

Summary
Aims
Neurodegenerative changes observed in Alzheimer's disease (AD) have been suggested to begin at the entorhinal cortex and hippocampus and then to propagate in a stereotypical fashion. Using diffusion‐weighted imaging, we test whether disruption of structural connectivity in AD is centered on these “epicenters of disease”.
Methods
Fifteen healthy controls, 14 amnestic mild cognitive impairment (aMCI), 13 mild, and 15 moderate patients with AD were enrolled. The percentages of affected connections directly linking to the epicenter (named first ring) and to nodes with topological distance 2 from the epicenter (named second ring) were calculated.
Results
For the group of aMCI patients, just 5.3% of the first ring (n.s.) and 2.9% of the second ring (n.s.) connections were affected. However, for mild AD there was disruption involving 20% of the first ring (P < 0.0001) and 10.3% of the second ring (P < 0.0001) connections. In the moderate AD group, a stronger effect was observed, with 38.0% of the first ring (P < 0.0001) connections and 17.9% of the second ring (P < 0.0001) connections affected.
Conclusion
Our results favor an epicentral disruption of structural connectivity in aMCI and AD around entorhinal and hippocampal regions, consistent with the transneuronal spread hypothesis.
Keywords: Alzheimer's Disease, Connectivity, Connectome, Diffusion‐weighted imaging, Epicentral disruption, Mild cognitive impairment
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, known as the most common form of dementia 1. The two neuropathological hallmarks of AD are neurofibrillary tangles (NFTs), related to intraneuronal formation of hyperphosphorylated tau protein, and neuritic plaques (NPs) composed of extracellular deposits of amyloid beta (Aβ) protein 2, 3, 4. Pathology studies have shown that AD‐related neuropathological changes, especially NFTs, begin at specific sites of the human brain and then propagate from affected to unaffected areas in a systematic and stereotypical fashion 2, 3, 4, 5, 6. Indeed, the first brain regions affected by NFTs are the transentorhinal and the entorhinal cortex (EC), followed by the hippocampus (HIP) 2, 3, 4, 5, 6. It is until a later phase of the disorder that the neocortex and especially the associative areas are thought to become involved in the disease process related to NFTs 2, 3, 4, 5, 6. Structural MRI studies have shown supporting evidence that EC and HIP are the first affected brain regions in subjects with mild cognitive impairment (MCI) and AD, and, furthermore, that the atrophy pattern in EC and HIP can predict the conversion from MCI to AD 7, 8.
There is a growing body of research showing that network analyses contribute greatly to the understanding of brain dysfunction 9. Abnormal structural connectivity has indeed been revealed in several psychiatric 10 and neurological disorders, including AD 11. Four major models of network‐based neurodegeneration spread have been proposed. In the ‘‘nodal stress’’ model, regions with high neural activity are damaged due to a ‘‘wear and tear’’ mechanism 12, 13, 14. The ‘‘trophic failure’’ model assumes connectivity impairment to cause failure of inter‐nodal trophic factor, promoting neurodegeneration 15, 16. In the “shared vulnerability’’ model, a common disease‐specific gene or protein expression underlies the susceptibility of specific regions of a network 17. Last, the ‘‘transneuronal spread’’ model is based on the hypothesis that toxic agents propagate along brain connections with a prion‐like behavior 18, 19. Recently, Zhou and colleagues used graph theoretical analysis to investigate how intrinsic functional connectivity in health predicts the brain's vulnerability to neurodegeneration 17. Their findings suggested the transneuronal model as the most likely to underlie the changes observed in neurodegenerative diseases including AD 17.
Here, using graph theoretical analysis of diffusion‐weighted imaging (DWI) data, we tested the hypothesis that structural connectivity of AD and amnestic mild cognitive impairment (aMCI) patients might be explained by the transneuronal spread model, focusing specifically on the topological distance of nodal connectivity from the EC/HIP, a priori selected as a potential disease epicenter. Disease effects of reduced structural connectivity were expected to be stronger at shorter topological distance from the EC/HIP epicenter. Correlations between connectivity measures and Mini Mental State Examination (MMSE) scores were assessed, to test a possible relationship between neurodegenerative effects of global, EC, and HIP connectivity disruption and cognitive performance in patients.
Materials and Methods
Subjects and Clinical Evaluation
The study was approved by the local ethical committee and was conducted in accordance with the Declaration of Helsinki. All subjects or caregivers provided their written informed consent to use their anonymized data for research purposes.
In total, 57 subjects, of which 15 healthy controls (HC), 14 aMCI, 13 mild AD, and 15 moderate AD, were included in this study. Demographics and clinical characteristics are listed in Table 1. All subjects were recruited from the outpatient clinic of Neurology Unit at Università Campus Bio‐Medico di Roma (Italy). Each subject underwent medical, neurological, cognitive, and laboratory screening together with an MRI scan of the brain, reviewed by two independent and experienced neuroradiologists to rule out structural abnormalities. A detailed description of the administered neuropsychological test battery is provided in the Data S1.
Table 1.
Demographic and clinical features of the diagnostic groups
| HC (N = 15) | aMCI (N = 14) | Mild AD (N = 13) | Moderate AD (N = 15) | χ 2 or F | P value | |
|---|---|---|---|---|---|---|
| Gender (men, women) | 7, 8 | 10, 4 | 8, 5 | 8, 7 | 0.57 | n.s. |
| Age years (SD) | 73.1 (6.1) | 74.4 (5.9) | 74.5 (9.7) | 74.9 (4.8) | 0.21 | n.s. |
| Range | 61–84 | 63–85 | 58–89 | 63–82 | ||
| Education years (SD) | 6.9 (3.2) | 8.8 (4.3) | 10.2 (4.7) | 9.0 (4.9) | 1.15 | n.s. |
| Range | 5–15 | 4–18 | 5–17 | 4–18 | ||
| MMSE raw score (SD) | 28.6 (1.1) | 26.9 (2.2) | 23.6 (1.7) | 17.9 (3.1) | 61.9 | <10−7 |
| Range | 27–30 | 23–30 | 22–28 | 13–23 | ||
| Corrected score (SD) | 28.2 (1.1) | 26.3 (2.5) | 22.5 (1.1) | 17.2 (2.5) | 83.3 | <10−7 |
| Range | 26.7–30.0 | 22.3–30.0 | 21.0–24.4 | 12.9–21.3 | ||
| CDR | 0 | 0.5 | 1 | 2 |
HC, healthy controls; aMCI, amnestic mild cognitive impairment; AD, Alzheimer's disease; MMSE, Mini‐Mental‐State Examination; CDR, Clinical Dementia Rating; SD, standard deviation.
The diagnosis of AD or aMCI was made by two independent, experienced neurologists according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) 20 and to the criteria of Petersen 21, respectively. Patients with AD were further classified into two groups based on the corrected MMSE scores (adjusted for age and education), being mild AD (22–27), and moderate AD (10–20) 22, 23. Severity differences were confirmed using the Clinical Dementia Rating (CDR) scale 24, 25.
For aMCI patients exclusion criteria included mild AD, concomitant dementia, reversible dementias, fluctuations in cognitive performance, mixed dementias, concomitant extra‐pyramidal symptoms, depression with Geriatric Depression Scale (GDS) scores >13, other psychiatric diseases, epilepsy, drug or alcohol addiction, use of psychoactive drugs, systemic diseases, traumatic brain injuries, and use of cholinergic drugs. For patients with AD, exclusion criteria included frontotemporal dementia 26, vascular dementia 27, extra‐pyramidal syndromes, reversible dementias (including pseudodementia of depression and normotensive hydrocephalus) and Lewy body dementia 28. Healthy subjects were recruited among non‐consanguineous relatives of the patients and through advertisements. All healthy subjects met the criteria of having a GDS score <13 and a MMSE score ≥27 and did not have a history of neurological or psychiatric diseases, chronic systemic illnesses or use of psychoactive drugs. In total, this resulted in the inclusion of 57 subjects divided into 4 groups: aMCI (14 patients), mild AD (13 patients), moderate AD (15 patients), and the healthy control group (15 subjects).
Imaging Acquisition
Images were acquired using a 1.5 Tesla MRI system (Avanto B13, Siemens, Erlangen, Germany), configured with a 12‐element designed Head Matrix Coil. The imaging protocol included the following: structural T1‐weighted MPRAGE (Magnetization Prepared Rapid Acquisition with Gradient Echo) sequences (TR 2400 ms, TE 3.61 ms, TI 1000 ms, FOV read 240 mm, FOV phase 100.0%, flip angle 8◦, slice thickness 1.20 mm, averages 1, slice for slab 160, slab 1, bandwidth 180 Hz/Px) and T2‐/PD‐weighted TSE (Turbo Spin Echo) sequences (TR 3000 ms, TE1 12 ms, TE2 92 ms, FOV read 240 mm, FOV phase 89.1%, flip angle 150◦, slice thickness 3 mm, averages 1, slices 48, bandwidth 163 Hz/Px), according to the Italian Alzheimer Disease Neuroimaging Initiative (ADNI) harmonization protocol. Diffusion‐weighted images (DWI) were acquired with a weighting 1000 mm2/s and with gradients applied in 12 different directions, together with a b0 scan (slice group 1, slices 38, dist factor 30%, phase enc. dir. A ≫ P, phase oversampling 0%, FOV read 250 mm, FOV phase 100%, slice thickness 3 mm, TR 7600 ms, TE 103 ms, concatenations 1, coil elements BO1.2.SP4.5). Four complete sets (each of 12 directions) were acquired, providing a total of four b0 and 48 diffusion‐weighted volumes.
Construction of Structural Brain Networks
Individual structural brain networks were reconstructed on the basis of a parcellation of the brain into 83 distinct brain regions combined with white matter fibers resulting from tractography (see Data S1 for details including T1 and DWI preprocessing and tractography). The networks, mathematically described as a graph consisting of a collection of nodes (brain regions) and a collection of edges between nodes (white matter tracts), were represented by 83 × 83 structural connectivity matrices storing information regarding the existence, absence, and weight of a connection between node i and j in cell c(i,j). Fractional anisotropy (FA) was chosen as a measure of microstructural fiber directionality and the number of streamlines (NOS) as a measure to quantify the absolute number of connection fibers. Streamline density (NOS divided by the mean volume of the two interconnected nodes 29, 30) was added in an attempt to account for potential effects of cortical atrophy on NOS metrics.
Global Connectivity Strength
The Jonckheere–Terpstra test was used to assess whether average global connectivity strength, that is, the sum of all connection strengths per subject, decreased from the HC group to the aMCI, mild AD, and moderate AD groups, effectively testing for potential ordering of three or more population medians. Global connectivity strength levels were controlled for age, gender, and education effects using a linear regression model.
Connectivity Ring Analysis
To test whether disease progression from aMCI to mild and moderate AD could be reflected by structural damage to the epicenter, the percentage of connections to the epicenter that were affected was computed for all three groups of patients and was compared to the control population (Figure 1). First, individual connectivity matrices were masked to keep those connections that were present in at least 50% of the healthy control subjects 31, 32. Second, two rings of nodes were defined. A first ring of nodes directly linked by one connection to either one of the epicenter nodes, and a second ring of nodes at topological distance two from the epicenter 33. Third, the strengths of connections between the epicenter and the first ring and between first and second ring were compared in each of the three groups of patients with those observed in the control population. In each patient group separately, connections that showed a lower average strength compared to the HC group (t‐test, P‐value <0.05) were labeled as “affected” following an approach similar as defined by Network Based Statistics (NBS) 34. As for the global analysis, connection strengths were controlled for age, gender, and education effects using a linear regression model. Next, the percentage of affected connections was computed for the two rings in each patient group. This procedure was carried out for both hemispheres separately, and percentages of affected connections were subsequently averaged. Fourth, significance of the findings was assessed by means of permutation testing. For each of the three patient groups, the analysis was repeated for 10,000 random permutations of group assignments (i.e., HC or patient) to obtain a distribution of results under the null hypothesis.
Figure 1.
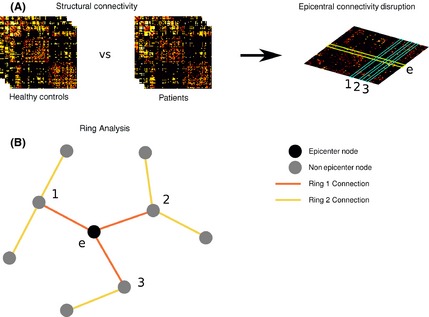
(A) Structural connectivity analysis of patients compared with healthy controls using matrices. (B) Two classes of connections were defined relative to the epicenter, referred to as first and second ring connections. First ring connections are those directly linking to the epicenter (orange lines in (B)), second ring connections are those with topological distance 2 from the epicenter (yellow lines in (B)).
Examining Specificity of the a priori Selected Epicenter
In addition, to assess the merit of selecting the EC and HIP as the epicenter, the analysis described above was carried out for all possible epicenters consisting of two homologous node pairs (41 × 40/2 = 820) according to the used gray matter parcellation. For each of the three patient groups, the share of all possible epicenters yielding higher percentages of affected connections than the original EC and HIP epicenter in both the first and second ring denoted the P‐value.
Relationship between Structural Connectivity and MMSE
To assess a potential link between global structural connectivity and cognition, for each patient the strengths of all connections present in the connectivity matrices were summed and examined for a correlation with corrected MMSE scores. To analyze the correlation between MMSE scores and structural connectivity of the epicenter regions specifically, strength values were summed of all connections linking to the HIP or EC regions. Global and epicenter connectivity strengths were controlled for age, gender, and education effects using a linear regression model. Pearson's correlation coefficient was used to assess correlations between connectivity strengths and MMSE scores.
Results
Demographic and Clinical Features
When the global cognitive status was assessed by means of MMSE, the four groups (HC, aMCI, mild AD, and moderate AD) differed significantly (F3,53 = 61.9; P < 0.0001). Post hoc comparisons (Fisher LSD test) showed that all groups differed significantly between each other (P < 0.0001). A similar effect was observed for corrected MMSE scores (F3,53 = 83.3; P < 0.0001). No significant difference among groups was observed in age (F3,53 = 0.21; p = n.s.) and education level (F3,53 = 1.15; p = n.s.).
Global Connectivity Strength
Two‐sample t‐tests on NOS global strengths of each patient group versus the HC group showed no significant global effects for aMCI (n.s.; P = 0.88) and mild AD (n.s.; P = 0.17), while a significant global effect was observed in moderate AD (P = 0.03). These findings suggest that only in moderate AD the connectivity disruption is widespread to an extent that enables detection of a global effect. Similar results were found using FA: aMCI (n.s.; P = 0.85), mild AD (n.s.; P = 0.43), and moderate AD (P = 0.05) as compared to controls. No significant global effect was observed for any of the groups using streamline density (aMCI n.s.; P = 0.45; mild AD n.s.; P = 0.98; moderate AD n.s.; P = 0.58).
Additional nonparametric Jonckheere–Terpstra testing revealed a significant group ordering effect for NOS (P < 0.01) and FA (P = 0.02), indicative of a progressive reduction of structural connectivity across patients groups as compared to controls (Figure 2). No significant group ordering effect was found using streamline density (n.s.; P = 0.32).
Figure 2.
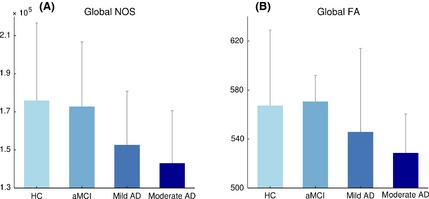
Global effect evaluated as the sum of all connection strengths per subject for NOS (A) and FA (B). A significant group ordering effect for global NOS (A) as well as for FA (B), showing progressive reduction of global structural connectivity across groups. Error bars represent standard deviations.
Connectivity Ring Analysis
To investigate whether pathways linking to nodes with lower topological distance to the selected EC/HIP were characterized by greater structural damage, the percentage of affected connections was calculated for connections directly linking to the EC/HIP and connections between first and second ring nodes. The total number of connections and nodes belonging to the first and second ring is listed in Table 2.
Table 2.
Regions and total number of connections of each ring centered on the epicenter
| LH Ring 1 | RH Ring 1 | LH Ring 2 | RH Ring 2 | |
|---|---|---|---|---|
| Number of connections | 21 | 19 | 167 | 142 |
| Node names | Thalamus | Thalamus | Accumbens area | Accumbens area |
| Caudate | Caudate | Caudal anterior cingulate | Caudal anterior cingulate | |
| Putamen | Putamen | |||
| Pallidum | Pallidum | Caudal middle frontal | Cuneus | |
| Amygdala | Amygdala | Cuneus | Inferior parietal | |
| Fusiform | Fusiform | Inferior parietal | Lateral occipital | |
| Inferior temporal | Inferior temporal | Lateral occipital | Lateral orbitofrontal | |
| Isthmus cingulate | Isthmus cingulate | Lateral orbitofrontal | Medial orbitofrontal | |
| Lingual | Lingual | Medial orbitofrontal | Middle temporal | |
| Parahippocampal | Parahippocampal | Middle temporal | Paracentral | |
| Pericalcarine | Precuneus | Paracentral | Pars opercularis | |
| Precuneus | Superior parietal | Pars opercularis | Pars orbitalis | |
| Superior frontal | Temporal pole | Pars orbitalis | Pars triangularis | |
| Superior parietal | Pars triangularis | Pericalcarine | ||
| Temporal pole | Postcentral | Postcentral | ||
| Insula | Posterior banks of superior temporal | Posterior banks of superior temporal | ||
| Posterior cingulate | Posterior cingulate | |||
| Precentral | Precentral | |||
| Rostral anterior cingulate | Rostral anterior cingulate | |||
| Rostral middle frontal | Rostral middle frontal | |||
| Superior temporal | Superior frontal | |||
| Supramarginal | Superior temporal | |||
| Frontal pole | Supramarginal | |||
| Transverse temporal | Frontal pole | |||
| Brain Stem | Transverse temporal | |||
| Insula | ||||
| Brain Stem |
LH, Left Hemisphere; RH, Right Hemisphere.
Number of Streamlines
Connectivity ring analysis based on NOS yielded a total of 15, 50, and 94 affected connections of which 13%, 16%, and 16% were first ring connections, and 60%, 62%, and 58% were second ring connections, in the aMCI, mild, and moderate AD groups, respectively.
Among all connections of the first ring, 5.3% were found to be affected (i.e., t‐test, P < 0.05) (n.s.; P = 0.11) for the aMCI group, together with just 2.9% in the second ring (n.s.; P = 0.18). For mild AD, 20.0% of connections in the first ring (P < 0.0001) and 10.3% in the second ring were found affected (P < 0.0001), and more severely, for the moderate AD group 38.0% of connections were found affected in the first ring (P < 0.0001) and 17.9% in the second ring (P < 0.0001) (Figure 3). Moreover, controlling for cortical atrophy effects, streamline density analysis revealed supportive effects for reduced EC/HIP connectivity. Using streamline density, a total of 31, 38, and 68 affected connections of which 8%, 13%, and 16% were first ring connections, and 54%, 68%, and 56% were second ring connections, in the aMCI, mild, and moderate AD groups, respectively.
Figure 3.
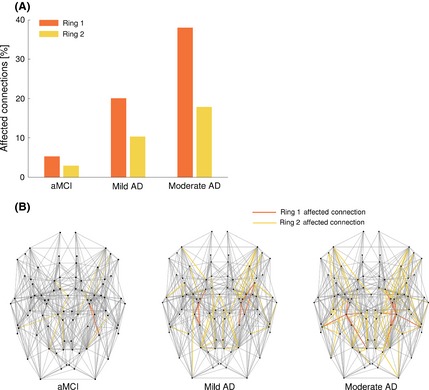
(A) Bar plot showing the percentage of affected connections for the first and second ring in each group of patients. (B) NOS‐weighted brain network for each group of patients showing the affected connections colored in orange for the first ring and in yellow for the second ring. The locations of homologous nodes, based on anatomy, have been symmetrized. For all groups, a greater percentage of connections were affected in the first ring as compared to the second (A) and a progressive increase of affected connections was observed across groups (B).
In the aMCI group, 2.6% of connections in the first ring (n.s.; P = 0.34) were found affected, with 2.3% in the second ring (n.s.; P = 0.32). For mild AD, 12.7% of connections in the first ring (P = 0.0038) and 8.6% in the second ring (P < 0.0001) were affected, while in the moderate AD, 28.2% of first ring connections (P < 0.0001) and 12.5% second ring connections (P < 0.0001) were found to be affected.
Fractional Anisotropy
Examining FA, a total of 30, 53, and 77 affected connections of which 3%, 13%, and 14% were first ring connections, and 80%, 72%, and 60% were second ring connections, in the aMCI, mild, and moderate AD groups, respectively. For the aMCI group, 2.6% connections in the first ring (n.s.; P = 0.43) and 7.9% in the second ring (P = 0.0003) were affected; for mild AD, 17.4% of connections in the first ring (P = 0.0003) and 12.3% in the second ring (P < 0.0001) were found affected; for moderate AD, 27.9% of connections in the first ring (P = 0.0001) and 14.9% in the second ring (P < 0.0001) were found to be affected.
Significant overlap was observed between the affected connections in the three patient groups for all investigated diffusion measures (NOS, streamline density, and FA), in support of the a priori selected EC/HIP epicenter, see Data S1.
Examining Specificity of the a priori Selected Epicenter
The reported proportions of affected connections, based on NOS and FA, were found to be specific for our epicenter selection consisting of the EC and HIP nodes (P < 0.02). The analysis of streamline density confirmed a specific effect for the selected epicenter in terms of affected connections in both patient with AD groups (P < 0.01), but not in the aMCI patients (n.s., P = 0.13). Less than 2% (NOS and FA) of all other possible combinations of nodes (n = 41 × 40/2 = 820 unique combinations of two nodes out of the 41 that make up one hemisphere) showed an equal or stronger effect than the EC and HIP combination, confirming the appropriateness of the a priori selected epicenter. Importantly, no node combination scored higher in both rings and in all three patient groups than our chosen EC/HIP epicenter. Furthermore, the majority of possible alternative epicenters overlapped with the original EC/HIP epicenter, containing either the EC or HIP, or included the parahippocampal gyrus which is directly connected to the HIP 35 and known to be involved in AD 36. Apart from the EC, HIP, or parahippocampal gyrus, the cingulate and middle frontal regions show strong involvement in patients with aMCI and AD, consistent with the literature 37, 38.
Relationship between Structural Connectivity and MMSE
A significant correlation between corrected MMSE scores and global connectivity strength based on NOS (r = 0.36; P = 0.02) as well as global connectivity strength based on FA (r = 0.34; P = 0.03) was observed (Figure 4A–B). For left and right HIP together, we found significant correlations with MMSE for both NOS (r = 0.43; P < 0.01), FA (r = 0.47; P < 0.01) and streamline density (r = 0.34; P = 0.03, correcting for regional volume effects) (Figure 4C–D). No significant correlation was observed between MMSE and structural connectivity strength (FA, NOS, and streamline density) of the left and right EC, nor between MMSE and global connectivity strength based on streamline density (r = 0.02; P = 0.90). Also, no significant correlations were observed between MMSE score and structural connectivity strength (FA, NOS, and streamline density) within any group taken separately.
Figure 4.
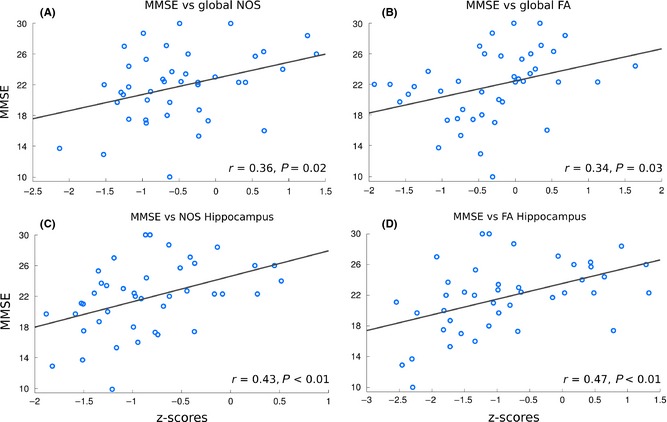
Scatter plots indicating significant correlations between cognitive status (i.e., MMSE) and structural connectivity metrics: global NOS (A), global FA (B), hippocampus NOS (C), and hippocampus FA (D).
Conclusion
Our findings demonstrate reduced structural connectivity in AD and aMCI patients with respect to controls, with a topological pattern that it is centered on the EC and HIP. White matter involvement surrounding the epicenter regions entorhinal cortex and hippocampus was found to increase with worsening of disease status from aMCI to moderate AD. In this study, graph theoretical analysis was used to assess the structural connectivity changes in AD and aMCI and our findings support the transneuronal model of disease spread.
Zhou and colleagues recently investigated four models of neurodegeneration (“nodal stress,” “trophic failure,” “shared vulnerability,” and “transneuronal spread”) and defined, for each of five different neurodegenerative disorders, critical “epicenters” as the regions whose seed‐based intrinsic functional connectivity map in healthy subjects best match the disease‐related atrophy pattern 17. They reported the vulnerability of a node to be best predicted by its greater total connectional flow and by a shorter functional path to the disease‐related epicenters. Furthermore, they put forward the transneuronal spread as the best model to fit processes of neurodegeneration 17, a conclusion consistent with our currently presented findings of epicentral structural connectivity disruptions around the EC/HIP.
The EC and HIP are two brain regions located in the medial temporal lobes. Spatially, EC and HIP are regions located close to each other. EC, indeed, is known to merge with structures belonging either to the hippocampal formation or the parahippocampal region on its medial side and with the perirhinal and the parahippocampal cortex on its lateral and posterior side 39. As it is known from early anatomical studies, EC and HIP are strongly inter connected, thus suggesting a common physiological significance of the two structures 39. Moreover, HIP is noted to be involved in declarative memory processes 40 and the EC is believed to be involved in memory functions 39. Although its specific functional contributions remain to be clearly determined, EC is considered to be the nodal point of connection between the hippocampal formation and the multimodal association areas 39. The above anatomical notions support the biological rationale of this study to focus on a disease epicenter based on EC and HIP, two brain regions closely linked and related to each other.
From a neuropathological point of view, intracellular neurofibrillary tangles (NFTs) are first found in the EC, then in the HIP and later on in other regions 2, 3, 4, 5, 6. As the selected epicenter is noted to be more involved in tau protein than beta accumulation, especially in early stages of AD 41, 42, where a random distribution of cortical amyloid beta deposits has been observed, the results of our study possibly relate to the effects of tau protein. The distribution of NFTs in the human cerebral cortex is noted to closely match, on a laminar and regional basis, that of pyramidal neurons enabling cortico‐cortical connections between and within hemispheres 43, 44, 45, 46, observations consistent with a latent interplay between cytoarchitectural features of cortical regions and their global cortico‐cortical connectivity fingerprint 47. Furthermore, neuritic plaques (NPs) and NFTs have been observed to be localized in the same cortical layers reached by the terminal axons of the perforant pathways both within the hippocampal formation and the dentate gyrus 6, 48. Thus, a connectivity‐based approach, as used in our study, provides more insight into the widespread involvement of brain regions in AD and, selecting the HIP and EC as the epicenter of connectivity disruption in AD allows for exploring the association of neuropathological changes with neurodegenerative processes.
Our current study hypothesized that AD pathology may propagate stepwise along white matter pathways and that the clinical expression of AD could be supported by selective vulnerability of subtypes of pyramidal neurons and consequent loss of cortico‐cortical connections, resulting in a so‐called disconnection syndrome 3, 4, 5, 6, 23. Results indicate that the progressive structural connectivity impairment (aMCI <mild AD <moderate AD) of pathways linking to nodes with lower topological distance to the epicenter begins in circumscribed sites and then gradually involves connection‐wise neighboring regions. Indeed, the connectivity ring analysis showed the same pattern of reduction for all metrics used (FA, NOS, streamline density) as disease severity increased. Although it may be argued that the percentage of affected tracts may be influenced by the different number of connections between ring 1 and ring 2 (i.e., ring 1 < ring 2), the progressive increase of affected connections in the first ring with the severity of the disease supports the notion that shorter topological distance from the epicenter of disease is associated with higher percentage of affected connections. The global connectivity analysis showed a significant reduction of FA and NOS only for the moderate AD group, suggesting that the connectivity ring analysis centered on the HIP/EC is more sensitive than a global approach to detect early connectivity changes associated to aMCI and AD.
The major aim of the study was to test a transneuronal spread model by examining potential epicentral structural connectivity disruptions. However, in this context, it is critical to specifically assess whether the total connectional flow (“nodal stress” model) may better explain the observed effects. Thus, to examine the “nodal stress” hypothesis, in a post hoc analysis, the connectivity ring analysis was performed for different selections of hub regions (see Data S1) that have been demonstrated to show strong connectional flow 12, 29, 49 and that have been suggested to form an anatomical substrate for integration among functional networks 30. Taking these “hub nodes” as potential disease epicenters did not result in differences between connectivity effects in the first and second ring (Figure S1) as was observed for the EC/HIP epicenter (Figure 3), suggesting that the total connectional flow did not affect our results and that the ‘‘wear and tear’’ mechanism is less likely to explain observed patterns of structural connectivity disruptions.
Global NOS and FA measures significantly correlated with MMSE, revealing a relationship between the level of dysconnectivity and cognitive status in patients. The regions belonging to the epicenter were also explored separately: Significant correlations between HIP structural connectivity and MMSE were observed, while no significant correlations were found with EC; these results suggest a significant impact of the HIP structural connectivity on the patient's cognitive status.
It is important to note that our current study does not include longitudinal data. Such data would be of great interest in order to strengthen findings and to confirm a spatiotemporal connection‐driven disease spread in AD. Moreover, although our results are consistent with transneuronal disease spread, they do not allow for a direct demonstration of transneuronal transfer of potential toxic agents. In addition, on a more technical note, a single‐tensor deterministic approach was used in our connectome reconstruction (as the data described only 12 diffusion directions), which might yield inaccurate fiber reconstructions in voxels of crossing fibers 50, 51. Future studies examining probabilistic fiber tracking and/or multi‐tensor approaches in AD structural connectivity would be of particular interest to elucidate the impact of crossing fibers voxels on our results. Using different imaging modalities, for example, functional MRI and FDG‐PET, other regions, most prominently in the default mode network including the posterior cingulate gyrus and the precuneus, have been shown to be early involved in MCI and AD 52, 53, 54, 55. These diverging functional and structural findings call for future integrative studies providing more insight into the structure–function relationship in AD.
The results of this study support an epicentral disruption of structural connectivity in aMCI and AD, consistent with the effects of a potential transneuronal spread of disease. EC and HIP together represent a good target to be considered the epicenter of anatomical connectivity impairment in aMCI and AD.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1. Methodological details and additional analyses.
Figure S1. Bar plot showing the percentage of affected connections (in terms of NOS) for the first and second ring in each group of patients when the four strongest hub nodes, having the highest number of connections, (superior parietal and putamen, left and right) were taken as the epicenter. Connectivity disruptions were not found to be stronger in the first ring compared to the second ring, suggesting that hub nodes are not specifically targeted in aMCI and AD as hypothesized by the “nodal stress” model of neurodegeneration. Proportions of affected connections observed in the aMCI/mild AD/moderate AD patient groups were: 3.7 / 8.4 / 17.5% (first ring); 2.8 / 10.4 / 18.8% (second ring).
Acknowledgments
We are thankful to Federica Scrascia, MD for her assistance in the recruitment of the subjects enrolled in this study. MPvdH was supported by a VENI grant (#451‐12‐001) of the Netherlands Organization for Scientific Research (NWO) and by a Fellowship of the Brain Center Rudolf Magnus.
The first two authors contributed equally to this work.
References
- 1. Kukull WA, Bowen JD. Dementia epidemiology. Med Clin North Am 2002;86:573–590. [DOI] [PubMed] [Google Scholar]
- 2. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Del Tredici K. Alzheimer's pathogenesis: Is there neuron‐to‐neuron propagation? Acta Neuropathol 2011;121:589–595. [DOI] [PubMed] [Google Scholar]
- 4. Reid AT, Evans AC. Structural networks in Alzheimer's disease. Eur Neuropsychopharmacol 2013;23:63–77. [DOI] [PubMed] [Google Scholar]
- 5. Delbeuck X, Van Der LM, Collette F. Alzheimer's disease as a disconnection syndrome? Neuropsychol Rev 2003;13:79–92. [DOI] [PubMed] [Google Scholar]
- 6. De Lacoste MC, White CL 3rd. The role of cortical connectivity in Alzheimer's disease pathogenesis: A review and model system. Neurobiol Aging 1993;14:1–16. [DOI] [PubMed] [Google Scholar]
- 7. Devanand DP, Pradhaban G, Liu X, et al. Hippocampal and entorhinal atrophy in mild cognitive impairment: Prediction of Alzheimer disease. Neurology 2007;68:828–836. [DOI] [PubMed] [Google Scholar]
- 8. Pennanen C, Kivipelto M, Tuomainen S, et al. Hippocampus and entorhinal cortex in mild cognitive impairment and early AD. Neurobiol Aging 2004;25:303–310. [DOI] [PubMed] [Google Scholar]
- 9. Tijms BM, Wink AM, de Haan W, et al. Alzheimer's disease: Connecting findings from graph theoretical studies of brain networks. Neurobiol Aging 2013;34:2023–2036. [DOI] [PubMed] [Google Scholar]
- 10. Van den Heuvel MP, Mandl RCW, Stam CJ, Kahn RS, Hulshoff Pol HE. Aberrant frontal and temporal complex network structure in schizophrenia: A graph theoretical analysis. J Neurosci 2010;30:15915–15926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lo C‐Y, Wang P‐N, Chou K‐H, Wang J, He Y, Lin C‐P. Diffusion tensor tractography reveals abnormal topological organization in structural cortical networks in Alzheimer's disease. J Neurosci 2010;30:16876–16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci 2009;29:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011;71:35–48. [DOI] [PubMed] [Google Scholar]
- 14. de Haan W, Mott K, van Straaten E, Scheltens P, Stam CJ. Activity dependent degeneration explains hub vulnerability in Alzheimer's Disease. PLoS Comput Biol 2012;8:e1002582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Appel SH. A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Ann Neurol 1981;10:499–505. [DOI] [PubMed] [Google Scholar]
- 16. Salehi A, Delcroix J‐D, Belichenko PV, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 2006;51:29–42. [DOI] [PubMed] [Google Scholar]
- 17. Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 2012;73:1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frost B, Diamond MI. Prion‐like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 2010;11:155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S‐J, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell‐to‐cell transmission of non‐prion protein aggregates. Nat Rev Neurol 2010;6:702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol 2001;58:1985–1992. [DOI] [PubMed] [Google Scholar]
- 22. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 23. Scrascia F, Curcio G, Ursini F, et al. Relationship among diffusion tensor imaging, EEG activity, and cognitive status in mild cognitive impairment and Alzheimer's Disease patients. J Alzheimers Dis 2013;38:939–950. [DOI] [PubMed] [Google Scholar]
- 24. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 25. Morris JC. The clinical dementia rating (CDR): Current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 26. Englund B, Brun A, Gustafson L, et al. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 1994;57:416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Román GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS‐AIREN International Workshop. Neurology 1993;43:250–260. [DOI] [PubMed] [Google Scholar]
- 28. McKeith I, O'Brien J, Ballard C. Diagnosing dementia with Lewy bodies. Lancet 1999;354:1227–1228. [DOI] [PubMed] [Google Scholar]
- 29. Hagmann P, Cammoun L, Gigandet X, et al. Mapping the structural core of human cerebral cortex. PLoS Biol 2008;6:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van den Heuvel MP, Sporns O. Rich‐Club organization of the human connectome. J Neurosci 2011;31:15775–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Reus MA, van den Heuvel MP. Estimating false positives and negatives in brain networks. NeuroImage 2013;70:402–409. [DOI] [PubMed] [Google Scholar]
- 32. Verstraete E, Veldink JH, van den Berg LH, Van den Heuvel MP. Structural brain network imaging shows expanding disconnection of the motor system in amyotrophic lateral sclerosis. Hum Brain Mapp 2014;35:1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt R, Verstraete E, de Reus MA, Veldink JH, van den Berg LH, van den Heuvel MP. Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum Brain Mapp 2014;35:4386–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zalesky A, Fornito A, Bullmore ET. Network‐based statistic: Identifying differences in brain networks. NeuroImage 2010;53:1197–1207. [DOI] [PubMed] [Google Scholar]
- 35. Powell HWR, Guye M, Parker GJM, et al. Noninvasive in vivo demonstration of the connections of the human parahippocampal gyrus. NeuroImage 2004;22:740–747. [DOI] [PubMed] [Google Scholar]
- 36. Van Hoesen GW, Augustinack JC, Dierking J, Redman SJ, Thangavel R. The parahippocampal gyrus in Alzheimer's disease: Clinical and preclinical neuroanatomical correlates. Ann N Y Acad Sci 2000;911:254–274. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y, Schuff N, Jahng GH, et al. Diffusion tensor imaging of cingulum fibers in midl cognitive impairment and Alzheimer disease. Neurology 2007;68:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davatzikos C, Fan Y, Wu X, Shen D, Resnick SM. Detection of prodromal Alzheimer's disease via pattern classification of magnetic resonance imaging. Neurobiol Aging 2008;29:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Canto CB, Wouterlood FG, Witter MP. What does the anatomical organization of the entorhinal cortex tell us? Neural Plast 2008;2008:381243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mallio CA, Assenza G, Occhicone F, Errante Y, Zobel BB, Quattrocchi CC. Transient global amnesia and hippocampal foci of restricted diffusion. BMJ Case Rep 2013;10:2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hatashita S, Yamasaki H. Diagnosed mild cognitive impairment due to Alzheimer's Disease with PET biomarkers of beta amyloid and neuronal dysfunction. PLoS ONE 2013;8:e66877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prescott JW, Guidon A, Doraiswamy PM, Choudhury KR, Liu C, Petrella J. The alzheimer structural connectome: Changes in cortical network topology with increased amyloid plaque burden. Radiology 2014;273:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gómez‐Isla T, Price J. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 1996;16:4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lewis DA, Campbell MJ, Terry RD, Morrison JH. Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: A quantitative study of visual and auditory cortices. J Neurosci 1987;7:1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci U S A 1985;82:4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Hoesen GW. Ventromedial temporal lobe anatomy, with comments on Alzheimer's disease and temporal injury. J Neuropsychiatry Clin Neurosci 1997;9:331–341. [DOI] [PubMed] [Google Scholar]
- 47. Scholtens LH, Schmidt R, de Reus MA, van den Heuvel MP . Linking macroscale graph analytical organization to microscale neuroarchitectonics in the macaque connectome. J Neurosci 2014;34:12192–12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hyman BT, Kromer LJ, Van Hoesen GW. A direct demonstration of the perforant pathway terminal zone in Alzheimer's disease using the monoclonal antibody Alz‐50. Brain Res 1988;450:392–397. [DOI] [PubMed] [Google Scholar]
- 49. Honey CJ, Sporns O, Cammoun L, et al. Predicting human resting‐state functional connectivity from structural connectivity. Proc Natl Acad Sci U S A 2009;106:2035–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johansen‐Berg H, Rushworth MF. Using diffusion imaging to study human connectional anatomy. Annu Rev Neurosci 2009;32:75–94. [DOI] [PubMed] [Google Scholar]
- 51. Jbabdi S, Johansen‐Berg H. Tractography: Where do we go from here? Brain Connect 2011;1:169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Greicius MD, Srivastava G, Reiss AL, Menon V. Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: Evidence from functional MRI. Proc Natl Acad Sci U S A 2004;101:4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yao Z, Zhang Y, Lin L, Zhou Y, Xu C, Jiang T. Alzheimer's Disease Neuroimaging Initiative. Abnormal cortical networks in mild cognitive impairment and Alzheimer's disease. PLoS Comput Biol 2010;6:e1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petrella JR, Sheldon FC, Prince SE, Calhoun VD, Doraiswamy PM. Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology 2011;76:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chhatwal JP, Schultz AP, Johnson K, et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 2013;81:736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methodological details and additional analyses.
Figure S1. Bar plot showing the percentage of affected connections (in terms of NOS) for the first and second ring in each group of patients when the four strongest hub nodes, having the highest number of connections, (superior parietal and putamen, left and right) were taken as the epicenter. Connectivity disruptions were not found to be stronger in the first ring compared to the second ring, suggesting that hub nodes are not specifically targeted in aMCI and AD as hypothesized by the “nodal stress” model of neurodegeneration. Proportions of affected connections observed in the aMCI/mild AD/moderate AD patient groups were: 3.7 / 8.4 / 17.5% (first ring); 2.8 / 10.4 / 18.8% (second ring).