

Summary
Aims
Connexin 43 (Cx43) has been reported to be involved in neuropathic pain, but whether it contributes to morphine antinociceptive tolerance remains unknown. The present study investigated the role of spinal Cx43 in the development of morphine tolerance and its mechanisms in rats.
Methods
Morphine tolerance was induced by intrathecal (i.t.) administration of morphine (15 μg) daily for seven consecutive days. The analgesia effect was assessed by hot‐water tail‐flick test. Expression of proteins was detected by Western blot and immunohistochemistry assay.
Results
Chronic morphine markedly increased the expression of spinal Cx43. Gap26, a specific Cx43 mimic peptide, attenuated not only morphine antinociceptive tolerance, but also the up‐regulation of spinal Cx43 expression, the activation of astrocytes, and N‐methyl‐D‐aspartic acid (NMDA) receptors (NR1 and NR2B subunits), as well as the decreased GLT‐1 expression induced by chronic morphine. MK‐801, a noncompetitive NMDA receptors antagonist, suppressed the chronic morphine‐induced spinal Cx43 up‐regulation, astrocytes activation and decline of GLT‐1 expression.
Conclusions
The spinal astrocytic Cx43 contributes to the development of morphine antinociceptive tolerance by activating astrocytes and NMDA receptors, and inhibiting GLT‐1 expression. We also demonstrate that the role of interaction between the spinal astrocytic Cx43 and neuronal NMDA receptors is important in morphine tolerant rats.
Keywords: Astrocyte, Connexin 43, Glutamate transporter‐1, Morphine tolerance, N‐methyl‐D‐aspartic acid receptor
Introduction
Although opioids, including morphine, are commonly used in clinical management of acute and chronic pain, the development of tolerance to its analgesia properties significantly hampers its clinical utility. The molecular and cellular mechanisms responsible for this phenomenon remain elusive. Increasing evidence has shown that astrocytes play important roles in the development of morphine antinociceptive tolerance 1, 2, 3. Chronic morphine induces the activation of astrocytes in the spinal cord, posterior cingulated cortex and hippocampus. Inhibiting astroglial activation by a glial inhibitor (fluorocitrate or propentofylline) significantly attenuated morphine antinociceptive tolerance 1, 2.
Astrocytes are characterized by forming gap junction (GJ)‐coupled networks 4. In the central nervous system (CNS), glial GJs play important roles in the molecular exchanges and informational communications between adjacent cells, which are involved in spatial buffering of extracellular potassium ions, glutamate and other signal molecules, energy sources, and mediation of intercellular calcium waves. 5. GJs are consisted of connexins (Cxs), with the name of respective molecular weights 6. In the mammalian nervous system, at least 11 Cxs (Cx26, Cx29, Cx30, Cx32, Cx36, Cx43, Cx31, Cx37, Cx45, Cx47, and Cx57) have been identified 7, and different cell populations contain distinctive sets of Cxs. In the spinal cord, Cx43 is the most abundant Cx, mainly expressed in astrocytes 7.
Recently, a number of studies have demonstrated the potential involvement of GJs and Cxs in neuropathic pain 8, 9, 10, 11, 12, 13, 14, 15, 16. The Cx43 expression is increased in several neuropathic pain models, such as spinal cord injury (SCI), chronic constriction injury (CCI), and neuroinflammation 9, 10, 11. Intrathecal delivery of carbenoxolone, a GJ decoupler 12, 13, or inhibiting Cx43 expression using RNA interference 14, 15, ameliorates neuropathic pain induced by sciatic nerve inflammation or CCI, suggesting that the spinal Cx43 and GJs play important roles in neuropathic pain. However, the potential role of the spinal Cx43 in morphine antinociceptive tolerance has not been investigated. As it is hypothesized that opioid tolerance and pathological pain may share common cellular mechanisms 17, we speculate that the spinal Cx43 might mediate morphine antinociceptive tolerance. To confirm this hypothesis, in the present study, we examined the effect of chronic morphine on the spinal Cx43 expression and roles of Cx43 in the development of morphine antinociceptive tolerance in rats.
It is well‐known that glutamate 18, 19 and N‐methyl‐D‐aspartic acid (NMDA) receptors participate in the development of morphine tolerance 20, 21, 22, 23, 24, 25. Both in patients and in experimental animals with long‐term use of morphine, morphine tolerance was developed accompanied by an increase in glutamate level in the cerebrospinal fluid 18, 19. The extracellular glutamate is cleared by glutamate transporters (GTs), which strictly regulate the homeostasis of the extracellular glutamate level 26. There are at least five indentified GTs. Among them, glutamate transporter‐1 (GLT‐1) mediates 90% extracellular glutamate uptake in the CNS. Several lines of research have shown that GTs play an important role in the development of morphine antinociceptive tolerance 27, 28, 29. Moreover, it is reported that GJ blockage prevents neuropathic pain by preventing the activation of NMDA receptors 13, and the expression of GLT‐1 is up‐regulated when Cx43 is knocked out in astrocytes 30. Therefore, we further investigated the potential roles of NMDA receptors and GLT‐1 in the Cx43‐mediated morphine antinociceptive tolerance in rats.
Materials and methods
Animals
Adult male Sprague‐Dawley rats (250–280 g) were housed individually with free access to water and food in a temperature (22 ± 2°C) and light control (12 h light/dark cycle) quiet room. All experimental procedures were approved by the local animal care committee and were carried out strictly in compliance with the NIH guidelines for the care and use of laboratory animals.
Intrathecal Catheter Implantation
For i.t. delivery of drugs, rats were implanted with i.t. catheters according to the methods described previously 31. Briefly, rats were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally), and then, a sterile polyethylene (PE‐10) tube filled with 0.9% saline (NS) was inserted through L5/L6 intervertebral space, and the tip of the tube was placed at the spinal lumbar enlargement level.
Induction of Morphine Antinociceptive Tolerance and Behavioral Test
Morphine antinociceptive tolerance was induced by i.t. injection of morphine (15 μg daily, Qing‐hai Pharmaceutical Factory, Xining, China) for 7 days. To investigate the effect of Gap26 or MK‐801 on morphine tolerance, Gap26 (15 μg, Anaspec, Fremont, CA, USA), MK‐801 (3.3 μg, Sigma, St. Louis, MO, USA), or vehicle (NS) was given intrathecally 30 min before each morphine administration. The drug or vehicle solution was i.t. injected in a volume of 10 μL followed by additional 10 μL of NS to flush the cather.
On odd days, morphine antinociception was evaluated by hot‐water tail‐flick test according to the methods described previously 31. Briefly, rats were restricted in a plastic container (22 × 6 cm), and distal one‐third of the tail was immersed into the water maintained at 50 ± 0.2°C. The latency response was defined by rapid removal of tail from hot water. A cutoff time of 15 second was used to avoid tissue damage. The percentage of maximal possible antinociceptive effect (%MPE) was calculated by comparing the tail‐flick latency before (baseline latency, BL) and after drug injection (test latency, TL) using the equation: %MPE = [(TL − BL)/(cut‐off time − BL)] × 100.
Immunohistochemistry
Immediately after behavioral test on the indicated days, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused through the ascending aorta with 4% paraformaldehyde. The spinal cord lumbar enlargements were quickly removed and postfixed in the same fixative for 2 h and then transferred into 30% sucrose until sectioning. Transverse spinal sections (20 μm) were cut on a cryostat. The procedure for immunohistochemistry was performed as described previously 31. In brief, the sections were incubated with the antibody for Cx43 (1:1000; Santa Cruz, Heidelberg, Germany), or GFAP (astrocyte marker, 1:800; Millipore, Billerica, MA, USA) overnight at 4°C. Then, the sections were incubated with corresponding Cy3‐ or FITC‐conjugated secondary antibody (1:800, ZSGB‐BIO, Beijing, China) for 2 h at room temperature (RT). For double immunofluorescence, sections were incubated with a mixture antibodies for anti‐Cx43 and GFAP, or NeuN (neuronal marker, 1:400, Millipore) or OX42 (microglial marker, 1:100, Millipore) followed by a mixture of Cy3‐ and FITC‐conjugated secondary antibodies. The stained sections were examined with an Olympus fluorescence microscope, and images were captured with a CCD spot camera. Nonspecific staining was determined by omitting the primary antibodies.
Western Blot Assay
On odd days, immediately after behavioral test, rats were killed and the spinal cord lumbar enlargements were rapidly removed. The preparation of proteins and Western blot were performed as described previously 25. In brief, tissues were homogenized in ice‐cold lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) containing protease and phosphatase inhibitors (Sigma, USA). Protein samples were separated on SDS–PAGE gradient gel (8–12%) and electrotransfered onto PVDF membranes. The membranes were probed with the antibody for Cx43 (1:5000), GFAP (1:10000), NR1 (1:800, Cell Signaling Technology), NR2B (1:2000, Abcam, Cambridge, UK), p‐NR1 (1:1000, Millipore), p‐NR2B (1:1000, Millipore), or GLT‐1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), and with GAPDH antibody as loading controls. Then, the membranes were incubated with HRP‐conjugated secondary antibody, developed in ECL solution and exposed onto X‐ray film. For quantification, the films were scanned, and the density of specific bands was measured and normalized with the control band (NS group).
Statistical Analysis
All data were presented as mean ± SEM. Data were analyzed using repeated measures ANOVA (for the behavioral data) or one‐way ANOVA (for Western blot and immunohistochemical data). Whenever applicable, post hoc analysis (LSD) was performed for comparisons between specific groups. The significance level was defined as P < 0.05.
Results
Chronic Morphine Treatment Up‐regulates the Expression of Spinal Astrocytic Cx43
The results from Western blot assay showed that in morphine‐treated rats, i.t. administration of morphine for seven consecutive days induced a significant increase in the expression of spinal Cx43 compared with control rats (Figure 1A). The expression of spinal Cx43 began to increase significantly following 3 days of repeated morphine treatment (P < 0.001) and gradually enhanced from 5 to 7 days.
Figure 1.
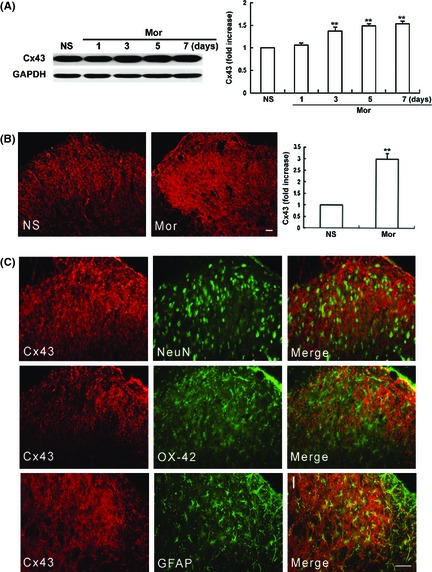
Chronic morphine treatment up‐regulates the expression and immunoreactivity of the spinal connexin 43 (Cx43). (A) Western blot assay showed that the spinal Cx43 expression was increased progressively after exposure to morphine (15 μg, intrathecal [i.t.]). n = 5. (B) Intrathecal injection of morphine for 7 days markedly increased the spinal Cx43 immunoreactivity (IR), n = 6. (C) Double immunostaining indicates that the spinal Cx43 was colocalized with GFAP (astrocytes marker), but not with NeuN (neuron marker) or OX‐42 (microglia marker). Scale bar: 50 μm.
Similarly, immunohistochemistry results showed that the Cx43‐immunoreactivity (IR) was up‐regulated markedly after chronic morphine treatment compared with the NS group (Figure 1B). Moreover, double immunofluorescent staining showed that the spinal Cx43 was exclusively colocalized with astrocytes (GFAP), but not with neurons (NeuN) or microglial cells (Ox‐42) (Figure 1C), which was consistent with the previous reports 32.
Gap26 Attenuates the Development of Morphine Antinociceptive Tolerance and Chronic Morphine‐Induced Increase in Spinal Cx43 Expression
To explore whether the inhibition of Cx43 would prevent the development of morphine antinociceptive tolerance, Gap26 (15 μg, i.t.), a mimetic peptide of Cx43, served as a relatively specific blocker of Cx43 33, 34, 35, was administered 30 min before each morphine treatment for consecutive 7 days. As shown in Figure 2A, morphine injection produced robust analgesia to noxious stimuli on day 1. Repeated morphine treatment produced a progressive decline of antinociception, indicating the development of tolerance to morphine analgesia. Gap26 alone did not affect the basal pain threshold. However, coadministration of Gap26 with morphine considerably delayed the decline of morphine antinociception. On day 7, the %MPE in Gap‐Mor group (45.83 ± 4.17) was markedly higher than the one in NS‐Mor group (23.88 ± 2.81) (P < 0.001). Moreover, the Western blot data showed that coadministration of Gap26 with morphine inhibited the chronic morphine‐induced increase in the spinal Cx43 expression (Figure 2B). These data suggested that Cx43 is involved in the development of morphine antinociceptive tolerance.
Figure 2.
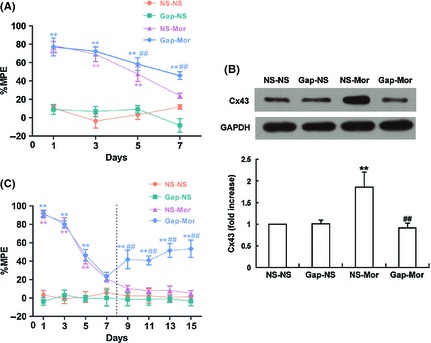
Effects of Gap26 on the development of morphine antinociceptive tolerance (A), chronic morphine‐induced increase in the spinal connexin 43 (Cx43) expression (B), and the established morphine antinociceptive tolerance (C). Gap26 (15 μg), a specific Cx43 mimic peptide, or vehicle (10 μL) was intrathecally injected 30 min before daily morphine (15 μg) treatment. The vertical dashed line in (C) indicated that Gap26 administration was initiated on day 8 following the development of morphine tolerance and continued for the next 8 days. MPE%, percentage of maximal possible antinociceptive effect. **P < 0.001 versus NS‐NS group; ## P < 0.001 versus NS‐Mor group. n = 6.
Gap26 Ameliorates the Established Morphine Antinociceptive Tolerance
To investigate whether Gap26 would reverse the established morphine antinociceptive tolerance, Gap26 (15 μg, i.t.) was administrated 30 min before morphine injection on day 8 and continued for the next 8 consecutive days. As shown in Figure 2C, on day 9, 11, 13, and 15, the analgesic effect of i.t. morphine in NS‐Mor group were further declined, having no statistical difference as compared with NS‐NS group (P > 0.05). However, morphine produced significant analgesic efficacy in Gap26‐Mor group (P < 0.001 vs. NS‐Mor group) on day 9, 11, 13, and 15. These results indicated that Gap26 could partly ameliorate the established morphine antinociceptive tolerance.
Gap26 Suppresses Chronic Morphine‐Induced Activation of Astrocytes
As the spinal astrocytes have been found to be activated by chronic morphine treatment and Cx43 is mainly expressed in astrocytes, we examined the effect of Gap26 on morphine‐induced astrocyte activation. Immunoblotting assay (Figure 3A) indicated that the spinal GFAP expression was obviously increased after 7 days of morphine exposure (P < 0.05), indicating that astrocytes are activated by chronic morphine treatment. The increased expression of GFAP was blocked by coadministration of Gap26 with morphine (P < 0.001). Similarly, the immunofluorescence staining showed that Gap26 also considerably attenuated chronic morphine‐induced increase in the spinal GFAP‐IR level (Figure 3B).
Figure 3.
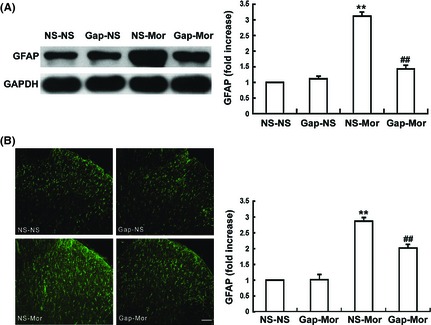
Gap26 suppresses chronic morphine‐induced activation of the spinal astrocytes. (A) Western blot showed that coadministration of Gap26 (15 μg, intrathecal [i.t.]) with morphine significantly prevented the chronic morphine‐induced spinal GFAP up‐regulation. (B) Immunofluroescence assay showed that chronic morphine induced an increase in spinal GFAP‐IR. Gap26 attenuated the chronic morphine‐induced increase in spinal GFAP‐IR. Scale bar: 50 μm. Data are the mean ± SEM (n = 6) **P < 0.001 versus NS‐NS group. ## P < 0.001 versus NS‐Mor group.
Gap26 Inhibits Chronic Morphine‐Induced Down‐Regulation of Spinal GLT‐1
Figure 4 shows an obvious decrease in the spinal GLT‐1 expression after 7 days of continuous morphine infusion (P < 0.001), indicating that GLT‐1 is down‐regulated by chronic morphine treatment. However, this inhibitory effect was distinctly attenuated by coadministration of Gap26 with morphine (P < 0.001). Gap26 alone did not alter the basal expression of spinal GLT‐1.
Figure 4.
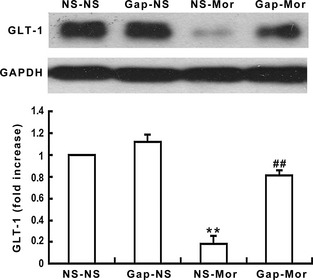
Gap26 inhibits chronic morphine‐induced down‐regulation of spinal GLT‐1 expression. The spinal GLT‐1 expression was decreased after chronic morphine administration. Gap26 significantly suppressed the decreased GLT‐1 expression. Data are presented as mean ± SEM (n = 4). **P < 0.001 versus NS‐NS group. ## P < 0.001 versus NS‐Mor group.
Gap26 Prevents Chronic Morphine‐Induced Phosphorylation of NR1 and NR2B Subunits of NMDA Receptors
As it has reported that the GJ blockage prevents neuropathic pain by preventing the activation of NMDA receptors 13, 36, the effects of Gap26 on chronic morphine‐induced alterations in NR1 and NR2B expressions were therefore examined. As shown in Figure 5, chronic morphine not only up‐regulated the expressions of NR1 and NR2B (Figure 5A and B), but also increased the phosphorylations of NR1 and NR2B (Figure 5C and D) (P < 0.001) in the spinal cord compared with the NS group (P < 0.001). These increases in the expression levels and the phosphorylations of the two subunits were inhibited by Gap26 coadministration (P < 0.001). Gap26 or NS alone did not induce changes in the basal expression levels and phosphorylations of NR1 and NR2B.
Figure 5.
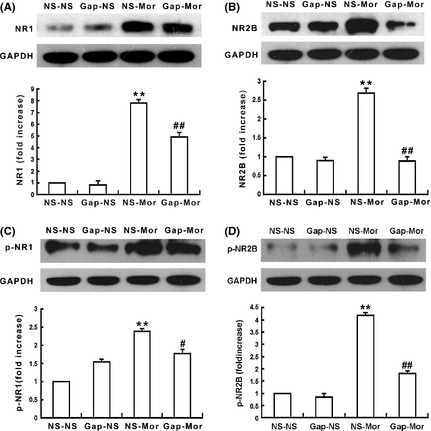
Gap26 prevents chronic morphine‐induced increases in the expressions and phosphorylations of spinal N‐methyl‐D‐aspartic acid (NMDA) receptor NR1 and NR2B subunits. Chronic morphine treatment up‐regulated the expressions of spinal NR1 (A) and NR2B (B), and increased phosphorylate (p) NR1 (p‐NR1) (C) and NR2B (p‐NR2B) (D); Gap26 significantly prevented the chronic morphine‐induced up‐regulation of NR1, p‐NR1, NR2B, and p‐NR2B expressions, but Gap26 alone did not affect the basal levels of them. Data are presented as mean ± SEM (n = 4). **P < 0.001 versus NS‐NS group. # P < 0.05, ## P < 0.001 versus NS‐Mor group.
MK‐801 Attenuates Chronic Morphine‐induced Up‐regulation of the Spinal Cx43 and GFAP Expression, and Down‐regulation of the GLT‐1 Expression
Interestingly, we also observed that MK‐801, a noncompetitive antagonist of NMDA receptors, not only prevented the development of morphine antinociceptive tolerance (Figure 6A), but also significantly attenuated chronic morphine‐induced increases in the spinal Cx43 (Figure 6B) (P < 0.001) and GFAP (Figure 6C) (P < 0.05) expressions and the decrease in the spinal GLT‐1 expression (Figure 6D) (P < 0.001) when coadministrated with morphine. MK‐801 alone did not affect the basic levels of their expressions.
Figure 6.
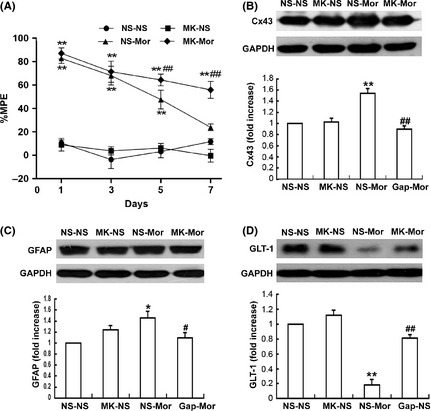
Effects of MK‐801 on morphine antinociceptive tolerance and chronic morphine‐induced changes of the spinal Cx43, GFAP and GLT‐1. (A) MK‐801 (3.3 μg, intrathecal [i.t.]) was injected 30 min before daily morphine (15 μg, i.t.) treatment. MK‐801 distinctly reduced the chronic morphine‐induced increase in levels of the spinal Cx43 (B) and GFAP expression (C), and the decrease in GLT‐1 expression (D). Data are presented as mean ± SEM (n = 4). *P < 0.05, **P < 0.001 versus NS‐NS group. # P < 0.05, ## P < 0.001 versus NS‐Mor group.
Discussions
Glial cells are now recognized as important contributors in the development of morphine antinociceptive tolerance. In the present study, we provide a novel mechanism that the spinal astrocytic Cx43 mediates morphine antinociceptive tolerance by activating astrocytes and NMDA receptors (including NR1 and NR2B subunits), and inhibiting GLT‐1 expression.
In the CNS, astrocytes are the most abundant cells which communicate with each other mainly through GJs 37. Cx43 is the major component of astrocytic GJs 7. Accumulating evidence indicates that Cx43 and GJs are involved in neuropathic pain. Wu et al. 38 reported that the expression levels of GFAP (a marker of astroglia) and Cx43 in the spinal cord are significantly increased after CCI of the sciatic nerve in rats. Similarly, Cx43 expression is up‐regulated in the trigeminal ganglion in rats with a CCI of the infraorbital nerve and genetic inactivation of Cx43 by RNAi attenuates the pain‐like behavior in this model 14, 15. Intrathecal or intracisternal treatment with carbenoxolone, a GJ decoupler, markedly suppresses subcutaneous formalin‐induced thermal hyperalgesia 39 and thrombotic ischemia‐induced mechanical allodynia in rats 40. Collectively, the above results suggest that Cx43 and GJs contribute to the development of neuropathic pain. However, whether the spinal Cx43 participates in the development of morphine antinociceptive tolerance is unknown.
In the present study, we provided the first evidence that the spinal astrocytic Cx43 contributes to morphine antinociceptive tolerance in rats. This is supported by the following results: (1) the spinal Cx43 expression and Cx43‐IR are significantly enhanced by chronic morphine treatment; (2) the spinal Cx43 is exclusively colocalized with astrocytes (GFAP), which is consistent with the previous studies 7; and (3) i.t. treatment with Gap26, a specific Cx43 mimic peptide, markedly attenuates the development of morphine antinociceptive tolerance, and partly attenuates the established morphine antinociceptive tolerance. As it was shown that the spinal astrocytic Cx43 is involved in the development of neuropathic pain 12, 13, 14, 15, 38, 39, 40, this study provides another novel evidence to confirm the well‐known hypothesis that opioid tolerance and phathological pain may share common cellular mechanisms 17.
As the spinal astrocytes have been shown to interconnect by Cx43 and to be colocalized with Cx43, we explore the effect of chronic morphine‐induced increase in Cx43 on the spinal astrocytic activation. Consistent with the previous studies 1, 2, 3, we found that chronic morphine induced astrocyte activation, as evidenced by increased GFAP expression and astrocyte hypertrophy in the spinal dorsal horn. Noteworthily, we found that Gap26 also markedly reduced chronic morphine‐induced increases in GFAP expression and GFAP‐IR level in the spinal cord, suggesting the stimulatory effect of Cx43 on the spinal astrocyte activation. Astrocytes, the principal glial cell type, have been shown to participate in the development of morphine tolerance 1, 2, 3, 41, 42, 43. However, the biological molecules by which chronic morphine induces the activation of astrocytes are still incompletely clear. Based on our above results, it was suggested that the spinal astrocytic Cx43 may be an inducer for the activation of astrocytes in the chronic morphine‐treated rats. Notably, increasing studies confirmed that the activated glia can release some proinflammatory cytokines, such as interleukin (IL)‐1β, IL‐6, monocyte chemoattractant protein‐1 (MCP‐1), and tumor necrosis factor (TNF)‐α, which participate in the development of morphine tolerance 2, 3, 44, 45, 46. Therefore, we speculated that chronic morphine‐induced up‐regulation of the spinal Cx43 expression activates astrocytes, which in turn trigger release of these mediators, leading to the tolerance to morphine analgesia. To confirm this hypothesis, further studies are needed.
Abundant evidence suggests that glutamate and NMDA receptors participate in the development of morphine tolerance. The level of extracellular glutamate is actively and tightly regulated by GTs 26. GLT‐1 mediates 90% extracellular glutamate uptake in the CNS. The spinal GTs are down‐regulated in chronic morphine‐treated rats 27, 28, 29. Pharmacological inhibition of GT by trans‐pyrrolidine‐2‐4‐dicarboxylate potentials, whereas activation of GT by riluzole reduces the development of morphine antinociceptive tolerance 27. In addition, ceftriaxone inhibits morphine antinociceptive tolerance through activating GLT‐1, increasing glutamate uptake, and attenuating extracellular glutamate levels 47. GLT‐1 is mainly expressed in astrocytes, which is in accordance with Cx43 7, 48. Noteworthily, a recent study demonstrated that GLT‐1 expression in cerebral cortex is increased in the conditional Cx43 knockout mice, suggesting that Cx43 may have a regulatory effect on GLT‐1 30. However, it was unclear the relationship between spinal Cx43 and GLT‐1 in the chronic morphine‐treated rats. Therefore, the present study explored the effect of spinal Cx43 activation on GLT‐1 in morphine tolerant rats. In agreement with the previous studies 27, 28, 29, 47, our data showed that chronic morphine treatment induced down‐regulation of the spinal GLT‐1 expression. Importantly, blocking Cx43 by Gap26 markedly depressed the chronic morphine‐induced decrease in the spinal GLT‐1 expression, suggesting that the spinal Cx43 may regulate GLT‐1 expression, and the decreased GLT‐1 may be implicated in the spinal Cx43‐mediated morphine antinociceptive tolerance. Conceivably, the chronic morphine‐induced down‐regulation of GLT‐1 expression would enhance the availability of extracellular glutamate and then would increase the probability of excitatory amino acid receptors, such as NMDA receptors. The activation of NMDA receptors and downstream signal transduction pathway plays a critical role in tolerance to morphine analgesia 20, 49, 50, 51.
Due to the above‐mentioned findings that the spinal astrocytic Cx43 regulates GLT‐1 expression which regulates the functional activity of NMDA receptors 36, we further investigated the effect of spinal Cx43 on the activation of NMDA receptor subunits (NR1 and NR2 subunits) and the possible interaction between them in the chronic morphine‐treated rats. Consistent with the previous studies 20, 21, 22, 23, 24, 25, this study showed that both NMDA receptor NR1 and NR2B subunits participated in morphine antinociceptive tolerance. Importantly, we found that blocking the spinal Cx43 with Gap26 not only attenuated morphine antinociceptive tolerance, but also inhibited chronic morphine‐induced increases in expressions and phosphorylations of NR1 and NR2B subunits, indicating that the spinal Cx43 may contribute to the increased activation of NMDA receptors by chronic morphine. Moreover, MK‐801 coadministration with morphine attenuated the chronic morphine‐induced increase in spinal Cx43 expression, suggesting the involvement of NMDA receptor activation in the increased expression of the spinal Cx43 by chronic morphine.
As the spinal Cx43 is mainly colocalized with astrocytes and NMDA receptors are mainly expressed on the membrane of postsynaptic neurons, we proposed that the spinal Cx43‐mediated astrocyte–neuron signaling may represent a novel mechanism underlying the development of morphine antinociceptive tolerance in rats. We speculated that there might be a feedback mechanism to modulate the astrocyte–neuron signaling communication. That is, chronic morphine‐induced increase in Cx43 activates astrocytes and reduces extracellular glutamate uptake by down‐regulating the GLT‐1 expression, and accelerates extracellular glutamate accumulation, resulting in activation of NMDA receptors (including NR1 and NR2B subunits); and the activated NMDA receptors enhance the Cx43 expression, reduce the GLT‐1 expression, and activate astrocytes. This interaction between the spinal astrocytic Cx43 and neuronal NMDA receptors might form a positive feedback and aggravate the tolerance to morphine analgesia in chronic morphine‐treated rats. However, the mechanisms of the neuron–astrocyte interaction in the development of morphine antinociceptive tolerance are complicated and require further studies.
In conclusion, the present study demonstrated for the first time that the spinal astrocytic Cx43 contributes to the development of morphine antinociceptive tolerance by activating astrocytes and NMDA receptors (NR1 and NR2B subunits) and inhibiting GLT‐1 expression in the spinal cord of rats. Although the role of the spinal Cx43 in the tolerance to morphine analgesia may involve multiple pathways, the spinal Cx43‐NMDA receptors pathway may be an important one. The interaction between the spinal astrocytic Cx43 and neuronal NMDA receptors may reflect the functional significance of astrocyte–neuron signaling in the development of morphine antinociceptive tolerance in rats.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No. 81100827), the Medical Scientific Research Foundation of Guangdong Province (Grant No. A2011153), and the Guangdong Natural Science Foundation (Grant No. S2011040002763). This study has never been published elsewhere.
The first two authors contributed equally to this work.
References
- 1. Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res 2001;39:281–286. [DOI] [PubMed] [Google Scholar]
- 2. Raghavendra V, Tanga F, Rutkowski MD, et al. Anti‐hyperalgesic and morphine‐sparing actions of propentofylline following peripheral nerve injury in rats: Mechanistic implications of spinal glia and proinflammatory cytokines. Pain 2003;104:655–664. [DOI] [PubMed] [Google Scholar]
- 3. Chen ML, Cao H, Chu YX, et al. Role of P2X7 receptor‐mediated IL‐18/IL‐18R signaling in morphine tolerance: Multiple glial‐neuronal dialogues in the rat spinal cord. J Pain 2012;13:945–958. [DOI] [PubMed] [Google Scholar]
- 4. Giaume C, McCarthy KD. Control of gap‐junctional communication in astrocytic networks. Trends Neurosci 1996;19:319–325. [DOI] [PubMed] [Google Scholar]
- 5. De Pina‐Benabou MH, Srinivas M, Spray DC, Scemes E. Calmodulin kinase pathway mediates the K+‐induced increase in Gap junctional communication between mouse spinal cord astrocytes. J Neurosci 2001;21:6635–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Söhl G, Willecke K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes 2003;10:173–180. [DOI] [PubMed] [Google Scholar]
- 7. Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev 2004;47:191–215. [DOI] [PubMed] [Google Scholar]
- 8. Contreras JE, Sánchez HA, Véliz LP, Bukauskas FF, Bennett MV, Sáez JC. Role of connexin‐based gap junction channels and hemichannels in ischemia‐induced cell death in nervous tissue. Brain Res Brain Res Rev 2004;47:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee IH, Lindqvist E, Kiehn O, Widenfalk J, Olson L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J Comp Neurol 2005;489:1–10. [DOI] [PubMed] [Google Scholar]
- 10. O'Carroll SJ, Alkadhi M, Nicholson LF, Green CR. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun Adhes 2008;15:27–42. [DOI] [PubMed] [Google Scholar]
- 11. Haupt C, Witte OW, Frahm C. Up‐regulation of Connexin43 in the glial scar following photothrombotic ischemic injury. Mol Cell Neurosci 2007;35:89–99. [DOI] [PubMed] [Google Scholar]
- 12. Spataro LE, Sloane EM, Milligan ED, et al. Spinal gap junctions: Potential involvement in pain facilitation. Pain J 2004;5:392–405. [DOI] [PubMed] [Google Scholar]
- 13. Roh DH, Yoon SY, Seo HS, et al. Intrathecal injection of carbenoxolone, a gap junction decoupler, attenuates the induction of below‐level neuropathic pain after spinal cord injury in rats. Exp Neurol 2010;224:123–132. [DOI] [PubMed] [Google Scholar]
- 14. Ohara PT, Vit JP, Bhargava A, Jasmin L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J Neurophysiol 2008;100:3064–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jasmin L, Vit JP, Bhargava A, Ohara PT. Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol 2010;6:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T. Neuroinflammation leads to region‐dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci 2011;31:414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao JR, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: A current view of their possible interactions. Pain 1995;62:259–274. [DOI] [PubMed] [Google Scholar]
- 18. Wen ZH, Chang YC, Cherng CH, Wang JJ, Tao PL, Wong CS. Increasing of intrathecal CSF excitatory amino acids concentration following morphine challenge in morphine‐tolerant rats. Brain Res 2004;995:253–259. [DOI] [PubMed] [Google Scholar]
- 19. Wong CS, Chang YC, Yeh CC, Huang GS, Cherng CH. Loss of intrathecal morphine analgesia in terminal cancer patients is associated with high levels of excitatory amino acids in the CSF. Can J Anaesth 2002;49:561–565. [DOI] [PubMed] [Google Scholar]
- 20. Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK‐801. Science 1991;251:85–87. [DOI] [PubMed] [Google Scholar]
- 21. Mao J. NMDA and opioid receptors: Their interactions in antinociception, tolerance and neuroplasticity. Brain Res Brain Res Rev 1999;30:289–304. [DOI] [PubMed] [Google Scholar]
- 22. Lin SL, Tsai RY, Shen CH, et al. Co‐administration of ultra‐low dose naloxone attenuates morphine tolerance in rats via attenuation of NMDA receptor neurotransmission and suppression of neuroinflammation in the spinal cords. Pharmacol Biochem Behav 2010;96:236–245. [DOI] [PubMed] [Google Scholar]
- 23. Shimoyama N, Shimoyama M, Davis AM, Monaghan DT, Inturrisi CE. An antisense oligonucleotide to the N‐methyl‐D‐aspartate (NMDA) subunit NMDAR1 attenuates NMDA‐induced nociception, hyperalgesia, and morphine tolerance. J Pharmacol Exp Ther 2005;312:834–840. [DOI] [PubMed] [Google Scholar]
- 24. South SM, Kohno T, Kaspar BK, et al. A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury‐induced pain. J Neurosci 2003;23:5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo RX, Zhang M, Liu W, et al. NMDA receptors are involved in upstream of the spinal JNK activation in morphine antinociceptive tolerance. Neurosci Lett 2009;467:95–99. [DOI] [PubMed] [Google Scholar]
- 26. Danbolt NC, Pines G, Kanner BI. Purification and reconstitution of the sodium‐ and potassium‐coupled glutamate transport glycoprotein from rat brain. Biochemistry 1990;29:6734–6740. [DOI] [PubMed] [Google Scholar]
- 27. Mao JR, Sung B, Ji RR, Lim G. Chronic morphine induces down‐regulation of spinal glutamate transporters: Implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen ZH, Wu GJ, Chang YC, Wang JJ, Wong CS. Dexamethasone modulates the development of morphine tolerance and expression of glutamate transporters in rats. Neuroscience 2005;133:807–817. [DOI] [PubMed] [Google Scholar]
- 29. Shen CH, Tsai RY, Tai YH, Lin SL, Chien CC, Wong CS. Intrathecal etanercept partially restores morphine's antinociception in morphine‐tolerant rats via attenuation of the glutamatergic transmission. Anesth Analg 2011;13:184–190. [DOI] [PubMed] [Google Scholar]
- 30. Unger T, Bette S, Zhang J, Theis M, Engele J. Connexin‐deficiency affects expression levels of glial glutamate transporters within the cerebrum. Neurosci Lett 2012;506:12–16. [DOI] [PubMed] [Google Scholar]
- 31. Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen‐activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res 2006;1069:235–243. [DOI] [PubMed] [Google Scholar]
- 32. Ochalski PA, Frankenstein UN, Hertzberg EL, Nagy JI. Connexin‐43 in rat spinal cord: Localization in astrocytes and identification of heterotypic astro‐oligodendrocytic gap junctions. Neuroscience 1997;76:931–945. [DOI] [PubMed] [Google Scholar]
- 33. Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive‐like behaviors in rats. Neuropsychopharmacology 2012;37:1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang N, De Bock M, Decrock E, et al. Connexin targeting peptides as inhibitors of voltage‐ and intracellular Ca(2 + )‐triggered Cx43 hemichannel opening. Neuropharmacology 2013;75:506–516. [DOI] [PubMed] [Google Scholar]
- 35. Desplantez T, Verma V, Leybaert L, Evans WH, Weingart R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol Res 2012;65:546–552. [DOI] [PubMed] [Google Scholar]
- 36. Nie H, Weng HR. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol 2009;101:2041–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aldskogius H, Kozlova EN. Central neuron‐glial and glial‐glial interactions following axon injury. Prog Neurobiol 1998;55:1–26. [DOI] [PubMed] [Google Scholar]
- 38. Wu XF, Liu WT, Liu YP, Huang ZJ, Zhang YK, Song XJ. Reopening of ATP‐sensitive potassium channels reduces neuropathic pain and regulates astroglial gap junctions in the rat spinal cord. Pain 2011;152:2605–2615. [DOI] [PubMed] [Google Scholar]
- 39. Qin M, Wang JJ, Cao R, et al. The lumbar spinal cord glial cells actively modulate subcutaneous formalin induced hyperalgesia in the rat. Neurosci Res 2006;55:442–450. [DOI] [PubMed] [Google Scholar]
- 40. Seo HS, Kim HW, Roh DH, et al. A new rat model for thrombus‐induced ischemic pain (TIIP); development of bilateral mechanical allodynia. Pain 2008;139:520–532. [DOI] [PubMed] [Google Scholar]
- 41. DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist 2004;10:40–52. [DOI] [PubMed] [Google Scholar]
- 42. Watkins LR, Hutchinson MR, Ledeboer A, Wieseler‐Frank J, Milligan ED, Maier SF. Norman cousins lecture. Glia as the “bad guys”: Implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun 2007;21:131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu N, Lu XQ, Yan HT, et al. Aquaporin 4 deficiency modulates morphine pharmacological actions. Neurosci Lett 2008;448:221–225. [DOI] [PubMed] [Google Scholar]
- 44. Berta T, Liu T, Liu YC, Xu ZZ, Ji RR. Acute morphine activates satellite glial cells and up‐regulates IL‐1β in dorsal root ganglia in mice via matrix metalloprotease‐9. Mol Pain 2012;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010;7:482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao CM, Guo RX, Hu F, et al. Spinal MCP‐1 contributes to the development of morphine antinociceptive tolerance in rats. Am J Med Sci 2012;344:473–479. [DOI] [PubMed] [Google Scholar]
- 47. Rawls SM, Zielinski M, Patel H, Sacavage S, Baron DA, Patel D. Beta‐lactam antibiotic reduces morphine analgesic tolerance in rats through GLT‐1 transporter activation. Drug Alcohol Depend 2010;107:261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jhamandas KH, Marsala M, Ibuki T, Yaksh TL. Spinal amino acid release and precipitated withdrawal in rats chronically infused with spinal morphine. J Neurosci 1996;16:2758–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim G, Wang S, Zeng Q, Sung B, Yang L, Mao J. Expression of spinal NMDA receptor and PKCgamma after chronic morphine is regulated by spinal glucocorticoid receptor. J Neurosci 2005;25:11145–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wong CS, Hsu MM, Chou R, Chou YY, Tung CS. Intrathecal cyclooxygenase inhibitor administration attenuates morphine antinociceptive tolerance in rats. Br J Anaesth 2000;85:747–751. [DOI] [PubMed] [Google Scholar]
- 51. Tai YH, Cheng PY, Tsai RY, Chen YF, Wong CS. Purinergic P2X receptor regulates N‐methyl‐D‐aspartate receptor expression and synaptic excitatory amino acid concentration in morphine‐tolerant rats. Anesthesiology 2010;113:1163–1175. [DOI] [PubMed] [Google Scholar]