) were perfused into the left ventricle. The spinal cord sections from L1 (1 cm) were harvested and assigned to three segments. The first segment was fixed with 4% paraformaldehyde for 24 h to 48 h (4°C) and dehydrated with 30% sucrose overnight (4°C). The samples were then cut into serial sections at a thickness of 10 μm for TUNEL and immunofluorescence methods. The second segment was used for ultrastructure observation. The third segment was fixed with 4% paraformaldehyde again for osmic acid staining.
Osmic Acid Staining of the Myelin Sheath
The specimens were removed from 4% paraformaldehyde and rinsed with 0.01 mol/L PBS (pH 7.4) thrice for 20 min each. The specimens were then transferred to 1% osmic acid for 5 days or so (not less than 3 days or longer than 7 days). After this period, the specimens were rinsed again with 0.01 mol/L PBS (pH 7.4) and stored in 75% ethanol. Afterward, these specimens were dehydrated by gradient alcohol series and cut into sections with a thickness of 10 μm using a conventional microtome (Leica CM1900, Leica Camera AG, Oskar‐Barnack, Germany). Five micrographs from the posterior funiculus of each section were obtained under an Olympus microscope with an objective lens of 40× and an Olympus camera (Olympus‐45, Tokyo, Japan) with a magnification lens of 400×. These micrographs were further analyzed by a blinded investigator. ImagePro Plus (IPP; Media Cybernetics, Inc., Rockville, MD, USA) analysis software was used to measure the total number of myelinated fibers from the posterior funiculus of each section.
Transmission Electron Microscopy
A portion of each specimen from the rats perfused with 4% paraformaldehyde was immediately fixed in 2.5% glutaraldehyde for 2 days, postfixed in 2% osmic acid for 1 h, serially dehydrated in alcohol and propylene oxide, and embedded in araldite for morphometric analyses of the semithin sections. Ultrathin sections mounted on copper grids were stained with uranyl acetate and lead citrate. Images with a magnification of 15,000× were obtained using a transmission electron microscopy (TEM; Hitachi‐7500, Hitachi Ltd., Tokyo, Japan). These images were analyzed using the MIAS image analysis system to determine the ratio of myelin thickness to axon diameter (G‐ratio) 36.
Immunofluorescence
To identify MBP expression, caspase‐12 and CNPase (mature oligodendrocyte marker) coexpression, cytochrome c and CNPase coexpression, caspase‐12 and cytochrome c coexpression, Id2 and CNPase coexpression in the spinal cord sections, we used the primary antibodies listed in Table 1.
Table 1.
Antibodies used in immunofluorescence in this study
| Antibody name | Manufacturer | Dilution | Host | Labeling |
|---|---|---|---|---|
| Myelin basic protein (MBP) | Santa Cruz, USA | 1:200 | Mouse | Myelin |
| Caspase‐12 | Santa Cruz, USA | 1:200 | Goat | Caspase‐12 |
| Cytochrome c | Santa Cruz, USA | 1:200 | Rabbit | Cytochrome c |
| Inhibitor of DNA binding 2 (Id2) | Santa Cruz, USA | 1:50 | Goat | An oligodendrocyte lineage gene |
| CNPase | Abcam, Cambridge, UK | 1:200 | Mouse | Mature oligodendrocyte |
Tissue sections were rewarmed, rinsed, blocked with 5% donkey serum (Jackson ImmunoResearch, Lancaster, PA, USA) for 1 h at 37°C in a humidified atmosphere, and incubated with the primary antibodies overnight (4°C). These tissue sections were rinsed again with 0.01 mol/L PBS and incubated with secondary antibodies (mouse, goat, or rabbit IgG conjugated with Alex647/594/488, 1:200, Jackson ImmunoResearch) for 1 h at 37°C in a humidified atmosphere in the dark. To facilitate proper photographic orientation, we counterstained the sections with a nuclear dye (4′,6‐diamidino‐2‐phenylindole, 1:20; Bestbio Inc., China) for 5 min. The tissue sections were then washed and mounted in 50% glycerol dissolved in PBS. The samples were observed under a confocal microscope (Leica TCS SP2, Germany). Control samples were prepared by omitting either one or both primary antibodies. All of the control samples yielded negative results without detectable labeling.
TUNEL Staining and Apoptotic Cell Quantification
Tissue sections were blocked with 5% donkey serum (Jackson ImmunoResearch) for 1 h at 37°C in a humidified atmosphere and incubated with a primary antibody against CNPase overnight (4°C). These tissue sections were rinsed with 0.01 mol/L PBS and incubated with fluorescein‐labeled secondary antibodies (mouse IgG conjugated with Alex647; Jackson ImmunoResearch) for 1 h at 37°C in a humidified atmosphere. The sections were then washed and incubated with a TUNEL reaction mixture (one‐step TUNEL apoptosis assay kit; Beyotime Institute of Biotechnology, Jiangsu, China) for 60 min at 37°C in a humidified atmosphere in the dark. The slides were rinsed thrice with 0.01 mol/L PBS to terminate the reaction. After the slides were washed, the tissue sections were mounted in 50% glycerol dissolved in 0.01 mol/L PBS. The samples were observed under a confocal microscope (Leica TCS SP2). All of the digital images were captured in a double‐blind manner from four random fields per section of the injured epicenter of the cross sections in rats. The images were obtained at 20× magnification for TUNEL‐positive oligodendrocyte counting. We counted the digital images of TUNEL‐positive oligodendrocytes by using ImageJ software (National Institutes of Health [NIH], Bethesda, MD, USA). The average values of four random fields were obtained to provide single value per rat. The results were expressed as the number of TUNEL‐positive oligodendrocytes.
Western Blot
Tissue specimens were homogenized at 4°C in a homogenization buffer containing 50 mmol/L ethylenediaminetetraacetic acid, 2 μg/mL of leupeptin, 2 μg/mL of pepstatin A, 2 mmol/L phenylmethylsulfonyl fluoride, and 200 KIE/mL aprotinin. The homogenates were then centrifuged at 10,000 × g for 20 min at 4°C. The supernatant was collected, and protein concentration was determined using a Bradford assay kit (Bio‐Rad, Hercules, CA, USA). The samples were separated by 12% SDS‐PAGE and then transferred to polyvinylidene difluoride membrane. Blotted membranes were blocked with 5% skim milk. For immunoblotting, the following primary antibodies were used: polyclonal goat anticaspase‐12 antibody (1:500; Santa Cruz Biotechnology, Inc., Dallas, TX , USA); a monoclonal rabbit anticytochrome c antibody (1:500; Santa Cruz); a polyclonal goat anti‐Id2 antibody (1:500; Santa Cruz); and a monoclonal mouse anti‐MBP (1:2000; Santa Cruz). The secondary antibodies used were alkaline phosphatase–conjugated anti‐IgG antibodies (Santa Cruz). Immunoreactive bands were visualized using a chemiluminescent substrate (Pierce Inc., Rockford, IL, USA). We quantified Western bands by gel densitometry (Bio‐Rad). We divided the value of an individual protein band by the value of beta‐actin of the same sample. A ratio of protein–beta‐actin for each sample was obtained (each point was repeated in triplicate).
The bands were normalized with a beta‐actin loading control, and each group was normalized to the ratio of the corresponding sham control for analysis.
Statistical Analysis
Statistical analyses were performed using SPSS 11.0 software package (Chicago, IL, USA). Caspase‐12, cytochrome c, MBP, and Id2 protein expression levels for the specimens were expressed as means ± SD. Differences between individual groups were initially compared using one‐way ANOVA. Data were then analyzed with LSD multiple‐comparison post hoc test. All of the reported P values were two‐sided, and P < 0.05 was considered statistically significant.
Results
Neurological Function Assessment and TTC Staining after CSCI
To validate the custom‐designed model of CSCI as a suitable model system for functional studies of clinical CSCI, we performed BBB and TTC staining. We found that the rats in the CSCI group became spastic, paralyzed, and incontinent. The mean BBB scores in the CSCI group were lower than those in normal and sham groups (CSCI group vs. normal group P = 0.000 or P < 0.05; CSCI group vs. sham group P = 0.000 or P < 0.05). The BBB scores of the rats were remarkably decreased at 1 day and continued to decrease with time until the minimum score was reached at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05, Figure 1). TTC staining was performed to evaluate the infarction of the spinal cord after compressed injury. After compressing, spinal cord slices were divided into two regions, the white region, which was ischemic, and the red region, which was normal based on the TTC staining. The white border expanded along with the duration of time (Figure 2A–E). TTC is a colorless salt that can be converted to a red formazan product in the presence of a functioning mitochondrial electron transport chain. Thus, a normal spinal cord tissue was dyed to become bright red; the infarcted region appeared white.
Figure 1.
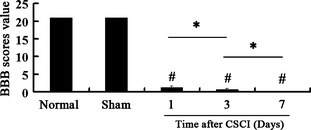
Neurological function assessment. Locomotor scores assessed based on the Basso, Beattie, and Bresnahan (BBB) rating scale. Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD post hoc tests where appropriate. # P < 0.05, compared with the sham group; *P < 0.05, compared with Compressed spinal cord injury (CSCI) group.
Figure 2.
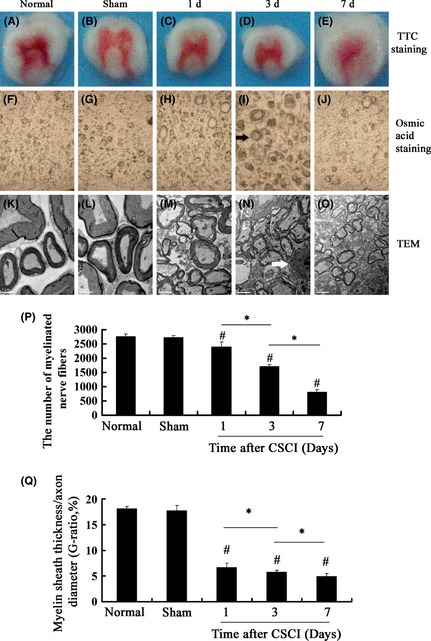
Compressed spinal cord injury (CSCI)‐induced ischemia of the spinal cord was detected by triphenyltetrazolium chloride (TTC) staining, and the pathological changes in myelinated nerve fibers at the posterior funiculus in the white matter were detected by osmic acid staining and transmission electron microscopy (TEM). (A–E) A representative image showing TTC staining of a spinal cord section, which was divided into two regions: white border and red core region from the rats at 1, 3, and 7 days after CSCI. (F–J): Representative images (×400) showing osmic acid staining. (K–O) The ultrastructural features of myelinated nerve fibers at the posterior funiculus in the white matter, scale bar: 1 μm. ( ): The degenerated nerve fiber; (
): The degenerated nerve fiber; ( ): oligodendrocytes. (P) The number of myelinated axons at the corresponding time after CSCI was remarkably lower than that of normal and sham groups. The number of myelinated fibers reached the minimum at 7 days after CSCI. (Q) The G‐ratios in 3 CSCI groups were lower than the sham group and reached the minimum on the 7th day after injury. Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD
post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group.
): oligodendrocytes. (P) The number of myelinated axons at the corresponding time after CSCI was remarkably lower than that of normal and sham groups. The number of myelinated fibers reached the minimum at 7 days after CSCI. (Q) The G‐ratios in 3 CSCI groups were lower than the sham group and reached the minimum on the 7th day after injury. Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD
post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group.
Demyelination after CSCI
Pathological changes in axonal myelinated fibers were investigated by osmic acid staining and TEM to determine whether or not demyelination occurs after CSCI. MBP was also detected by immunofluorescence and Western blot assays, given that MBP is one of the major myelin proteins, and MBP breakdown is a characteristic of demyelination.
Under a light microscope, darkly stained myelin sheaths were intact and clearly visible with the background showing little or no staining in the normal group, but were a little swollen in the sham group (Figure 2F,G). Degeneration of myelinated nerve fibers occurred immediately at 1 day after compression (Figure 2H). The nerve fibers became swollen, the space between fibers were reduced, and the number of myelinated axons was remarkably decreased compared with the normal group (1‐day CSCI group vs. normal group, P = 0.00 or P < 0.05; Figure 2P). At 3 days after compression, the degeneration of myelinated nerve fibers became more severe than that in the normal group (Figure 2I). At 7 days after compression, numerous fibers were lost (Figure 2J) and the number of myelinated nerve fibers reached the minimum (7‐day CSCI group vs. normal group, P = 0.000 or P < 0.05; 7‐day CSCI group vs. 1‐day CSCI group, P = 0.000 or P < 0.05; 7‐day CSCI group vs. 3‐day CSCI group, P = 0.000 or P < 0.05; Figure 2P).
We investigated the ultrastructural features of myelinated nerve fibers at the posterior funiculus of the white matter by TEM and measured the G‐ratio after CSCI with the duration of time. In the normal group, extensively distributed myelinated fibers were routinely observed. The myelinated axons showed a normal axoplasm with well‐preserved cellular structures such as intact microtubules, normal mitochondria, and appropriately oriented neurofilaments. The multiple layers of myelin sheaths were also noted in the normal group: dense major line and intraperiod line were preserved (Figure 2K). The ultrastructural features of fibers in the sham group were normal except a few myelin sheaths that became swollen (Figure 2L). At 1 day after CSCI, the myelinated axons became swollen and exhibited an axoplasm with a reduced neurofilament space, and the myelin sheaths were swollen as well (Figure 2M). At 3 days after compression, the axons showed an abnormal axoplasm with a reduced axonal space. In the axoplasm, the mitochondria became swollen and enlarged; low mitochondrial density was observed in the cross sections. The layers of the myelin sheath shrank, folded, and wrinkled exhibiting an onion‐like appearance. The myelin lamellae were disorganized and loose. Oligodendrocytes exhibited chromatin condensation, indicating oligodendrocyte apoptosis (Figure 2N) 4. The ultrastructural features of myelinated nerve fibers at 7 days after CSCI showed that the axons contained an abnormal axoplasm with reduced organelles, such as varying degrees of neurofilaments and microtubules. The mitochondria were disintegrated. Infiltrating macrophages were identified as large, darkly stained, and debris‐laden cells. The myelin sheaths became thin (Figure 2O). The G‐ratios in 3 CSCI groups were lower than those in the sham group and reached the minimum on the 7th day after injury, and the difference was statistically significant (7‐day CSCI group vs. normal group, P = 0.000 or P < 0.05; 7‐day CSCI group vs. 1‐day CSCI group, P = 0.000 or P < 0.05; 7‐day CSCI group vs. 3‐day CSCI group, P = 0.000 or P < 0.05, Figure 2Q).
Considering that demyelination occurred after CSCI, we determined MBP expression. Single‐fluorescent immunostaining using antibodies against MBP was performed, and it revealed that MBP immunoreactivity was affected mostly by myelin sheaths in the sham group (Figure 3A). However, low immunoreactivity was detected for MBP after CSCI (Figure 3B).
Figure 3.
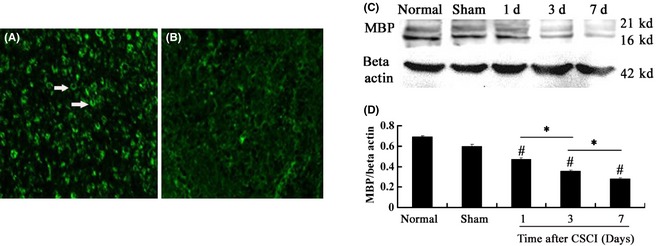
(A, B) Immunofluorescence of MBP in the posterior funiculus of the white matter. Scale bar = 75 μm. (C, D) MBP expression at the corresponding time examined by Western blot. MBP level in the compressed region at the corresponding time was significantly lower than that in normal and sham groups and reached the minimum at 7 days after compressed spinal cord injury (CSCI). Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD
post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group. (): MBP, myelin basic protein.
Western blot was performed to analyze the MBP expression quantitatively and detect the MBP levels in normal, sham, and CSCI specimens. The MBP expression in the CSCI group was significantly lower than that in the normal group and the sham group (CSCI group vs. normal group, P = 0.000, P < 0.05; CSCI group vs. sham group, P = 0.000, P < 0.05). This minimum expression level was reached at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000, P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; Figure 3C,D).
Oligodendrocyte Apoptosis after CSCI
An oligodendrocyte spiral wrapping produces myelin sheaths that function as a protective layer of axons and facilitates the conduction velocity of an electrical impulse. Demyelination, hypomyelination, or delayed myelination occurs if oligodendrocytes are damaged 37, 38, 39. To investigate whether or not oligodendrocyte apoptosis occurs and the spatial and temporal features of oligodendrocyte apoptosis are delineated after CSCI, we examined the white matter at the lesion site in different groups by double‐labeling experiments combining TUNEL staining and immunofluorescence. TUNEL labeling of oligodendrocytes at the lesion site (immunofluorescence staining for oligodendrocytes using CNPase antibody) revealed oligodendrocyte apoptosis. The results exhibited that few TUNEL‐positive oligodendrocytes were randomly distributed in normal and sham spinal cords (Figure 4A). The number of TUNEL‐positive oligodendrocytes at the corresponding time after CSCI was significantly higher than that in normal and sham groups (CSCI group vs. normal group, P = 0.000 or P < 0.05; CSCI group vs. sham group, P = 0.000 or P < 0.05). These TUNEL‐positive oligodendrocytes reached the maximum at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05, Figure 4C). The TUNEL‐positive oligodendrocytes were widely distributed in the white matter at the lesion site (Figure 4B).
Figure 4.
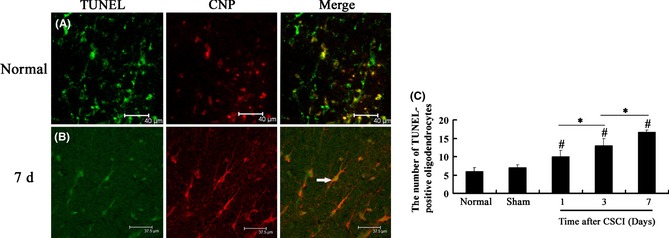
(A, B) TUNEL‐positive oligodendrocyte distribution in the white matter of the spinal cord in different groups by TUNEL staining. Scale bar = 40 μm. (C) The number of TUNEL‐positive oligodendrocytes at the corresponding time examined after compressed spinal cord injury (CSCI) was significantly higher than that in normal and sham groups. This number reached the maximum at 7 days after CSCI. Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD
post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group. (): TUNEL‐positive oligodendrocytes.
ER–Mitochondria Interactions and Id2 Expression Involved in Oligodendrocyte Apoptosis
Endoplasmic reticulum–mitochondria interaction mediates apoptotic cell death 40. To investigate the apoptotic pathway of oligodendrocytes, we examined the function of ER stress by assessing caspase‐12, which corresponds to ER stress, and mitochondrial dysfunction by assessing cytochrome c, which indicates mitochondrial involvement. We determined the localization of caspase‐12 by double‐labeling immunofluorescence using antibodies against caspase‐12 and CNPase, an oligodendrocyte marker. The results revealed that the majority of caspase‐12‐immunoreactive cells in the white matter of the normal group were oligodendrocytes (Figure 5A). Furthermore, the distribution of caspase‐12‐immunoreactive oligodendrocytes expanded at 1 day after CSCI and was found in the whole white matter with the duration of time (Figure 5B). We further performed Western blot to analyze caspase‐12 protein expression quantitatively and detect caspase‐12 protein levels in normal, sham, and CSCI specimens. Caspase‐12 protein expression in CSCI group at the corresponding time was significantly higher than that in normal and sham groups (CSCI group vs. normal group, P = 0.000 or P < 0.05; CSCI group vs. sham group, P = 0.000 or P < 0.05) and reached the maximum at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; Figure 5G,H).
Figure 5.
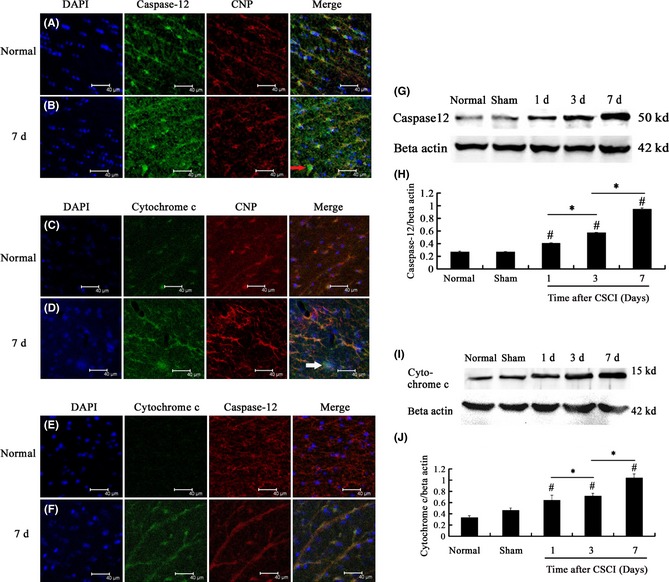
(A, B) Double‐labeling immunofluorescence of caspase‐12 and CNPase, an oligodendrocyte marker, in the white matter of the spinal cord of rats in different groups. Scale bar = 40 μm. (C, D) Double‐labeling immunofluorescence of cytochrome c and CNPase in the white matter of rats in different groups. Scale bar = 40 μm. (E, F) Double‐labeling immunofluorescence of caspase‐12 and cytochrome c in the white matter of the spinal cord at the lesion. Scale bar = 40 μm. (G, H) Caspase‐12 protein expression at the corresponding time examined by Western blot. Caspase‐12 protein expression at the corresponding time in the compressed spinal cord injury (CSCI) group was significantly higher than that in normal and sham groups and reached the maximum on the 7th day after CSCI. (I, J) Cytochrome c protein expression at the corresponding time examined by Western blot. Cytochrome c protein expression at the corresponding time in CSCI group was significantly higher than that in normal and sham groups. This expression reached the maximum at 7 days after CSCI. Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD
post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group. ( ): Caspase‐12‐positive oligodendrocytes. (): Cytochrome c‐positive oligodendrocytes.
): Caspase‐12‐positive oligodendrocytes. (): Cytochrome c‐positive oligodendrocytes.
In addition to ER stress pathway, mitochondrial dysfunction is an important pathway in apoptosis 41. Cytochrome c is an apoptotic factor released from the mitochondria and has also been observed in ER stress‐induced apoptosis of several cell lines, including mouse embryonic fibroblast cells 42. To investigate whether or not cytochrome c is required during oligodendrocyte apoptosis and can be observed in oligodendrocytes, we examined the localization of cytochrome c by double‐labeling immunofluorescence using antibodies against cytochrome c and CNPase. In normal and sham groups, a small number of cytochrome c‐immunoreactive oligodendrocytes were observed (Figure 5C). However, the distribution of cytochrome c‐immunoreactive oligodendrocytes significantly expanded over time after CSCI (Figure 5D). Western blot results also showed that cytochrome c expression at the corresponding time in the CSCI group was significantly higher than that in the normal and sham groups (CSCI group vs. normal group, P = 0.000 or P < 0.05; CSCI group vs. sham group, P = 0.000 or P < 0.05). This expression reached the maximum at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; Figure 5I,J).
Studies have focused on the functional interaction between ER and mitochondria for several decades. ER stress and mitochondrial dysfunction can apparently function together to activate caspase‐3, a caspase that induces apoptosis 29, 40. To detect the interaction between ER stress and mitochondrial dysfunction in the regulation of oligodendrocyte apoptosis after CSCI, we determined the localization of caspase‐12 and cytochrome c by double‐labeling immunofluorescence using antibodies against caspase‐12 and cytochrome c. The results revealed that caspase‐12 and cytochrome c immunoreactivities corresponded to oligodendrocyte apoptosis in the white matter (Figure 5E,F).
Id2 promotes cell death in IL‐3‐dependent 32D.3 myeloid progenitors and U2OS osteosarcoma cells 30. Id2 also functions in myelination and terminal maturation of oligodendrocytes. Furthermore, Id2 regulates MBP negatively and modulates the oligodendrocyte developmental timer that couples the proliferation of oligodendrocyte precursor cells to leave the cell cycle and the early oligodendrocyte terminal differentiation in normal animals 32. To investigate whether or not Id2 is involved in oligodendrocyte apoptosis and negatively regulates MBP after CSCI, we determined the characteristics of Id2 protein in oligodendrocytes in the white matter subjected to CSCI. We also identified the localization of Id2 by double‐labeling immunofluorescence using antibodies against Id2 and CNPase. The results revealed that Id2 was colocalized with oligodendrocyte‐positive profiles. Id2‐immunoreactive oligodendrocytes were randomly distributed in the white matter of the normal group (Figure 6A) and expanded at 1 day after CSCI. Id2‐immunoreactive oligodendrocytes were found in the whole white matter at 7 days after CSCI (Figure 6B). Western blot was performed to detect the levels of Id2 proteins in normal, sham, and CSCI specimens. The Id2 protein level in the compressed region at the corresponding time was significantly higher than that in normal and sham spinal tissues (CSCI group vs. normal group, P = 0.000 or P < 0.05; CSCI group vs. sham group, P = 0.000 or P < 0.05). The maximum Id2 protein level was reached at 7 days after CSCI (1‐day CSCI group vs. 7‐day CSCI group, P = 0.000 P < 0.05; 3‐day CSCI group vs. 7‐day CSCI group, P = 0.000 or P < 0.05; Figure 6C,D).
Figure 6.
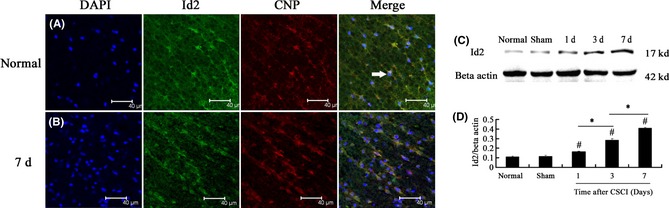
(A, B) Double‐labeling immunofluorescence of Id2 and CNPase, an oligodendrocyte marker, in the white matter of the spinal cord of rats in different groups. Scale bar = 40 μm. (C, D) Id2 protein expression at the corresponding time examined by Western blot. Id2 protein level in the compressed region at the corresponding time was significantly higher than that in normal and sham groups. Id2 expression reached the maximum at 7 days after compressed spinal cord injury (CSCI). Data represent mean ± SEM (n = 3 per group). Two‐way ANOVA was performed followed by LSD post hoc tests where appropriate. #
P < 0.05 compared with the sham group; *P < 0.05, compared with CSCI group. (): Id2‐positive oligodendrocytes.
Discussion
Compressed spinal cord injury causes sequential pathological changes, including ischemia 43, edema 44, immune cell transmigration 45, loss of neurons 43, 46, 47, infiltration of peripheral inflammatory cells, and activation of astrocytes and microglia 48 along with demyelination 46, 49, axonal degeneration 46, or “spongioform” degeneration in the white matter.
Neuronal loss in a compressed spinal cord is considered as significant evidence of profound and irreversible motor paresis, which has been observed in autopsy cases 50, 51 and several animal models 51, 52. Dysfunction of neuronal soma and its consecutive degeneration in the course of neurological disorders have dominated the perception in neuropathology. This conception has been accepted for many years 53. Information about demyelination after spinal cord injury has been scarcely reported compared with neuronal loss, although early pathological studies suggested that demyelination functions in CSCI pathology 10. Emerging evidence also suggests that demyelination is an important pathological factor of spinal cord injury 6. Given that myelin is important in normal nerve impulse conduction, the loss of myelin sheaths around axons may significantly contribute to neurological deficits after CSCI 7.
In general, individuals consider the pathological characteristics of spinal cord injury because of toxin‐ and virus‐induced as well as EAE demyelination. By contrast, very little is known about the pathological characteristics of demyelination induced by mechanical spinal cord injury. A suitable model of the morphology and mechanism studies of clinical CSCI is seldom provided. Therefore, a custom‐designed model of CSCI was used in the study to define the pathological characteristics of CSCI. This model could provide a basis for future research designed to determine mechanisms of demyelination caused by CSCI. In this study, the neurological function was assessed to investigate quantitatively the disease progression in rats after CSCI. BBB locomotor rating scale sensitively detected neurological functions, indicating the motor impairments that occurred at the onset of acute phase in CSCI. The hindlimbs of the rats in CSCI group became spastic and paralyzed. Their motor functions were abnormal. Incontinence appeared as early as 1 day and then became aggravated progressively over time. All of the neurological deficits were consistent with the manifestation of clinical CSCI. In addition, the TTC results revealed that the infarction of the spinal cord after compressed injury expanded with time. Thus, BBB locomotor scale and TTC staining results suggested that the custom‐designed model of CSCI in rats was validated as a suitable, objective, and reproducible model system that could be used to study CSCI‐induced demyelination.
Pathological Characteristics of Demyelination
We observed the pathological characteristics of lesion regions by osmic acid staining and TEM. The alteration of MBP expression was also investigated to reveal the possibility of demyelination following CSCI with the duration of time. Various techniques, including a classic silver impregnation method, modern immunohistochemistry, and molecular staining, have been used to determine the structure of nerve tissues 54, 55, 56, 57. However, these methods are limited in terms of qualitative assessment. Morphometric and quantitative analyses, particularly involving myelin sheaths, are urgently required 58. Studies have shown that osmium tetroxide is a contrasting agent of lipids, which are the main components of myelin sheaths. Myelin sheaths can be oxidized and stained by osmium tetroxide producing a black appearance 58. Hence, osmium tetroxide staining applied in this study effectively demonstrated the significant reduction in myelinated nerve fibers in the CSCI group compared with normal and sham groups. Light microscopy revealed that the lesion regions where the loss of myelinated nerve fibers was observed contained little or no osmium tetroxide stain. In TEM, myelin sheaths became swollen and degenerated and were broken down gradually. In addition to myelin sheath degeneration, the axolemma was separated from the myelin sheath. The axoplasm in lesion regions contained varying degrees of reduction in the mitochondria and microtubules. Oligodendrocyte chromatin condensation was also observed, indicating oligodendrocyte apoptosis 4. Macrophage infiltration was observed by TEM. Both oligodendrocyte apoptosis and macrophage infiltration are characteristics of demyelination 16, 59.
Myelin basic protein is essential for myelin sheath formation 21, 22. In animal models of MS or EAE, the cleavage of MBP to smaller peptides sensitizes T cells to cause further damage to the myelin; as a result, demyelination occurs 60, 61, 62. In the present study, single‐fluorescent immunostaining revealed that the majority of immunoreactive MBPs were observed in the myelin sheaths of the sham group; lower amounts of immunoreactive MBPs were detected after CSCI. Western blot also showed that MBP expression was decreased as compression time increased. Minimum MBP expression was observed in the compressed segments at 7 days after CSCI. The findings suggested that the breakdown of MBP occurred after CSCI, which was essential and responsive to demyelination.
These results indicated that the methods were very suitable to detect demyelination qualitatively and quantitatively. Demyelination occurred in the white matter of the injured spinal cord and was aggravated with the duration of time. Demyelination also contributed to neurological deficits after CSCI. The amount of demyelination indicated the severity of motor dysfunction after CSCI 9. This study provided information about the mechanisms of demyelination for future research.
Function and Mechanism of Oligodendrocyte Apoptosis in CSCI‐Induced Demyelination
Evidence has shown that neuronal apoptosis contributes to demyelination and Wallerian degeneration, thereby affecting neurological function 63. However, other studies have revealed that oligodendrocyte apoptosis is an important event in demyelination 45, 64, 65. Primary demyelination in MS or several other inflammatory diseases are mediated by different pathological mechanisms accompanied by myelin‐forming oligodendrocyte apoptosis 66.
In the study, TUNEL staining showed that oligodendrocyte apoptosis immediately occurred after CSCI. The number of oligodendrocyte apoptosis was remarkably increased at the compressed segments in the CSCI group compared with normal and sham groups (P < 0.05). This finding is consistent with the extent of demyelination, suggesting that oligodendrocyte apoptosis occurs after compression and contributes to CSCI‐induced demyelination 67, 68.
In metazoans, oligodendrocyte apoptosis is considered as a very complex process. Studies have demonstrated that apoptosis is correlated with ER–mitochondria interaction and enhanced Id2 expression 29, 40.
Endoplasmic reticulum is a cell organelle where secretory, membrane‐bound, and some organelle‐targeted proteins are synthesized and folded. Correctly folded proteins are transported from the ER to the Golgi and other destinations within the cell, but misfolded proteins or proteins that fail to fold properly are retained in the ER. The accumulation of misfolded proteins may induce stress to the cell, that is, ER stress. Cells that may be unnecessary or harmful for organisms are possibly eliminated in the ER pathway 69. Under ER stress, Ca2+ is released from the ER, and caspase‐12 is triggered by calpains 70. Caspase‐12, which belongs to the family of cysteine proteases and is widely expressed in various cells, is identified as the first ER‐associated member of the caspase family and a representative molecule involved in cell death–executing mechanisms relevant to ER stress 71, 72. The activation of caspase‐12 in the ER cleaves procaspase‐9, leading to caspase‐9‐dependent activation of caspase‐3, which executes apoptosis 40, 42. To determine the impact factor closely related to oligodendrocyte apoptosis and clarify ER stress, we investigated the expression of caspase‐12 in oligodendrocytes. In this study, caspase‐12‐immunoreactive oligodendrocytes were primarily distributed in compressed segments. Caspase‐12 expression at the corresponding time in the CSCI group was significantly higher than that in the normal group (P < 0.05). The maximum caspase‐12 expression was reached at 7 days after CSCI (P < 0.05). These results strongly suggested that the enhanced caspase‐12 expression was induced by ER stress. Caspase‐12 expression likely functioned as an inducible cell death effector and contributed to oligodendroctye apoptosis, which caused demyelination after CSCI.
In addition to ER stress pathway, mitochondrial dysfunction is an important pathway in apoptosis 41. Mitochondria function as power stations involved in ATP production and regulation of other cell functions such as lipid oxidation, oxygen radical production, and hormone metabolism 73. For several years, studies have also focused on two additional functions of mitochondria: Ca2+ homeostasis and release of apoptotic factors in response to death signals 74. Although the classical functions between ER and mitochondria are distinct, emerging evidence emphasizes the importance of reciprocal structural and functional interactions of these organelles for some key integrated cellular functions 29. At the onset of ER stress, Ca2+‐ATPase 1 (S1T), a truncated form of the sarcoendoplasmic reticulum, is induced 75. S1T transfers Ca2+ from the ER to the mitochondria to promote Ca2+ overload in the mitochondria and activate the mitochondrial apoptotic pathway via the PERK/ATF4 pathway 69. As a result, mitochondrial membrane potential is decreased and cytochrome c is released into the cytoplasm. Cytochrome c then binds to apoptotic protease and triggers the formation of an apoptosome that activates caspase‐9 and caspase‐3, thereby leading to apoptosis 76. Our results revealed that the distribution of cytochrome c‐immunoreactive oligodendrocytes expanded with the duration of time. The expression of cytochrome c at the corresponding time in CSCI group was significantly higher than that in normal and sham groups (P < 0.05) and reached the maximum at 7 days after CSCI (P < 0.05). The enhanced cytochrome c expression results suggested that cytochrome c seems to be released from the intermembrane spaces of the mitochondria into the cytoplasm. The release of cytochrome c is also a key event in oligodendrocyte apoptosis 77. Furthermore, the coexistence of caspase‐12 and cytochrome c in cells demonstrates that ER and mitochondria are closely associated, establishing a dynamic ER–mitochondria interconnection that has an important function in apoptosis 29, 76.
It is reported that some factors may provide protection against both apoptosome assembly and cleaved caspases. For example, the upregulation of heat‐shock protein 70 (HSP 70), which is the major inducible form of HSPs, and the neuroprotective protein can protect against apoptotic cell death after cytochrome c release in U937 human leukemic cells and caspase‐3 activation in the model of excitotoxic tolerance 78, 79, 80. In other words, cytochrome c release and caspase expression could be observed in the absence of cell death under some specific circumstances.
Id2 is one of the oligodendrocyte lineage genes that belong to the family of HLH transcription factors. Id2 lacks a basic region and fails to bind to DNA. This characteristic makes Id2 unique from other basic HLH transcription factors. One of the important functions of Id2 is observed in its dominant negative effect when heterodimers are formed with other DNA‐binding members of the HLH family, disrupting the protein–DNA interaction 81. Thus, Id2 and other partner HLH transcription factors are important negative regulators that determine biological events in cells, including cell proliferation, development, and specialization as well as prevention of these proteins from differentiation 82, 83. As a known modulator, Id2 may function not only to promote cellular proliferation and inhibit differentiation but also to augment apoptosis. The N‐terminal region of Id2 is required for its apoptotic functions. The N‐terminal domain of apoptotic targets could be apoptotic effectors such as the programmed cell death cascade, Bax protein. The Bax is a parameter for description of apoptotic state 18, 84. The level of expression of Bax is closely correlated with the level of Id2 expression. Id2 expression leads to upregulation of Bax levels 30. Increase in Bax will alter the mitochondrial membrane permeability, leading to the release of cytochrome c. Then the downstream effector caspase‐3 is activated, followed by chromatin condensation and DNA fragmentation 85. Id2 also regulates MBP negatively in vitro and modulates myelination in normal animals 31. In the study, double‐labeling immunofluorescence revealed that Id2‐immunoreactive oligodendrocytes were randomly distributed in the white matter in the normal group and then expanded at 1 day after CSCI. Id2‐immunoreactive oligodendrocytes were found in the whole white matter at 7 days after CSCI. Id2 expression level was increased at 1 day, and the maximum level was reached at 7 days. This finding suggested that the upregulated Id2 expression was associated with oligodendrocyte apoptosis and the downregulation of MBP expression, which was clearly correlated with oligodendrocyte apoptosis and CSCI‐induced demyelination.
We cannot exclude the possibility, however, that the demyelination may also be attributed in some part to necrosis besides oligodendrocyte apoptosis. The researcher found that oligodendrocytes are particularly susceptible to the toxicity of the acute lesion environment after CSCI. They undergo both necrosis and apoptosis acutely, which contribute to demyelination of myelinated nerve fibers and their electrophysiological defect 86.
Conclusion
The study demonstrated that demyelination occurred and was aggravated with the duration of time after CSCI, which functions in motor dysfunction. Demyelination might be partly caused by oligodendrocyte apoptosis, which is positively correlated with enhanced Id2 expression and ER–mitochondria interactions after CSCI in rats.
Conflict of Interest
The authors declare no conflict of interest.
Disclosure
The authors have not published or submitted the manuscript elsewhere.
Acknowledgments
This study is partially supported by the National Natural Science Foundation of China (Grant No. 81273870) and the Natural Science Foundation Project of CQ CSTC (Grant No. cstc2012jjA10020). The authors extend their gratitude to Mr. Zaiyun Long (the Third Military Medical University) for his technical support.
References
- 1. Lebl DR, Hughes A, Cammisa FP Jr, O'Leary PF. Cervical spondylotic myelopathy: Pathophysiology, clinical presentation, and treatment. HSS J 2011;7:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 1991;75:15–26. [DOI] [PubMed] [Google Scholar]
- 3. Breig A, Turnbull I, Hassler O. Effects of mechanical stresses on the spinal cord in cervical spondylosis. A study on fresh cadaver material. J Neurosurg 1966;25:45. [DOI] [PubMed] [Google Scholar]
- 4. Casha S, Yu W, Fehlings M. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 2001;103:203. [DOI] [PubMed] [Google Scholar]
- 5. Hukuda S, Wilson CB. Experimental cervical myelopathy: Effects of compression and ischemia on the canine cervical cord. J Neurosurg 1972;37:631–652. [DOI] [PubMed] [Google Scholar]
- 6. McGavern DB, Murray PD, Rodriguez M. Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J Neurosci Res 1999;58:492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang S, Khan MI, Lu Y, et al. Guanosine promotes myelination and functional recovery in chronic spinal injury. NeuroReport 2003;14:2463–2467. [DOI] [PubMed] [Google Scholar]
- 8. Lipton HL, Dal Canto MC. Chronic neurologic disease in Theiler's virus infection of SJL/J mice. J Neurol Sci 1976;30:201–207. [DOI] [PubMed] [Google Scholar]
- 9. McGavern DB, Zoecklein L, Drescher KM, Rodriguez M. Quantitative assessment of neurologic deficits in a chronic progressive murine model of CNS demyelination. Exp Neurol 1999;158:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holmes G. On the relation between loss of function and structural change in focal lesions of the central nervous system, with special reference to secondary degeneration. Brain 1907;29:514–523. [Google Scholar]
- 11. Blakemore W, Eames R, Smith K, McDonald W. Remyelination in the spinal cord of the cat following intraspinal injections of lysolecithin. J Neurol Sci 1977;33:31–43. [DOI] [PubMed] [Google Scholar]
- 12. Jeffery N, Blakemore W. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J Neurocytol 1995;24:775–781. [DOI] [PubMed] [Google Scholar]
- 13. Bai L, Lennon DP, Caplan AI, et al. Hepatocyte growth factor mediates mesenchymal stem cell‐induced recovery in multiple sclerosis models. Nat Neurosci 2012;15:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodriguez M, Oleszak E, Leibowitz J. Theiler's murine encephalomyelitis: A model of demyelination and persistence of virus. Crit Rev Immunol 1987;7:325. [PubMed] [Google Scholar]
- 15. Kim BS, Jin Y‐H, Meng L, et al. IL‐1 signal affects both protection and pathogenesis of virus‐induced chronic CNS demyelinating disease. J Neuroinflammation 2012;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akassoglou K, Bauer J, Kassiotis G, et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol 1998;153:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gong Y, Wang Z, Liang Z, et al. Soluble MOG35‐55/I‐Ab dimers ameliorate experimental autoimmune encephalomyelitis by reducing encephalitogenic T cells. PLoS ONE 2012;7:e47435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giacoppo S, Galuppo M, Iori R, et al. Protective role of (RS)‐glucoraphanin bioactivated with myrosinase in an experimental model of multiple sclerosis. CNS Neurosci Ther. doi: 10.1111/cns.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bieber AJ, Warrington A, Asakura K, et al. Human antibodies accelerate the rate of remyelination following lysolecithin‐induced demyelination in mice. Glia 2002;37:241–249. [DOI] [PubMed] [Google Scholar]
- 20. Yang Y, Lewis R, Miller RH. Interactions between oligodendrocyte precursors control the onset of CNS myelination. Dev Biol 2011;350:127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. D'Aversa TG, Eugenin EA, Lopez L, Berman JW. Myelin basic protein induces inflammatory mediators from primary human endothelial cells and blood‐brain‐barrier disruption:Implications for the pathogenesis of multiple sclerosis. Neuropathol Appl Neurobiol 2013;39:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boggs JM, Rangaraj G, Heng YM, Liu Y, Harauz G. Myelin basic protein binds microtubules to a membrane surface and to actin filaments in vitro: Effect of phosphorylation and deimination. Biochim Biophys Acta 2011;1808:761. [DOI] [PubMed] [Google Scholar]
- 23. Barbarese E, Pfeiffer S. Developmental regulation of myelin basic protein in dispersed cultures. Proc Natl Acad Sci USA 1981;78:1953–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bologa L, Bisconte JC, Joubert R, Margules S, Herschkowitz N. Proliferative activity and characteristics of immunocytochemically identified oligodendrocytes in embryonic mouse brain cell cultures. Exp Brain Res 1983;50:84–90. [DOI] [PubMed] [Google Scholar]
- 25. Omlin FX, Webster HD, Palkovits CG, Cohen SR. Immunocytochemical localization of basic protein in major dense line regions of central and peripheral myelin. J Cell Biol 1982;95:242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gavrilescu LC, Denkers EY. Apoptosis and the balance of homeostatic and pathologic responses to protozoan infection. Infect Immun 2003;71:6109–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. James ER, Green DR. Manipulation of apoptosis in the host‐parasite interaction. Trends Parasitol 2004;20:280–287. [DOI] [PubMed] [Google Scholar]
- 28. Moss JE, Aliprantis AO, Zychlinsky A. The regulation of apoptosis by microbial pathogens. Int Rev Cytol 1999;187:203–259. [DOI] [PubMed] [Google Scholar]
- 29. Pizzo P, Pozzan T. Mitochondria‐endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol 2007;17:511. [DOI] [PubMed] [Google Scholar]
- 30. Florio M, Hernandez MC, Yang H, Shu HK, Cleveland JL, Israel MA. Id2 promotes apoptosis by a novel mechanism independent of dimerization to basic helix‐loop‐helix factors. Mol Cell Biol 1998;18:5435–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jessen KR, Mirsky R. Negative regulation of myelination: Relevance for development, injury, and demyelinating disease. Glia 2008;56:1552–1565. [DOI] [PubMed] [Google Scholar]
- 32. Chen XS, Zhou DS, Yao ZX. The inhibitor of DNA binding 2 is mainly expressed in oligodendrocyte lineage cells in adult rat brain. Neurosci Lett 2007;428:93–98. [DOI] [PubMed] [Google Scholar]
- 33. Kondo T, Raff M. Basic helix‐loop‐helix proteins and the timing of oligodendrocyte differentiation. Development 2000;127:2989–2998. [DOI] [PubMed] [Google Scholar]
- 34. Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol 2011;93:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight‐drop device versus transection. Exp Neurol 1996;139:244–256. [DOI] [PubMed] [Google Scholar]
- 36. Pavelko KD, van Engelen BG, Rodriguez M. Acceleration in the rate of CNS remyelination in lysolecithin‐induced demyelination. J Neurosci 1998;18:2498–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leviton A, Gilles F. Ventriculomegaly, delayed myelination, white matter hypoplasia, and “periventricular” leukomalacia: How are they related? Pediatr Neurol 1996;15:127–136. [DOI] [PubMed] [Google Scholar]
- 38. Blumenthal I. Periventricular leucomalacia: A review. Eur J Pediatr 2004;163:435–442. [DOI] [PubMed] [Google Scholar]
- 39. Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int J Dev Neurosci 2011;29:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arduíno DM, Esteves AR, Cardoso SM, Oliveira CR. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: Relevance to Parkinson's disease. Neurochem Int 2009;55:341–348. [DOI] [PubMed] [Google Scholar]
- 41. Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicol Lett 2004;149:19–23. [DOI] [PubMed] [Google Scholar]
- 42. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress‐specific caspase cascade in apoptosis cytochrome c‐independent activation of caspase‐9 by caspase‐12. J Biol Chem 2002;277:34287–34294. [DOI] [PubMed] [Google Scholar]
- 43. Harkey HL, Al‐Mefty O, Marawi I, Peeler DF, Haines DE, Alexander LF. Experimental chronic compressive cervical myelopathy: Effects of decompression. J Neurosurg 1995;83:336–341. [DOI] [PubMed] [Google Scholar]
- 44. Siegal T, Siegal T, Shapira Y, Sandbank U, Catane R. Indomethacin and dexamethasone treatment in experimental neoplastic spinal cord compression: Part 1: Effect on water content and specific gravity. Neurosurgery 1988;22:328–333. [DOI] [PubMed] [Google Scholar]
- 45. Yu WR, Fehlings MG. Fas/FasL‐mediated apoptosis and inflammation are key features of acute human spinal cord injury: Implications for translational, clinical application. Acta Neuropathol 2011;122:747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maegele M, Riess P, Sauerland S, et al. Characterization of a new rat model of experimental combined neurotrauma. Shock 2005;23:476–481. [DOI] [PubMed] [Google Scholar]
- 47. Profyris C, Cheema SS, Zang D, Azari MF, Boyle K, Petratos S. Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol Dis 2004;15:415–436. [DOI] [PubMed] [Google Scholar]
- 48. Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol 2005;67:1385–1387. [DOI] [PubMed] [Google Scholar]
- 49. Hagg T, Oudega M. Degenerative and spontaneous regenerative processes after spinal cord injury. J Neurotrauma 2006;23:263–280. [DOI] [PubMed] [Google Scholar]
- 50. Mizuno J, Nakagawa H, Inoue T, Hashizume Y. Clinicopathological study of “snake‐eye appearance” in compressive myelopathy of the cervical spinal cord. J Neurosurg Spine 2003;99:162–168. [DOI] [PubMed] [Google Scholar]
- 51. Yamaura I, Yone K, Nakahara S, et al. Mechanism of destructive pathologic changes in the spinal cord under chronic mechanical compression. Spine 2002;27:21–26. [DOI] [PubMed] [Google Scholar]
- 52. Kim P, Haisa T, Kawamoto T, Kirino T, Wakai S. Delayed myelopathy induced by chronic compression in the rat spinal cord. Ann Neurol 2004;55:503–511. [DOI] [PubMed] [Google Scholar]
- 53. Lingor P, Koch JC, Tönges L, Bähr M. Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res 2012;349:289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. twlHe F, Fan Q, Cushway T, et al. Long‐term result of guided nerve regeneration with an inert microporous polytetrafluoroethylene conduit. Chin J Traumatol 2003;6:145. [PubMed] [Google Scholar]
- 55. Kurihara K, Cohen M, Schultz RC. Nerve staining with leucomethylene blue: An experimental study. Plast Reconstr Surg 1984;73:960–964. [DOI] [PubMed] [Google Scholar]
- 56. Gu X, Yan Z, Yan W, Chen C. Rapid immunostaining of live nerve for identification of sensory and motor fasciculi. Chin Med J (Engl) 1992;105:949. [PubMed] [Google Scholar]
- 57. Starega U. A silver impregnation technique for normal axons in the human central nervous system for celloidin and Epon sections, with substitutes for soft tap water. Stain Technol 1985;60:103–110. [DOI] [PubMed] [Google Scholar]
- 58. Wei LP, He FC, Chen XW, Lu SB, Lanzetta M, De Iongh R. Osmic acid staining of myelin sheath in normal and regenerated peripheral nerves. Chin J Traumatol 2007;10:86–89. [PubMed] [Google Scholar]
- 59. Dogan R‐NE, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF‐and iNOS‐expressing macrophages and myeloid dendritic cells. J Immunol 2008;180:7376–7384. [DOI] [PubMed] [Google Scholar]
- 60. Cross AH, Dolich S, Raine CS. Antigen processing of myelin basic protein is required prior to recognition by T cells inducing EAE. Cell Immunol 1990;129:22–31. [DOI] [PubMed] [Google Scholar]
- 61. D'Souza CA, Moscarello MA. Differences in susceptibility of MBP charge isomers to digestion by stromelysin‐1 (MMP‐3) and release of an immunodominant epitope. Neurochem Res 2006;31:1045–1054. [DOI] [PubMed] [Google Scholar]
- 62. Mastronardi FG, Moscarello MA. Molecules affecting myelin stability: A novel hypothesis regarding the pathogenesis of multiple sclerosis. J Neurosci Res 2005;80:301–308. [DOI] [PubMed] [Google Scholar]
- 63. Ackery A, Robins S, Fehlings MG. Inhibition of Fas‐mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma 2006;23:604–616. [DOI] [PubMed] [Google Scholar]
- 64. Emery E, Aldana P, Bunge MB, et al. Apoptosis after traumatic human spinal cord injury. J Neurosurg 1998;89:911–920. [DOI] [PubMed] [Google Scholar]
- 65. Yoshino O, Matsuno H, Nakamura H, et al. The role of Fas‐mediated apoptosis after traumatic spinal cord injury. Spine 2004;29:1394–1404. [DOI] [PubMed] [Google Scholar]
- 66. Lucchinetti CF, Brück W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity in pathogenesis. Brain Pathol 1996;6:259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 1997;50:798–808. [DOI] [PubMed] [Google Scholar]
- 68. Takagi T, Takayasu M, Mizuno M, Yoshimoto M, Yoshida J. Caspase activation in neuronal and glial apoptosis following spinal cord injury in mice. Neurol Med Chir 2003;43:20–30. [DOI] [PubMed] [Google Scholar]
- 69. Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009;14:996–1007. [DOI] [PubMed] [Google Scholar]
- 70. Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Ubiquitous calpains promote caspase‐12 and JNK activation during endoplasmic reticulum stress‐induced apoptosis. J Biol Chem 2006;281:16016–16024. [DOI] [PubMed] [Google Scholar]
- 71. Nakagawa T, Zhu H, Morishima N, et al. Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐β. Nature 2000;403:98–103. [DOI] [PubMed] [Google Scholar]
- 72. Zhang HY, Zhang X, Wang ZG, et al. Exogenous basic fibroblast growth factor inhibits ER stress–induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther 2013;19:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nicholls DG. Mitochondrial function and dysfunction in the cell: Its relevance to aging and aging‐related disease. Int J Biochem Cell Biol 2002;34:1372–1381. [DOI] [PubMed] [Google Scholar]
- 74. Hajnóczky G, Csordás G, Das S, et al. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006;40:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chami M, Oulès B, Szabadkai G, Tacine R, Rizzuto R, Paterlini‐Bréchot P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell 2008;32:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhao CQ, Zhang YH, Jiang SD, Jiang LS, Dai LY. Both endoplasmic reticulum and mitochondria are involved in disc cell apoptosis and intervertebral disc degeneration in rats. Age (Dordr) 2010;32:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bhattarai G, Lee YH, Lee NH, et al. Fomitoside‐K from fomitopsis nigra induces apoptosis of human oral squamous cell carcinomas (YD‐10B) via mitochondrial signaling pathway. Biol Pharm Bull 2012;35:1711–1719. [DOI] [PubMed] [Google Scholar]
- 78. Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: From adenosine receptor to KATP channel. Annu Rev Physiol 2000;62:79–109. [DOI] [PubMed] [Google Scholar]
- 79. Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E. HSP27 inhibits cytochrome c‐dependent activation of procaspase‐9. FASEB J 1999;13:2061–2070. [DOI] [PubMed] [Google Scholar]
- 80. McLaughlin B, Hartnett KA, Erhardt JA, et al. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA 2003;100:715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: A negative regulator of helix‐loop‐helix DNA binding proteins. Cell 1990;61:49–59. [DOI] [PubMed] [Google Scholar]
- 82. Bhattacharya A, Baker NE. A network of broadly expressed HLH genes regulates tissue‐specific cell fates. Cell 2011;147:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Niola F, Zhao X, Singh D, et al. Id proteins synchronize stemness and anchorage to the niche of neural stem cells. Nat Cell Biol 2012;14:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bilbault P, Lavaux T, Launoy A, et al. Influence of drotrecogin alpha (activated) infusion on the variation of Bax/Bcl‐2 and Bax/Bcl‐xl ratios in circulating mononuclear cells: A cohort study in septic shock patients. Crit Care Med 2007;35:69–75. [DOI] [PubMed] [Google Scholar]
- 85. Kumari A, Kakkar P. Lupeol prevents acetaminophen‐induced in vivo hepatotoxicity by altering the Bax/Bcl‐2 and oxidative stress‐mediated mitochondrial signaling cascade. Life Sci 2012;90:561–570. [DOI] [PubMed] [Google Scholar]
- 86. Almad A, Sahinkaya FR, McTigue DM. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics 2011;8:262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]