

Summary
The cellular mechanisms underlying the stereotypical progression of pathology in neurodegenerative diseases are incompletely understood, but increasing evidence indicates that misfolded protein aggregates can spread by a self‐perpetuating neuron‐to‐neuron transmission. Novel neuroimaging techniques can help elucidating how these disorders spread across brain networks. Recent knowledge from structural and functional connectivity studies suggests that the relation between neurodegenerative diseases and distinct brain networks is likely to be a strict consequence of diffuse network dynamics. Diffusion tensor magnetic resonance imaging also showed that measurement of white matter tract involvement can be a valid surrogate to assess the in vivo spreading of pathological proteins in these conditions. This review will introduce briefly the main molecular and pathological substrates of the most frequent neurodegenerative diseases and provide a comprehensive overview of neuroimaging findings that support the “network‐based neurodegeneration” hypothesis in these disorders. Characterizing network breakdown in neurodegenerative diseases will help anticipate and perhaps prevent the devastating impact of these conditions.
Keywords: Diffusion tensor MRI, Network‐based neurodegeneration, Neurodegenerative diseases, Prion‐like proteins, Resting‐state functional MRI
Introduction
Neurodegenerative diseases have an enormous diversity in clinical phenotypes, affecting distinct cerebral functions. In recent years, however, intense research has been made in the field, arising the knowledge that they also share some common features. One of these commonalities is the accumulation of disease‐specific proteins into insoluble aggregates 1, 2, such as amyloid β (Aβ) in plaques in Alzheimer disease (AD), tau in neurofibrillary tangles (NFTs) in AD and many cases of frontotemporal lobar degeneration (FTLD), TAR DNA‐binding protein 43 (TDP‐43) aggregates in amyotrophic lateral sclerosis (ALS) and cases of FTLD, and α‐synuclein (α‐syn) in Lewy bodies (LB) in Parkinson disease (PD) and Dementia with Lewy bodies (Figure 1). This evidence has allowed the diseases to be recategorized in proteinopathies based on their molecular traits. Second, pathological changes in various neurodegenerative diseases progress with time in a stepwise characteristic anatomical pattern. Neuropathological studies have shown that NFTs in AD 3, LB in PD 4, and, more recently, TDP‐43 aggregates in ALS 5 and the behavioral variant of frontotemporal dementia (bvFTD) 6 initiate very early in the disease in a circumscribed area of the brain and then progress in a topographically predicted manner through anatomical connections (Figure 1). Until recently, the causative mechanisms for this networked spread were thought to be passive, including secondary Wallerian degeneration, disconnection, loss of signaling, axonal reaction, and postsynaptic dendrite retraction 1, 2. The latest evidence, however, favors the hypothesis that the stereotypical and topographical patterns of pathological progression in the central nervous system (CNS) of patients with neurodegenerative diseases may be explained by a “prion‐like” transsynaptic or transneuronal spreading of misfolded proteins between different brain regions over years 1, 2. Understanding how and where pathological protein propagation is initiated and the characterization of the major factors playing a role in the modulation of intracerebral spreading will lead to the identification of new therapeutic targets aiming at slowing or stopping the disease progression.
Figure 1.
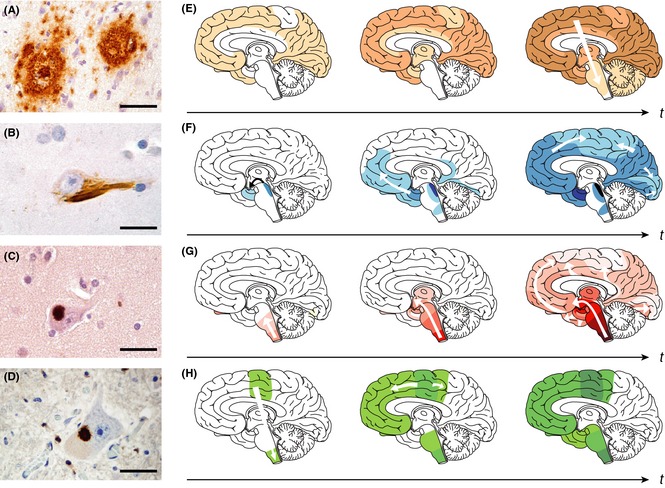
Protein aggregates show “prion‐like” self‐propagation and spreading in experimental settings, consistent with the progressive appearance of the lesions in the brain of patients with neurodegenerative diseases. (A) Aβ deposits in the neocortex of a patient with Alzheimer disease (AD). (B) Tau inclusion as a neurofibrillary tangle in a neocortical neuron of a patient with AD. (C) α‐Synuclein inclusion (Lewy body) in a neocortical neuron from a patient with Parkinson disease (PD)/Lewy body dementia. (D) TDP‐43 inclusion in a motor neuron of the spinal cord from a patient with amyotrophic lateral sclerosis (ALS). Scale bars are 50 μm in a and 20 μm in B–D. (E–H) Characteristic progression of specific proteinaceous lesions in neurodegenerative diseases over time (t, black arrows), inferred from postmortem analyses of brains. Aβ deposits and tau inclusions in brains of patients with AD (E and F), α‐synuclein inclusions in brains of patients with PD (G), and TDP‐43 inclusions in brains of patients with ALS (H). Three stages are shown for each disease, with white arrows indicating the putative spread of the lesions. Reproduced with permission from 2.
In parallel to the molecular and pathological advances, the idea that the pathological substrates of neurodegenerative diseases spread along discrete brain networks has also been increasingly strengthened by neuroimaging studies 7. It has been observed, indeed, that neurodegenerative diseases spatially affect patterns that reflect the healthy brain's network architecture 8. In this review, we will introduce briefly the main molecular and pathological substrates of the most frequent neurodegenerative diseases. Then, we will provide a comprehensive overview of neuroimaging findings that support the “network‐based neurodegeneration” hypothesis in patients with AD, bvFTD, ALS, and PD, bringing studies that range from the large‐scale brain networks alterations to the microscopic abnormalities of structural pathways.
Clinical Phenotypes, Molecules, and Pathology of Neurodegenerative Diseases
Alzheimer Disease
Alzheimer disease is the most common form of dementia. Typically, AD is characterized by an insidious onset of cognitive decline, starting with deficits in episodic memory. As the disease progresses, other deficits such as aphasia, apraxia, agnosia, visuospatial difficulties, and executive dysfunction arise gradually 9. The patient becomes increasingly dependent on others. Psychiatric and behavioral problems such as mood disorders, psychosis, agitation, and sleep disorders occur more frequently in the advanced phase of the disease. The term mild cognitive impairment (MCI) identifies those individuals who have subjective memory and/or cognitive symptoms accompanied by objective evidence of isolated memory and/or other cognitive impairment and whose activities of daily living are considered to be generally normal 10. Progression to clinically diagnosable dementia occurs at a higher rate from MCI than from normal (typically 10–15% per year—compared to rates of ~1% with normal aging), but is clearly not the invariable clinical outcome at follow‐up 10.
Besides the typical neuropsychological profile of AD presenting with early memory deficits, there is evidence from clinicopathological studies that patients with AD may present with different cognitive profiles. Atypical presentations are more often seen in patients with early‐onset AD (EOAD) (arbitrarily defined as before the age of 65). EOAD is often characterized by atypical manifestations with greater impairment in attention, executive, language, and visuospatial functions at the time of presentation. Furthermore, AD can present as relatively focal clinical syndromes, more frequently associated with early age‐of‐onset, that is, as posterior cortical atrophy (PCA) and logopenic variant (lv) of primary progressive aphasia (PPA) 11. PCA presents with visual and visuospatial impairment with less prominent memory loss 12, 13. Over time, patients with PCA can develop visual agnosia, topographical difficulty, optic ataxia, simultanagnosia, ocular apraxia (Balint syndrome), alexia, acalculia, right–left confusion, and agraphia (Gerstmann syndrome), and later a more generalized dementia. Patients with lvPPA present with language deficits, characterized by slow rate of speech, with long word‐finding pauses 14. Grammar and articulation are usually preserved in lvPPA, although phonological paraphasias could be present. Repetition and comprehension are impaired for sentences but preserved for single words, and naming is moderately affected 14.
Two abnormal protein aggregates characterize AD pathology: neuritic plaques and NFTs 15. Neuritic plaques are extracellular deposits and consist of a dense central core of Aβ fibrils with inflammatory cells and dystrophic neurites in its periphery. Aβ peptide is a normal proteolytic product of the Aβ precursor protein (APP) 16. Due to the ability of the protease γ‐secretase to cleave APP at multiple sites, Aβ peptides are 39–43 amino acid residues in length, but Aβ 40 and Aβ 42 are the predominant species in vivo. In contrast, plaques in AD are composed primarily of Aβ 42 and Aβ 43, which are more hydrophobic and aggregation‐prone than the slightly shorter and more polar (but very abundant) Aβ 40. The second major proteinopathy in AD is aggregated tau, which consists of intraneuronal polymers primarily composed of hyperphosphorylated tau in the form of NFTs 15. Tau is a natively unfolded cytoplasmic protein that normally helps microtubule stabilization 17. If hyperphosphorylated, tau becomes prone to aggregation. In AD, the pattern of tau pathology is highly regular, whereas Aβ plaque pathology is much more varied. NFTs follow a stereotypic topographical progression scheme as described by Braak and Braak 3, first appearing in the entorhinal cortex and closely related areas, then progressing to the hippocampus, to paralimbic and adjacent medial‐basal temporal cortex, to association cortex, and last to primary sensorimotor and visual cortical areas.
The initiating event in the molecular cascade that eventually leads to clinical and pathological AD has been controversial for decades. The amyloid cascade hypothesis, which posits that Aβ production and aggregation in the brain are the prime pathogenic drivers, leading to tau hyperphosphorylation and other histological and clinical features of AD, has dominated research for the past 20 years 18. The amyloid cascade hypothesis was reinforced by the identification of gene defects in APP, PSEN1, and PSEN2 in patients with an early‐onset, inherited form of the disease 19. The APP gene on chromosome 21 encodes the APP, from which Aβ is liberated after stepwise, amyloidogenic, proteolytic processing. The genes PSEN1 and PSEN2 encode presenilin 1 and presenilin 2, which are part of the γ‐secretase complex, the enzyme that carries out the second cleavage in APP processing. An alternative position is that tau hyperphosphorylation and Aβ accumulation are independent interacting pathophysiological processes 20, 21, 22. According to this second hypothesis, it is tau‐related neurodegeneration that is ultimately responsible for clinical symptoms 23.
Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration is the umbrella term encompassing a group of progressive proteinopathies, which are heterogeneous with regard to etiology and neuropathology, but share atrophy of the frontal and/or temporal cortex as a morphological feature and the deposition of abnormal, ubiquitinated protein inclusions in the cytoplasm and nucleus of neuronal and glial cells as major pathological constituent 24. FTLD includes three clinical syndromes and three major underlying neuropathological subtypes. The clinical syndromes, which are distinguished by the early and predominant symptoms, are as follows: a bvFTD; a language disorder (nonfluent and semantic PPA variants); and a motor disorder such as ALS, corticobasal syndrome, and progressive supranuclear palsy (PSP) syndrome 25. This review focused on evidence for the “network‐based neurodegeneration” hypothesis in bvFTD and ALS. bvFTD is characterized by a prominent change in personality and social behavior, with apathy and/or disinhibition, emotional blunting, stereotyped or ritualized behaviors, loss of empathy, alterations in appetite and food preference with limited or no insight 26. ALS, the most common form of motor neuron disease, is a relatively rare progressive degenerative condition affecting the lower motor neurons within the spinal cord and the brainstem, accompanied by degeneration of the upper motor neurons in the motor cortex 27. Up to 50% of patients with ALS have also cognitive and/or behavioral changes, ranging from an overt FTD to mild executive and/or nonexecutive cognitive impairment and behavioral deficits 28. The neuropathological subtypes are characterized by an abnormal accumulation of proteins 29: microtubule‐associated protein tau (MAPT), TDP‐43, and fused in sarcoma protein (FUS). FTLD‐tau, FTLD‐TDP, and FTLD‐FUS represent 45%, 50%, and 5% of all FTLD cases, respectively, at postmortem examination.
Frontotemporal lobar degeneration‐tau cases include those with the neuropathology of Pick disease, PSP, corticobasal degeneration (CBD), and cases of familial FTLD caused by mutations in the MAPT gene. FTLD‐tau subtypes are characterized by specific inclusions: Pick bodies in Pick disease, tufted astrocytes and numerous NFTs in subcortical nuclei in PSP, and astrocytic plaques and abundant thread pathology in CBD 29. In addition, the biochemical form of tau that accumulates in the inclusions varies among the different subtypes, with Pick bodies composed primarily of tau isoforms with three microtubule‐binding domains (3‐repeat), while the inclusions of PSP and CBD contain 4‐repeat tau 29.
In 2006, the majority of cases with tau‐negative inclusions that stained positive for ubiquitin in FTLD were found to contain TDP‐43 protein, as did the majority of sporadic and familial ALS cases 30. TDP‐43 is a highly conserved and widely expressed RNA‐binding protein that is a member of the heterogeneous nuclear ribonucleoprotein family of proteins 31. It is predominantly found in the nucleus, but shuttles between there and the cytoplasm, where it is present only at low levels. Pathological modifications of TDP‐43 in the disease state include a redistribution from the nucleus to the cytoplasm in cells with inclusions, hyperphosphorylation, ubiquitination, and N‐terminal truncation 31. Dominantly inherited genetic mutations within the gene that encodes TDP‐43 (TAR DNA‐binding protein, TARDBP) are linked with ALS and FTLD‐TDP phenotypes 24. Different patterns of FTLD‐TDP are now recognized, based on the cortical distribution and relative abundance of cytoplasmic inclusions compared to neurites, with each having fairly specific clinical and genetic correlations 29.
Most of the remaining tau‐/TDP‐negative FTLD subtypes are characterized by cytoplasmic inclusions that are immunoreactive for FUS 32. FUS is a 526 amino acid protein identified as a fusion oncogene causing human myxoid liposarcomas. When in the nucleus, FUS is thought to be involved in regulation of transcription and pre‐mRNA splicing. Cytoplasmic FUS in neurons appears to have a role in mRNA transport, where it can potentially facilitate local protein synthesis at synapse.
Recent pathological studies based upon the distribution patterns of phosphorylated TDP‐43 indicate that the disease progression in ALS and bvFTD cases with FTLD‐TDP pathology progresses in a sequential regional pattern possibly through axonal pathways 5, 6. ALS and FTLD‐TDP bvFTD are characterized by four neuropathological stages. In ALS 5, initial lesions (stage 1) develop in the frontal and sensorimotor cortex, brainstem motor nuclei, and in spinal cord α‐motor neurons, with beginning involvement of the prefrontal cortex, brainstem reticular formation, precerebellar nuclei, and red nucleus in stage 2; in stage 3, pathology progresses in the prefrontal and postcentral cortices, and striatum, followed by changes in anteromedial portions of the temporal lobe, including the hippocampal formation, during stage 4. FTLD‐TDP bvFTD cases with the lowest burden of pathology (pattern 1) are characterized by widespread phosphorylated TDP‐43 lesions in the orbitofrontal cortex and amygdala 6. With increasing burden of pathology (bvFTD pattern 2), TDP‐43 lesions emerged in the middle frontal and anterior cingulate gyrus as well as in anteromedial temporal lobe areas, the superior and medial temporal gyri, striatum, red nucleus, thalamus, and precerebellar nuclei. More advanced bvFTD cases show a third pattern (3) with involvement of the motor cortex, bulbar somatomotor neurons, and the spinal cord anterior horn, whereas cases with the highest burden of pathology (pattern 4) are characterized by TDP‐43 lesions in the visual cortex.
Parkinson Disease
Parkinson disease, the most common neurodegenerative movement disorder, is characterized clinically by four cardinal motor symptoms: rigidity, tremor, bradykinesia, and postural instability 33. Symptoms develop slowly and gradually progress over years. Superimposed on the classic motor symptoms, autonomic and sensory dysfunction, sleep disturbances, cognitive impairments and dementia are also common features in PD 34, 35.
The pathological hallmark of PD is the presence of intraneuronal proteinaceous intracytoplasmic inclusions called LB. One of the main protein components of the LB is α‐syn 36. α‐syn is a 14‐kDa natively unfolded protein, consisting of 140 amino acids, that binds lipids through its amino‐terminal repeat region. It is localized in the presynaptic terminals, nucleus, cytosol, and in some cellular membranes, such as the mitochondria‐associated membrane in the endoplasmic reticulum. Although the exact function of α‐syn remains unknown, substantial evidence suggests that α‐syn function is related to its capacity to interact directly with membrane phospholipids, particularly highly curved membranes such as vesicles 37. In particular, α‐syn seems to play a role in the vesicle trafficking during the neurotransmission release. In PD, this protein leaves its binding sites within synaptic boutons and, together with other components such as phosphorylated neurofilaments and ubiquitin, gradually adopts insoluble oligomeric and/or fibrillary conformations 38. α‐syn pathological species are toxic in vivo by several mechanisms including the disruption of normal α‐syn function in neurotransmission release and vesicular transport, and impairing mitochondrial structure and the efficiency of some protein‐degradation mechanism 39.
In 2003, Braak et al. 4 performed several longitudinal analyses to evaluate the neuroanatomical changes in the brain of patients with PD and proposed a model in which the disease stages are correlated with the regional distribution of LB in the CNS. According to the Braak's model, LB formation starts early in the disease (even before the motor symptoms emerge) and LB originate in the olfactory bulb and in the brainstem, specifically at the dorsal motor nucleus of the vagus nerve. In parallel to disease progression, LB are detected in other brain regions and appear to propagate through brain structures, in a stereotypic pattern, to reach the other regions including the midbrain and, at later stages, the cerebral cortex.
The “Prion‐Like” Transmission of Pathogenic Proteins in Neurodegenerative Diseases
Prion diseases are a unique group of neurodegenerative disorders in which the conformationally altered prion protein PrPSc constitutes the infectious agent that corrupts normal cellular PrP through “seeded” fibrillization 40. Although not being infectious, that is, transmissible between people, a rapidly growing body of literature has provided compelling evidence that a “prion‐like” self‐propagating mechanism may be applicable to a wide range of disease‐associated proteins, including Aβ, tau, TDP‐43, and α‐syn 1, 2. The self‐propagation of aggregates of Aβ was predicted decades ago 1, 2. More recently, the ability of tau to propagate transsynaptically through well‐established brain anatomical pathways has been reported, including AD and FTLD cases with argyrophilic grain pathology 17. Experimental support for the existence of a cell‐to‐cell transfer of α‐syn inclusions has come from the seminal research showing that misfolded intraneuronal α‐syn can transfer to neighboring cells both in culture and in the brains of patients with PD who had received fetal mesencephalic nerve cell transplants 11–16 years earlier revealing the presence of LB in the grafts 41, 42. Then, several in vitro and in vivo studies suggested that α‐syn can undergo a toxic template conformational change, spread from cell to cell and from region to region, and initiate the formation of LB‐like aggregates, contributing to the PD pathogenesis 41, 42. Whereas a cell‐to‐cell transmission of TDP‐43 has not been demonstrated conclusively, a recently discovered C‐terminal prion‐like domain has been implicated in the aggregation of TDP‐43 in cultured cells from diseased brains 31, 43. In addition, a notable feature shared by nearly all neurons involved in ALS is that they receive strong afferents from neocortical pyramidal cells, supporting a neuron‐to‐neuron propagation through corticofugal connections 5.
It seems likely that prion‐like aggregates are able to travel within the neuron to reach potential site for interneuronal transfer, to be released from the originating cell and taken up by neighboring cells, where they penetrate the cytoplasm and nucleate further aggregation 1, 2. Both tau and α‐syn aggregates can move anterogradely as well as retrogradely within a neuron, possibly by axonal transport. Among the potential mechanisms of the cell‐to‐cell spreading of proteins, endocytosis or receptor‐mediated endocytosis, transfer through exosomes or even by nanotubes that directly connect the cytoplasm of two cells, has been reported 1, 2. Regardless of the mechanism of transmission between cells and the consequent ability of self‐amplification, what triggers the initial conversion of normally produced proteins into abnormal aggregates remains unknown.
Functional and Structural Connectivity‐Based Findings in Neurodegenerative Diseases
Functional and Structural Connectivity‐Based Imaging Techniques
Resting‐state fMRI constitutes an advanced technique that measures the spontaneous low‐frequency (<0.001–0.001 Hz) fluctuations of the blood oxygen level‐dependent signal while the individual rests in the scanner without performing any task. Resting‐state fMRI allows to examine brain connectivity between functionally linked brain regions with no bias toward specific motor, visual and cognitive functions 44. Spatially distributed maps of temporal synchronization can be detected that characterize resting‐state networks 45. Resting‐state fMRI assessment has been focused primarily on a characteristic set of brain regions, including the posterior cingulate and precuneus, inferolateral parietal cortex, medial temporal lobe, and medial prefrontal cortex, which is deactivated during a broad range of cognitive tasks and is believed to support a default mode activity of the human brain (i.e., default mode network [DMN]) 46. Analysis of resting‐state fMRI data has more recently suggested the existence of other networks which are thought to subserve cognition, such as the salience, executive, frontoparietal, and associative visual networks 45.
Information on the microstructural integrity of the white matter (WM) pathways connecting the different structures of the human brain can be obtained in vivo using diffusion tensor (DT) MRI 47. DT MRI characterizes the three‐dimensional diffusion of water as a function of spatial location 47. The two most common DT MRI measures are mean diffusivity (MD) and fractional anisotropy (FA). MD is a measure of the magnitude of diffusion and is rotationally invariant. FA describes the degree of anisotropy of the diffusion tensor. The diffusion of water within the tissues will be altered by changes in the tissue microstructure and organization due to many pathologic processes of the CNS, including demyelination, axonal damage, edema, and ischemia 48.
Alzheimer Disease
Neurodegeneration in AD leads to a marked reduction of brain tissue. Indeed, typical late‐onset, amnestic AD is characterized by global atrophy on MRI. The medial temporal lobes, especially the hippocampus and entorhinal cortex, are among the earliest sites of structural damage 49. Other severely affected regions include the posterior part of the cingulate gyrus, precuneus, and splenium of the corpus callosum on the medial surface, and the parietal, posterior superior temporal, and frontal regions on the lateral cerebral surfaces 49.
Interestingly (yet probably not coincidently), there is a remarkable overlap between the pattern of Aβ pathology and atrophy in AD and the DMN 50. A decreased DMN connectivity has been described in patients with AD 51, 52 as well as in patients with amnestic MCI 51, 53, 54, 55 and in healthy elderly subjects harboring amyloid plaques (as measured by amyloid imaging) 56, 57 or carrying the apolipoprotein E4 allele 58. In addition, altered connectivity among the DMN nodes do occur regardless cortical damage 59, suggesting that functional deficits within the network may precede structural damage. As the disease progresses, DMN connectivity continues to decline as shown by cross‐sectional studies across successive disease stages 60 and a few longitudinal studies 61.
Other brain networks are inevitably affected with AD progression. However, the sequence of involvement of functional systems outside the DMN is not well known. Resting‐state fMRI studies demonstrated aberrant functional connectivity in the executive network and the salience network in patients with AD, along with loss of anticorrelation between the DMN and the executive network along the AD continuum 51, 62.
Another compelling evidence supporting the notion that neurodegenerative diseases spread along networks comes from recent studies in patients with atypical AD forms, such as PCA and lvPPA. Recent studies combining structural MRI from patients with resting‐state fMRI data from healthy subjects highlighted that the DMN is affected in all AD forms. In addition, there is a good anatomical correspondence between the patterns of atrophy in patients (i.e., of the visual network in PCA and language network in lvPPA), distinct brain functional networks in healthy subjects, and symptoms for each AD variant (Figure 2). Therefore, these recent multimodal analyses seem to suggest that atypical AD forms may reflect a different dissemination of pathology through specific interconnected neural networks relative to typical, late‐onset AD 63, 64.
Figure 2.
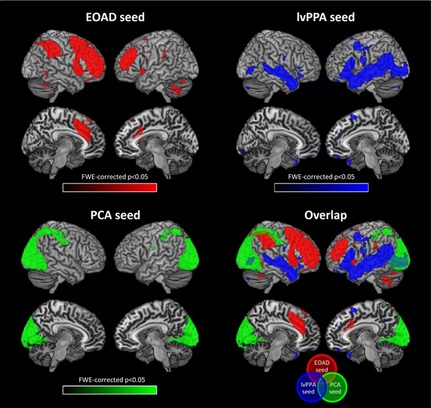
Resting‐state functional connectivity network maps in healthy individuals produced by seeding three regions that were specifically atrophied in Alzheimer disease (AD) variants, that is, early‐onset AD (EOAD), posterior cortical atrophy (PCA), and logopenic variant of primary progressive aphasia (lvPPA). Figure shows statistical P maps after correction for multiple comparisons (P < 0.05 family‐wise‐error corrected for multiple comparisons). Reproduced with permission from 64.
White matter tracts that connect regions of the DMN, such as the cingulum (linking the medial temporal lobe with the posterior cingulate cortex and the medial frontal regions) and the corpus callosum, are widely affected in patients with AD 65, 66. Damage to these WM regions correlates with cognitive impairment and disease progression in patients with AD 67 and may be related to secondary degeneration. Nevertheless, another major finding of DT MRI studies in AD is that WM damage is more severe and widely distributed than expected on the basis of cortical atrophy. In addition, in MCI and healthy subjects, WM damage can be detected even before the development of cortical atrophy and overt dementia 68, 69. To date, the causes of WM degeneration in AD are still unknown. However, converging data support the notion that WM damage has a central role in how the disease strikes and progresses. Here again, DT MRI findings may reflect the dissemination of pathology from early damaged to yet unaffected cortical regions in AD, thus supporting pathological transmission of Aβ and tau aggregates from neuron to neuron along WM connections 1, 2. In keeping with this hypothesis, a DT MRI study of patients with AD and MCI suggested that microglia activation, which produces neurotoxic and oligodendrotoxic oligomers in the presence of Aβ in excess, can contribute to disease spreading to neighboring and connected areas through WM tracts 70. In addition, one study investigating the patterns of WM damage in atypical AD variants suggested that the disease has targeted specific peripheral networks (memory, visual, language) at onset in different AD forms and then converged to medial and dorsal frontoparietal regions 71. The spread of pathology in AD would occur through the corpus callosum and the main long‐range WM fibers between the posterior and anterior brain regions 71. Together with functional connectivity studies, DT MRI findings suggest that clinical heterogeneity of AD may be related to the fact that pathology starts from different medial temporal or lateral neocortical hubs and then eventually progresses along the same WM network to converge to a similar pattern of involvement matching the key hubs of the DMN. Longitudinal studies are needed to confirm such a model clarifying in vivo the direction of the pathology spreading through brain networks in AD.
Frontotemporal Lobar Degeneration
Behavioral Variant FTD
In bvFTD, early atrophy occurs in orbitofrontal/subgenual, medial frontal cortex (including anterior cingulate cortex), frontoinsula, anterior temporal lobe, and basal ganglia 72. In bvFTD, atrophy maps strongly resemble a resting‐state fMRI network called salience network 73. This network is activated in tasks requiring attentional selection, task switching, and self‐regulation of behavior, that is, events where we determine which inputs are salient for processing 74. Within this network, two key nodes have been identified: the frontoinsula, an afferent hub which integrates inputs coming from other networks with the interoceptive ones; and the anterior cingulate cortex, an efferent hub, which detects information from the previous hub and mobilizes visceroautonomic, emotional, cognitive, and behavioral responses 75. Patients with bvFTD have reduced connectivity in the salience network when compared either with controls or with patients with AD 76, 77, 78. In patients with bvFTD, the functional disconnectivity between these key nodes has been correlated with clinical severity, apathy, and disinhibition scores 76, 78. In addition, measures of salience network connectivity involving the left insula predict behavioral changes in patients with bvFTD 79.
White matter tracts connecting the key regions of the salience network are also altered 80, 81, 82, such as the uncinate fasciculus and genu of the corpus callosum. However, studies have shown that WM alterations may also go beyond the regions of cortical atrophy in a more distributed manner 80, 81, 82. Indeed, with the disease progression, WM abnormalities involve the posterior temporal and parietal regions, reflecting distal propagation of the pathology 83. It is worth noting that presymptomatic FTLD gene carriers present the same functional network alterations observed in patients with bvFTD without cortical atrophy but with considerable WM abnormalities in frontotemporal regions 84. These results suggest that WM alterations might precede cortical tissue loss and that DT MRI metrics can be a marker of pathology spreading through WM tracts in FTLD cases.
Amyotrophic Lateral Sclerosis
MRI observations revealed cross‐sectional brain atrophy in the motor and/or premotor cortices of patients with ALS 85. Several resting‐state fMRI studies of ALS reported significantly decreased functional connectivity within the sensorimotor network 86, 87, 88, 89 in keeping with the structural damage. However, other studies have identified regions of increased functional connectivity in the somatosensory system 89, 90, 91, 92. Two scenarios have been described to explain increased connectivity patterns. First, increased functional connectivity might compensate for structural damage and exhaust with increasing burden of pathology 91, 93. Second, the high level of functional connectivity in ALS might be related to pathogenic loss of local inhibitory circuitry 94. Indeed, increased functional connectivity was found over a large area spanning sensorimotor, premotor, prefrontal, and thalamic regions overlapping areas abutting WM tracts showing loss of integrity at DT MRI 92, 95.
Diffusion tensor MRI studies of patients with ALS have consistently reported the involvement of the corticospinal tract and middle‐posterior parts of the corpus callosum, correlating with disease severity and rate of disease progression 85. Although diagnosed and classified on the basis of motor system involvement only, the growing body of evidence demonstrating a frontotemporal syndrome is undeniable. In keeping with pathological and clinical data, an altered (both decreased and increased) functional connectivity of brain networks associated with cognition and behavior was found in ALS, even in the absence of overt dementia 86, 88, 93. Patients with ALS also show abnormalities in extramotor WM regions, especially in frontotemporal areas, in relation to the occurrence of cognitive impairment or ALS‐FTD 96, 97, 98, 99.
A recent study used DT MRI tractography to assess the pathways that are prone to be involved in ALS according to the different pTDP‐43 stages 5, and revealed significant WM tract abnormalities in patients relative to controls in a sequential progression 100 (Figure 3), that is, the corticospinal tract (stage 1), the corticorubral and corticopontine tracts (stage 2), the corticostriatal pathway (stage 3), and the proximal portion of the perforant path (stage 4). These results mirror the proposed neuropathological propagation pattern of ALS 5, supporting in vivo the evidence of the progressive expansion of WM damage from the motor to the extramotor networks.
Figure 3.
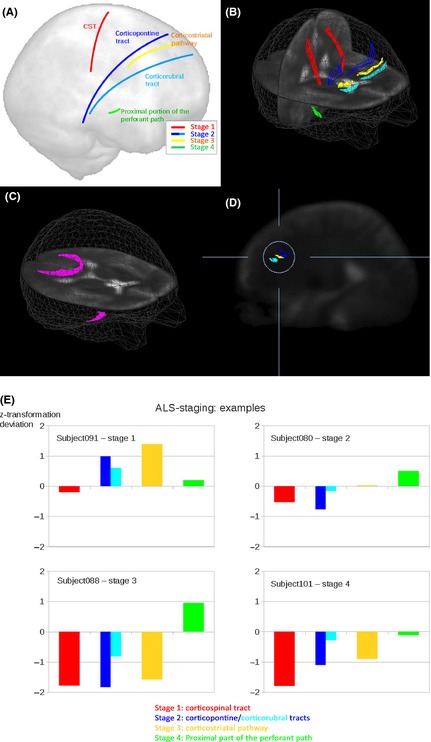
In vivo imaging of the disease stages in amyotrophic lateral sclerosis (ALS) using diffusion tensor tractography. (A) Schematic representation of the white matter tracts analyzed. (B) Three‐dimensional images of the corticospinal tract (CST, red) corresponding to ALS stage 1 5, corticopontine tract (dark blue) and corticorubral tract (light blue) corresponding to ALS stage 2 5, corticostriatal pathway (yellow) corresponding to ALS stage 3 5, and proximal portion of the perforant path (green) corresponding to ALS stage 4 5. (C) Reference paths (magenta) show starting points in the corpus callosum (area V) and starting points in the optic tract. (D) Sagittal slice for the illustration of the differences between the corticopontine tract (dark blue), corticorubral tract (light blue), and corticostriatal pathway (yellow). (E) Individual examples for the categorization of patients with ALS into ALS stages based upon deviations of z‐transformed fractional anisotropy values from controls’ values for different ALS stages. Modified with permission from 100.
Parkinson Disease
Although conventional structural MRI remains normal in PD until the late stage, advanced techniques have shown abnormalities in the substantia nigra and the cortex 101. Several studies assessed the resting‐state fMRI pattern of the corticostriatal–thalamic–cortical circuits in patients with mild to moderate PD, most of which report reduced functional connectivity in some regions and decreased functional connectivity in others relative to healthy controls 102, 103, 104, 105. A levodopa‐induced spatial remapping of the cortico‐striatal connectivity has been detected in chronically treated patients with PD 103, 104, suggesting that the clinical improvement associated with dopaminergic treatment could be related to the dopaminergic modulation of resting‐state functional connectivity. A modulation of thalamocortical functional connectivity by levodopa administration has been demonstrated to occur also in drug‐naïve PD cases 106, 107, 108.
Diffusion tensor MRI studies of patients with cognitively normal, early, idiopathic PD showed subtle WM alterations along the nigrostriatal projections, in the frontal regions, including premotor areas, and corpus callosum 109, 110, 111, 112. In early PD, diffusion changes precede atrophy that is detectable with conventional MRI, specifically within voxels containing the olfactory tracts 113. WM damage is emerging as an important pathological substrate of cognitive deficits in patients with PD 114, 115, 116, 117, 118. A large study of idiopathic nondemented PD cases at different disease stages showed that WM damage spreads predominantly to frontal and parietal regions with increasing PD severity and in association with the degree of cognitive impairment 115. DT MRI studies exploring WM tract abnormalities in patients with PD‐MCI showed a more severe involvement of the corpus callosum, cingulum, and major association WM tracts relative to those patients with no cognitive deficits 114, 116, 118.
Graph Theory and Network Properties in Neurodegenerative Diseases
Network‐based analysis of brain structural and functional connections has provided a novel instrument to study the human brain in healthy and diseased individuals 119. Using the theoretical framework of networks and graphs, the brain can be represented as a set of nodes (i.e., brain regions) joined by pairs by lines (i.e., structural or functional connectivity) 119. Graph analysis has revealed important features of brain organization, such as an efficient “small‐world” architecture (which combines a high level of segregation with a high level of global efficiency) and distributed, highly connected network regions, called “hubs” 119. In a small‐world network, a high clustering coefficient indicates that nodes tend to form dense regional cliques, implying high efficiency in local information transfer/processing 119. Path length and global efficiency are measures of network integration, which is the ability to combine specialized information rapidly from distributed brain regions 119. Distinct modifications of brain network topology have been identified during development and normal aging, whereas disrupted functional and structural network properties have been associated with several neurological and psychiatric conditions, including dementia, ALS, multiple sclerosis, and schizophrenia 119.
Many studies used graph theoretical analysis in AD using both structural and functional MRI 120, 121, pointing to a loss of highly connected areas in these patients 122. A correlation between the site of Aβ deposition in patients with AD and the location of major hubs as defined by graph theoretical analysis of functional connectivity in healthy adults has been demonstrated 50. These regions include the posterior cingulate cortex/precuneus, the inferior parietal lobule, and the medial frontal cortex, implying that the hubs are preferentially affected in the progression of AD. Although studies showed considerable variability in reported group differences of most graph properties, the average characteristic path length has been most consistently reported to be increased in AD, as a result of loss of connectivity, while the clustering coefficient is likely to be less affected by AD pathology 122. The global architecture of MCI networks was found to be intermediate between patients with AD and normal elderly controls 122. Additionally, compared with controls, patients with MCI retained their hub regions in the frontal lobe but lost those in the temporal lobe 123. Increased interregional correlations within the local brain lobes and disrupted long‐distance interregional correlations in MCI and AD were also detected 122. In patients with AD and MCI, altered graph theory patterns were associated with cognitive deficits 124, 125.
Graph theoretical analysis was recently applied to resting‐state fMRI data from patients with bvFTD 126. Global and local functional networks were altered in patients with bvFTD relative to normal subjects as indicated by reduced mean network clustering coefficient, and global efficiency and increased path length 126. Altered brain regions were located in structures that are closely associated with neuropathological changes in bvFTD, such as the frontotemporal lobes and subcortical regions 126 (Figure 4).
Figure 4.
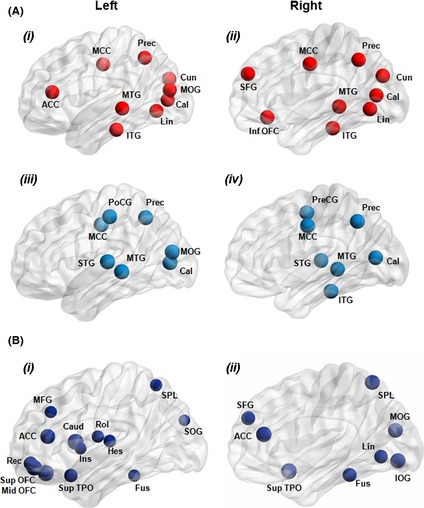
(A) Cortical hubs of brain functional networks in healthy controls (i, ii) and patients with the behavioral variant of frontotemporal dementia (bvFTD) (iii, iv). (B) Regions showing decreased integrated nodal degree (i, ii) in patients with bvFTD compared to healthy controls. Node size is proportional to the difference in the value of the integrated nodal parameters between the two groups. ACC, anterior cingulate cortex; Cal, calcarine cortex; Caud, caudate nucleus; Cun, cuneus; Fus, fusiform gyrus; Hes, Heschl gyrus; Ins, insula; IOG, inferior occipital gyrus; ITG, inferior temporal gyrus; Lin, lingual gyrus; MCC, middle cingulate cortex; MFG, middle frontal gyrus; MOG, middle occipital gyrus; MTG, middle temporal gyrus; OFC, orbitofrontal cortex; Prec, precuneus; PoCG, postcentral gyrus; PreCG, precentral gyrus; Rec, gyrus rectus; Rol, rolandic operculum; SFG, superior frontal gyrus; SOG, superior occipital gyrus; SPL, superior parietal lobule; STG, superior temporal gyrus; TPO, temporal pole. Reproduced with permission from 126.
Graph theoretical approach showed that overall functional organization of the motor network was unchanged in patients with ALS compared to healthy controls; however, the level of functional connectedness was correlated with disease progression rate, that is, stronger interconnected motor networks show a more progressive disease course 90. The effects of ALS on structural brain topology were assessed using DT MRI and graph theoretical analysis 127, 128. While the organization of the global brain network was intact in ALS, an impaired subnetwork of regions with reduced WM connectivity was detected 127 centered on primary motor regions, including secondary motor regions (frontal cortex and pallidum) as well as high‐order hub regions (posterior cingulate cortex and precuneus). A more recent study investigating the overlap between structural and functional connectivity abnormalities in patients with ALS showed coherent loss of structural and functional connections in the motor network 129.
Only two studies so far have investigated brain networks using graph analysis in patients with PD 130, 131, suggesting a decreased global and nodal functional efficiency relative to healthy controls. In addition, one study indicated that the topological properties of brain functional networks are severely impaired in PD patients with cognitive deficits 130. Patients with PD‐MCI had connectivity reductions predominantly affecting long‐range connections as well as increased local interconnectedness manifested as higher measures of clustering coefficient and small‐worldness 130. This latter measure also correlated negatively with cognitive performance in visuospatial and memory functions. Furthermore, normal hubs displayed reduced centrality and degree in these patients 130.
Recent graph theoretical MRI analyses tested various models of how neurodegenerative diseases spread across networks 128, 132, 133. Combining atrophy patterns of patients with five different neurodegenerative diseases with resting‐state fMRI data from healthy subjects, a first study revealed that, within each targeted network, neurodegenerative process spreads primarily between neurons according to the functional proximity of specific brain regions acting as critical hub‐like “epicenters,” rather than various alternative candidate mechanisms 133 (Figure 5). A second study modeled network diffusion based on brain structural connectivity networks obtained from DT MRI data of healthy subjects and derived robust spatial eigenmodes that correspond closely to known patterns of atrophy in patients with AD and bvFTD 132. A longitudinal study of patients with ALS demonstrated no progressive impairment of the initially affected connections of the motor system, but a propagating loss of brain connections over time to frontal and parietal regions 128. Therefore, all these sophisticated analyses best fit a transneuronal spread model of network‐based vulnerability from initial disease epicenters to directly connected neighboring nodes in patients with different neurodegenerative diseases.
Figure 5.
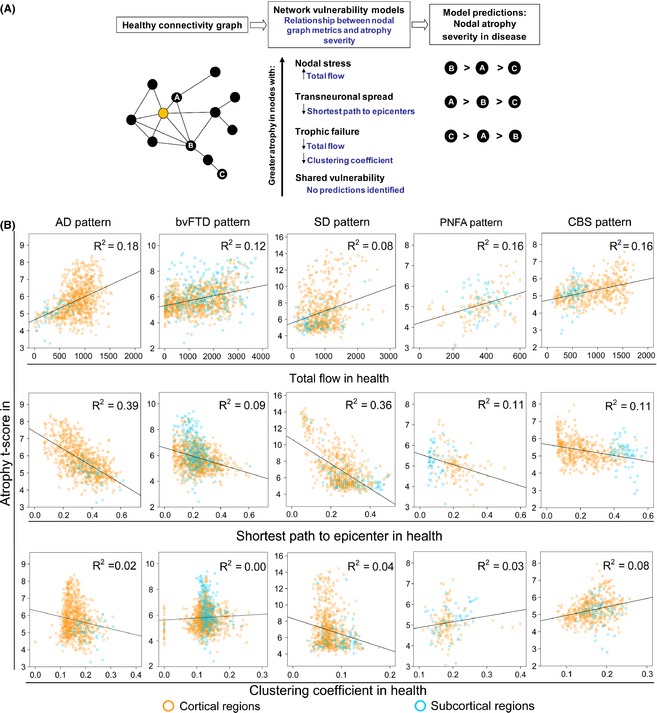
(A) Predictions made by network‐based degeneration models: effects of healthy intrinsic connectivity graph metrics on atrophy severity in neurodegenerative diseases. A simplified healthy connectivity graph is shown (far left) for illustration purposes only; circles represent nodes (brain regions), lines represent edges (a connection between two nodes), and edge lengths represent the connectivity strength between nodes, with shorter edges representing stronger connections. The orange node represents an epicenter. Three nodes, labeled as “A”, “B”, and “C”, feature contrasting graph theoretical properties to illustrate predictions made by the network‐based vulnerability models (far right). Listed in the center column are the relationships predicted by each model. For example, the transneuronal spread model predicts that nodes with shorter (↓) paths to the epicenter in health will be associated with greater (↑) atrophy severity in disease. (B) Regions with high total connectional flow (row 1) and shorter functional paths to the epicenters (row 2) showed significantly greater disease vulnerability (P < 0.05 family‐wise‐error corrected for multiple comparisons) in Alzheimer disease (AD), behavioral variant of frontotemporal dementia (bvFTD), semantic dementia (SD), progressive supranuclear palsy (PNFA), and corticobasal degeneration (CBS), whereas inconsistent weaker or nonsignificant relationships were observed between clustering coefficient and atrophy (row 3). Cortical regions = blue circles; subcortical regions = orange circles. Modified with permission from 133.
Conclusions
Neurodegenerative diseases feature characteristic patterns of early neuronal and regional vulnerability, with resulting neurological first symptoms. In turn, a common finding among neurodegenerative disease is that they show typical progressions of regional degeneration with associated downstream clinical disturbances. The cellular mechanisms underlying such a stereotypical progression of pathology in neurodegenerative diseases are incompletely understood, but increasing evidence indicates that misfolded protein aggregates can spread by a self‐perpetuating process that leads to amplification, templating, and neuron‐to‐neuron transmission of these pathologies. Novel neuroimaging techniques can help elucidating how these disorders spread across brain networks. Recent knowledge from structural and functional connectivity studies suggests that the relation between neurodegenerative diseases and separate brain networks is likely to be a strict consequence of diffuse network dynamics. Furthermore, in the majority of these conditions, measurement of WM tract involvement seems to be a valid surrogate to assess the in vivo spreading of pathological proteins. Therefore, characterizing network breakdown in neurodegenerative diseases will help anticipate and perhaps prevent the devastating impact of these disorders. However, the reviewed literature also arises several burning questions. First, the direction of pathology spreading in each neurodegenerative disease is still not completely understood. Longitudinal analyses of multimodal imaging datasets, involving subjects in the preclinical phase of the diseases, are currently being acquired to allow for more explicit testing of the hypothesis of predictable disease spread. In addition, new analyses techniques that relate those changes to underlying pathology, for example, tau imaging, will shed new light on how neurodegenerative diseases develop and spread. Finally, limited information is available about how selective vulnerability works and how pathological proteins interact with disease‐susceptible networks in these patients.
Conflict of Interest
F. Agosta serves on the editorial board of the Journal of Neurology; has received speaker honoraria from Biogen Idec and EXCEMED—Excellence in Medical Education; and receives research supports from the Italian Ministry of Health, and AriSLA (Fondazione Italiana di Ricerca per la SLA). M. Weiler reports no disclosures. M. Filippi is Editor‐in‐Chief of the Journal of Neurology; serves on scientific advisory boards for Teva Pharmaceutical Industries; has received compensation for consulting services and/or speaking activities from Bayer Schering Pharma, Biogen Idec, Merck Serono, Novartis, and Teva Pharmaceutical Industries; and receives research support from Bayer Schering Pharma, Biogen Idec, Merck Serono, Teva Pharmaceutical Industries, Novartis, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, Cure PSP, Alzheimer's Drug Discovery Foundation (ADDF), the Jacques and Gloria Gossweiler Foundation (Switzerland), and AriSLA (Fondazione Italiana di Ricerca per la SLA).
Acknowledgments
This review was partially supported by a grant from the Italian Ministry of Health (Grant #GR‐2011‐02351217).
References
- 1. Guo JL, Lee VM. Cell‐to‐cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014;20:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jucker M, Walker LC. Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013;501:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 5. Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013;74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP‐43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 2014;127:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greicius MD, Kimmel DL. Neuroimaging insights into network‐based neurodegeneration. Curr Opin Neurol 2012;25:727–734. [DOI] [PubMed] [Google Scholar]
- 8. Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large‐scale human brain networks. Neuron 2009;62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Migliaccio R, Agosta F, Rascovsky K, et al. Clinical syndromes associated with posterior atrophy: Early age at onset AD spectrum. Neurology 2009;73:1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mendez MF, Ghajarania M, Perryman KM. Posterior cortical atrophy: Clinical characteristics and differences compared to Alzheimer's disease. Dement Geriatr Cogn Disord 2002;14:33–40. [DOI] [PubMed] [Google Scholar]
- 13. Tang‐Wai DF, Graff‐Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174. [DOI] [PubMed] [Google Scholar]
- 14. Gorno‐Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008;71:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010;362:329–344. [DOI] [PubMed] [Google Scholar]
- 16. De Strooper B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol Rev 2010;90:465–494. [DOI] [PubMed] [Google Scholar]
- 17. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol 2013;12:609–622. [DOI] [PubMed] [Google Scholar]
- 18. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 19. Karch CM, Cruchaga C, Goate AM. Alzheimer's disease genetics: From the bench to the clinic. Neuron 2014;83:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Small SA, Duff K. Linking Abeta and tau in late‐onset Alzheimer's disease: A dual pathway hypothesis. Neuron 2008;60:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999;45:358–368. [DOI] [PubMed] [Google Scholar]
- 22. Moreno‐Trevino MG, Castillo‐Lopez J, Meester I. Moving away from amyloid Beta to move on in Alzheimer research. Front Aging Neurosci 2015;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron 2013;80:1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012;8:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Riedl L, Mackenzie IR, Forstl H, Kurz A, Diehl‐Schmid J. Frontotemporal lobar degeneration: Current perspectives. Neuropsychiatr Dis Treat 2014;10:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neary D, Snowden J, Mann D. Frontotemporal dementia. Lancet Neurol 2005;4:771–780. [DOI] [PubMed] [Google Scholar]
- 27. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955. [DOI] [PubMed] [Google Scholar]
- 28. Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: Nature of impairment and implications for assessment. Lancet Neurol 2013;12:368–380. [DOI] [PubMed] [Google Scholar]
- 29. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol 2010;119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 31. Lee EB, Lee VM, Trojanowski JQ. Gains or losses: Molecular mechanisms of TDP43‐mediated neurodegeneration. Nat Rev Neurosci 2012;13:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mackenzie IR, Rademakers R, Neumann M. TDP‐43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010;9:995–1007. [DOI] [PubMed] [Google Scholar]
- 33. Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet 2009;373:2055–2066. [DOI] [PubMed] [Google Scholar]
- 34. Chaudhuri KR, Schapira AH. Non‐motor symptoms of Parkinson's disease: Dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464–474. [DOI] [PubMed] [Google Scholar]
- 35. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol 2010;9:1200–1213. [DOI] [PubMed] [Google Scholar]
- 36. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 1998;95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha‐synuclein in neurological disorders. Lancet Neurol 2011;10:1015–1025. [DOI] [PubMed] [Google Scholar]
- 38. Baba M, Nakajo S, Tu PH, et al. Aggregation of alpha‐synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 39. Winner B, Jappelli R, Maji SK, et al. In vivo demonstration that alpha‐synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011;108:4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soto C. Prion hypothesis: The end of the controversy? Trends Biochem Sci 2011;36:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herva ME, Spillantini MG. Parkinson's disease as a member of Prion‐like disorders. Virus Res 2014. doi: 10.1016/j.virusres.2014.10.016. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 42. Recasens A, Dehay B. Alpha‐synuclein spreading in Parkinson's disease. Front Neuroanat 2014;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nonaka T, Masuda‐Suzukake M, Arai T, et al. Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep 2013;4:124–134. [DOI] [PubMed] [Google Scholar]
- 44. Biswal B, Yetkin FZ, Haughton VM, Hyde JS. Functional connectivity in the motor cortex of resting human brain using echo‐planar MRI. Magn Reson Med 1995;34:537–541. [DOI] [PubMed] [Google Scholar]
- 45. Smith SM, Fox PT, Miller KL, et al. Correspondence of the brain's functional architecture during activation and rest. Proc Natl Acad Sci U S A 2009;106:13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A 2001;98:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Basser PJ, Mattiello J, LeBihan D. Estimation of the effective self‐diffusion tensor from the NMR spin echo. J Magn Reson B 1994;103:247–254. [DOI] [PubMed] [Google Scholar]
- 48. Pierpaoli C, Barnett A, Pajevic S, et al. Water diffusion changes in Wallerian degeneration and their dependence on white matter architecture. NeuroImage 2001;13:1174–1185. [DOI] [PubMed] [Google Scholar]
- 49. Filippi M, Agosta F, Barkhof F, et al. EFNS task force: The use of neuroimaging in the diagnosis of dementia. Eur J Neurol 2012;19:e131–e140, 1487–1501. [DOI] [PubMed] [Google Scholar]
- 50. Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci 2009;29:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agosta F, Pievani M, Geroldi C, Copetti M, Frisoni GB, Filippi M. Resting state fMRI in Alzheimer's disease: Beyond the default mode network. Neurobiol Aging 2012;33:1564–1578. [DOI] [PubMed] [Google Scholar]
- 52. Greicius MD, Srivastava G, Reiss AL, Menon V. Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: Evidence from functional MRI. Proc Natl Acad Sci U S A 2004;101:4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sorg C, Riedl V, Muhlau M, et al. Selective changes of resting‐state networks in individuals at risk for Alzheimer's disease. Proc Natl Acad Sci U S A 2007;104:18760–18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weiler M, Fukuda A, Massabki LH, et al. Default mode, executive function, and language functional connectivity networks are compromised in mild Alzheimer's disease. Curr Alzheimer Res 2014;11:274–282. [DOI] [PubMed] [Google Scholar]
- 55. Petrella JR, Sheldon FC, Prince SE, Calhoun VD, Doraiswamy PM. Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology 2011;76:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hedden T, Van Dijk KR, Becker JA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci 2009;29:12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheline YI, Raichle ME, Snyder AZ, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 2010;67:584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Machulda MM, Jones DT, Vemuri P, et al. Effect of APOE epsilon4 status on intrinsic network connectivity in cognitively normal elderly subjects. Arch Neurol 2011;68:1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gili T, Cercignani M, Serra L, et al. Regional brain atrophy and functional disconnection across Alzheimer's disease evolution. J Neurol Neurosurg Psychiatry 2011;82:58–66. [DOI] [PubMed] [Google Scholar]
- 60. Zhang HY, Wang SJ, Liu B, et al. Resting brain connectivity: Changes during the progress of Alzheimer disease. Radiology 2010;256:598–606. [DOI] [PubMed] [Google Scholar]
- 61. Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer's disease. Neurobiol Aging 2012;33:828 e19–828 e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brier MR, Thomas JB, Snyder AZ, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer's disease progression. J Neurosci 2012;32:8890–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 2013;136:844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lehmann M, Madison CM, Ghosh PM, et al. Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer's disease. Proc Natl Acad Sci U S A 2013;110:11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pievani M, Agosta F, Pagani E, et al. Assessment of white matter tract damage in mild cognitive impairment and Alzheimer's disease. Hum Brain Mapp 2010;31:1862–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sexton CE, Kalu UG, Filippini N, Mackay CE, Ebmeier KP. A meta‐analysis of diffusion tensor imaging in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging 2011;32:2322 e5–2322 e18. [DOI] [PubMed] [Google Scholar]
- 67. Weiler M, de Campos BM, Nogueira MH, Pereira Damasceno B, Cendes F, Balthazar ML. Structural connectivity of the default mode network and cognition in Alzheimer's disease. Psychiatry Res 2014;223:15–22. [DOI] [PubMed] [Google Scholar]
- 68. Agosta F, Pievani M, Sala S, et al. White matter damage in Alzheimer disease and its relationship to gray matter atrophy. Radiology 2011;258:853–863. [DOI] [PubMed] [Google Scholar]
- 69. Maier‐Hein KH, Westin CF, Shenton ME, et al. Widespread white matter degeneration preceding the onset of dementia. Alzheimers Dement 2015;11:485–493.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Agosta F, Dalla Libera D, Spinelli EG, et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann Neurol 2014;76:813–825. [DOI] [PubMed] [Google Scholar]
- 71. Caso F, Agosta F, Mattavelli D, et al. White matter degeneration in atypical Alzheimer's disease. Radiology 2015; In press. [DOI] [PubMed] [Google Scholar]
- 72. Seeley WW, Crawford R, Rascovsky K, et al. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol 2008;65:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Seeley WW, Menon V, Schatzberg AF, et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci 2007;27:2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Uddin LQ. Salience processing and insular cortical function and dysfunction. Nat Rev Neurosci 2015;16:55–61. [DOI] [PubMed] [Google Scholar]
- 75. Zhou J, Seeley WW. Network dysfunction in Alzheimer's disease and frontotemporal dementia: Implications for psychiatry. Biol Psychiatry 2014;75:565–573. [DOI] [PubMed] [Google Scholar]
- 76. Farb NA, Grady CL, Strother S, et al. Abnormal network connectivity in frontotemporal dementia: Evidence for prefrontal isolation. Cortex 2013;49:1856–1873. [DOI] [PubMed] [Google Scholar]
- 77. Filippi M, Agosta F, Scola E, et al. Functional network connectivity in the behavioral variant of frontotemporal dementia. Cortex 2013;49:2389–2401. [DOI] [PubMed] [Google Scholar]
- 78. Zhou J, Greicius MD, Gennatas ED, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 2010;133:1352–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Day GS, Farb NA, Tang‐Wai DF, et al. Salience network resting‐state activity: Prediction of frontotemporal dementia progression. JAMA Neurol 2013;70:1249–1253. [DOI] [PubMed] [Google Scholar]
- 80. Agosta F, Scola E, Canu E, et al. White matter damage in frontotemporal lobar degeneration spectrum. Cereb Cortex 2012;22:2705–2714. [DOI] [PubMed] [Google Scholar]
- 81. Whitwell JL, Avula R, Senjem ML, et al. Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology 2010;74:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mahoney CJ, Ridgway GR, Malone IB, et al. Profiles of white matter tract pathology in frontotemporal dementia. Hum Brain Mapp 2014;35:4163–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lam BY, Halliday GM, Irish M, Hodges JR, Piguet O. Longitudinal white matter changes in frontotemporal dementia subtypes. Hum Brain Mapp 2014;35:3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dopper EG, Rombouts SA, Jiskoot LC, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology 2013;80:814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chiò A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M. Neuroimaging in amyotrophic lateral sclerosis: Systematic insight into structural and functional changes. Lancet Neurol 2014;13:1228–1240. [DOI] [PubMed] [Google Scholar]
- 86. Mohammadi B, Kollewe K, Samii A, Krampfl K, Dengler R, Munte TF. Decreased brain activation to tongue movements in amyotrophic lateral sclerosis with bulbar involvement but not Kennedy syndrome. J Neurol 2009;256:1263–1269. [DOI] [PubMed] [Google Scholar]
- 87. Jelsone‐Swain LM, Fling BW, Seidler RD, Hovatter R, Gruis K, Welsh RC. Reduced interhemispheric functional connectivity in the motor cortex during rest in limb‐onset amyotrophic lateral sclerosis. Front Syst Neurosci 2011;4:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tedeschi G, Trojsi F, Tessitore A, et al. Interaction between aging and neurodegeneration in amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:886–898. [DOI] [PubMed] [Google Scholar]
- 89. Fekete T, Zach N, Mujica‐Parodi LR, Turner MR. Multiple kernel learning captures a systems‐level functional connectivity biomarker signature in amyotrophic lateral sclerosis. PLoS One 2013;8:e85190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Verstraete E, van den Heuvel MP, Veldink JH, et al. Motor network degeneration in amyotrophic lateral sclerosis: A structural and functional connectivity study. PLoS One 2010;5:e13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Agosta F, Valsasina P, Absinta M, et al. Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cereb Cortex 2011;21:2291–2298. [DOI] [PubMed] [Google Scholar]
- 92. Douaud G, Filippini N, Knight S, Talbot K, Turner MR. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain 2011;134:3470–3479. [DOI] [PubMed] [Google Scholar]
- 93. Agosta F, Canu E, Valsasina P, et al. Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:419–427. [DOI] [PubMed] [Google Scholar]
- 94. Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler 2012;13:245–250. [DOI] [PubMed] [Google Scholar]
- 95. Agosta F, Canu E, Inuggi A, et al. Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol Aging 2014;35:916–925. [DOI] [PubMed] [Google Scholar]
- 96. Agosta F, Galantucci S, Riva N, et al. Intrahemispheric and interhemispheric structural network abnormalities in PLS and ALS. Hum Brain Mapp 2014;35:1710–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bede P, Bokde AL, Byrne S, et al. Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology 2013;81:361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Canu E, Agosta F, Galantucci S, et al. Extramotor damage is associated with cognition in primary lateral sclerosis patients. PLoS One 2013;8:e82017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lillo P, Mioshi E, Burrell JR, Kiernan MC, Hodges JR, Hornberger M. Grey and white matter changes across the amyotrophic lateral sclerosis‐frontotemporal dementia continuum. PLoS One 2012;7:e43993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kassubek J, Muller HP, Del Tredici K, et al. Diffusion tensor imaging analysis of sequential spreading of disease in amyotrophic lateral sclerosis confirms patterns of TDP‐43 pathology. Brain 2014;137:1733–1740. [DOI] [PubMed] [Google Scholar]
- 101. Pyatigorskaya N, Gallea C, Garcia‐Lorenzo D, Vidailhet M, Lehericy S. A review of the use of magnetic resonance imaging in Parkinson's disease. Ther Adv Neurol Disord 2014;7:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hacker CD, Perlmutter JS, Criswell SR, Ances BM, Snyder AZ. Resting state functional connectivity of the striatum in Parkinson's disease. Brain 2012;135:3699–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kwak Y, Peltier SJ, Bohnen NI, Muller ML, Dayalu P, Seidler RD. L‐DOPA changes spontaneous low‐frequency BOLD signal oscillations in Parkinson's disease: A resting state fMRI study. Front Syst Neurosci 2012;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wu T, Wang L, Chen Y, Zhao C, Li K, Chan P. Changes of functional connectivity of the motor network in the resting state in Parkinson's disease. Neurosci Lett 2009;460:6–10. [DOI] [PubMed] [Google Scholar]
- 105. Yang H, Zhou XJ, Zhang MM, Zheng XN, Zhao YL, Wang J. Changes in spontaneous brain activity in early Parkinson's disease. Neurosci Lett 2013;549:24–28. [DOI] [PubMed] [Google Scholar]
- 106. Choe IH, Yeo S, Chung KC, Kim SH, Lim S. Decreased and increased cerebral regional homogeneity in early Parkinson's disease. Brain Res 2013;1527:230–237. [DOI] [PubMed] [Google Scholar]
- 107. Esposito F, Tessitore A, Giordano A, et al. Rhythm‐specific modulation of the sensorimotor network in drug‐naive patients with Parkinson's disease by levodopa. Brain 2013;136:710–725. [DOI] [PubMed] [Google Scholar]
- 108. Agosta F, Caso F, Stankovic I, et al. Cortico‐striatal‐thalamic network functional connectivity in hemiparkinsonism. Neurobiol Aging 2014;35:2592–2602. [DOI] [PubMed] [Google Scholar]
- 109. Yoshikawa K, Nakata Y, Yamada K, Nakagawa M. Early pathological changes in the Parkinsonian brain demonstrated by diffusion tensor MRI. J Neurol Neurosurg Psychiatry 2004;75:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang K, Yu C, Zhang Y, et al. Voxel‐based analysis of diffusion tensor indices in the brain in patients with Parkinson's disease. Eur J Radiol 2011;77:269–273. [DOI] [PubMed] [Google Scholar]
- 111. Haller S, Badoud S, Nguyen D, Garibotto V, Lovblad KO, Burkhard PR. Individual detection of patients with Parkinson disease using support vector machine analysis of diffusion tensor imaging data: Initial results. AJNR Am J Neuroradiol 2012;33:2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gattellaro G, Minati L, Grisoli M, et al. White matter involvement in idiopathic Parkinson disease: A diffusion tensor imaging study. AJNR Am J Neuroradiol 2009;30:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Scherfler C, Schocke MF, Seppi K, et al. Voxel‐wise analysis of diffusion weighted imaging reveals disruption of the olfactory tract in Parkinson's disease. Brain 2006;129:538–542. [DOI] [PubMed] [Google Scholar]
- 114. Hattori T, Orimo S, Aoki S, et al. Cognitive status correlates with white matter alteration in Parkinson's disease. Hum Brain Mapp 2012;33:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Agosta F, Canu E, Stojkovic T, et al. The topography of brain damage at different stages of Parkinson's disease. Hum Brain Mapp 2013;34:2798–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Deng B, Zhang Y, Wang L, et al. Diffusion tensor imaging reveals white matter changes associated with cognitive status in patients with Parkinson's disease. Am J Alzheimers Dis Other Demen 2013;28:154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rae CL, Correia MM, Altena E, Hughes LE, Barker RA, Rowe JB. White matter pathology in Parkinson's disease: The effect of imaging protocol differences and relevance to executive function. NeuroImage 2012;62:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Agosta F, Canu E, Stefanova E, et al. Mild cognitive impairment in Parkinson's disease is associated with a distributed pattern of brain white matter damage. Hum Brain Mapp 2014;35:1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Filippi M, van den Heuvel M, Fornito A, et al. Assessing brain system dysfunction through MRI‐based connectomics. Lancet Neurol 2013;12:1189–1199. [DOI] [PubMed] [Google Scholar]
- 120. He Y, Chen Z, Evans A. Structural insights into aberrant topological patterns of large‐scale cortical networks in Alzheimer's disease. J Neurosci 2008;28:4756–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Supekar K, Menon V, Rubin D, Musen M, Greicius MD. Network analysis of intrinsic functional brain connectivity in Alzheimer's disease. PLoS Comput Biol 2008;4:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tijms BM, Wink AM, de Haan W, et al. Alzheimer's disease: Connecting findings from graph theoretical studies of brain networks. Neurobiol Aging 2013;34:2023–2036. [DOI] [PubMed] [Google Scholar]
- 123. Yao Z, Zhang Y, Lin L, Zhou Y, Xu C, Jiang T. Abnormal cortical networks in mild cognitive impairment and Alzheimer's disease. PLoS Comput Biol 2010;6:e1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lo CY, Wang PN, Chou KH, Wang J, He Y, Lin CP. Diffusion tensor tractography reveals abnormal topological organization in structural cortical networks in Alzheimer's disease. J Neurosci 2010;30:16876–16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang J, Zuo X, Dai Z, et al. Disrupted functional brain connectome in individuals at risk for Alzheimer's disease. Biol Psychiatry 2013;73:472–481. [DOI] [PubMed] [Google Scholar]
- 126. Agosta F, Sala S, Valsasina P, et al. Brain network connectivity assessed using graph theory in frontotemporal dementia. Neurology 2013;81:134–143. [DOI] [PubMed] [Google Scholar]
- 127. Verstraete E, Veldink JH, Mandl RC, van den Berg LH, van den Heuvel MP. Impaired structural motor connectome in amyotrophic lateral sclerosis. PLoS One 2011;6:e24239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Verstraete E, Veldink JH, van den Berg LH, van den Heuvel MP. Structural brain network imaging shows expanding disconnection of the motor system in amyotrophic lateral sclerosis. Hum Brain Mapp 2014;35:1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Schmidt R, Verstraete E, de Reus MA, Veldink JH, van den Berg LH, van den Heuvel MP. Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum Brain Mapp 2014;35:4386–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Baggio HC, Sala‐Llonch R, Segura B, et al. Functional brain networks and cognitive deficits in Parkinson's disease. Hum Brain Mapp 2014;35:4620–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Skidmore F, Korenkevych D, Liu Y, He G, Bullmore E, Pardalos PM. Connectivity brain networks based on wavelet correlation analysis in Parkinson fMRI data. Neurosci Lett 2011;499:47–51. [DOI] [PubMed] [Google Scholar]
- 132. Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron 2012;73:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 2012;73:1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]