

Summary
Aims
Epileptic seizures are well‐known neurological complications following stroke, occurring in 3% of patients. However, the intrinsic correlation of seizures with stroke remains largely unknown. Hydrogen sulfide (H2S) is a gas transmitter that may mediate cerebral ischemic injury. But the role of H2S in seizures has not been understood yet. We examined the effect of H2S on seizure‐like events (SLEs) and underlying mechanisms.
Methods and Results
Pentylenetetrazole (PTZ)‐ and pilocarpine‐induced rat epileptic seizure models were tested. Low‐Mg2+/high‐K+‐ and 4‐aminopyridine (4‐AP)‐induced epileptic seizure models were examined using patch‐clamp recordings in brain slices. It was found that NaHS aggravated both PTZ‐ and pilocarpine‐induced SLEs in rats, while both low‐Mg2+/high‐K+‐ and 4‐AP‐induced SLEs were also exacerbated by NaHS in brain slices, which may be due to its regulation on the voltage‐gated sodium channel, N‐methyl‐D‐aspartic acid receptor (NMDAR), and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPAR) function. Furthermore, these effects were reversed by blocking voltage‐gated sodium channel, NMDAR, and AMPAR.
Conclusions
These results suggest a pathological role of increased H2S level in SLEs in vivo and in vitro. Enzymes that control H2S biosynthesis could be interesting targets for antiepileptic strategies in poststroke epilepsy treatment.
Keywords: Epilepsy, Glutamate receptor, Hydrogen sulfide, Seizure, Voltage‐gated sodium channel
Introduction
Seizure is the clinical manifestation of a hyperexcitable network, in which the electrical balance underlying normal neuronal activity is pathologically altered, which affects ~1% of the global population 1. Although antiepileptic drugs (AEDs) are available, effective symptomatic relief achieved only in about two‐thirds of the patients 2. Acquired diseases including trauma, central nervous system (CNS) infections, hypoxic‐ischemic and metabolic disorders, tumors, and vascular abnormalities predict the development of epilepsy 3. Additionally, epileptic seizures are well‐known neurological complications following stroke, occurring in 3% of patients, particularly within the first 24 h after stroke. Approximately 1–2% of stroke patients present with status epilepticus 4. However, the pathological correlation of seizures with stroke remains largely unknown, and there is no effective drug to rescue the biological events leading to epileptogenesis after stroke.
Hydrogen sulfide (H2S) is known to be a toxic gas and an environmental hazard for many decades. However, it is recognized that H2S has been classified as a third “gasotransmitter” signaling molecule alongside nitric oxide (NO) and carbon monoxide (CO) 5. Several important biological actions of H2S have been identified in recent years, including regulation of blood pressure, insulin release, cytoprotection, muscle relaxation, and cellular excitability 5, 6. Moreover, H2S is involved in the pathophysiology of many diseases, including inflammation, atherosclerosis, hypertension, myocardial infarction, diabetes, sexual dysfunction, Alzheimer's disease (AD), Parkinson's disease (PD), and stroke 5, 7, 8, 9, 10, 11. Interestingly, a growing body of literature has documented a close link between endogenous H2S signal and cerebral ischemia. Cerebral ischemia causes an increase in H2S level in the lesioned cortex as well as an increase in the H2S synthesizing activity in a short time, indicating that H2S appears to act as a mediator of ischemic injury 12. In a small clinical trial, Wong et al. 13 found that high plasma cysteine (a precursor of H2S) level was correlated with poor clinical outcome 3 months after acute stroke. Ren et al. 14 found that the amount of H2S in the hippocampus was increased significantly at 12 h of reperfusion after cerebral ischemia in rats. On the other hand, H2S can maintain the excitatory/inhibitory balance in neurotransmission and regulate the activity of a number of targets involved in seizures such as ion channel and NMDA receptor (NMDAR) 15, 16. Moreover, one survivor of accidental H2S inhalation toxicity developed status epilepticus and severe neurotoxicity in Sri Lanka 17. All of the above evidence suggests that H2S may serve as a risk factor for seizures after stroke. However, the effect and underlying mechanisms of H2S on seizures have not been elucidated. Here, we demonstrated that pathophysiological concentration (200 μM) of NaHS, a donor of H2S 15, 18, aggravated seizure‐like events (SLEs) in rats in vivo and in vitro, which may be due to an increase in neuronal excitation.
Materials and methods
Slice Preparation
The research was conducted in accordance with the Declaration of Helsinki. All experimental protocols were approved by the Review Committee for the Use of Human or Animal Subjects of Huazhong University of Science and Technology.
Entorhinal cortex (EC) slices of rat were prepared as described previously with minor modification 19. In brief, Sprague‐Dawley rats (postnatal 15–25 day) were decapitated, and transverse EC slices (350 μm) were prepared using a Vibroslice (VT 1000S; Leica, Germany) in ice‐cold ACSF. The slice‐cutting solution contained 220 mM sucrose, 2.5 mM KCl, 1.3 mM CaCl2, 2.5 mM MgSO4, 1 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose, pH 7.4, whereas the ACSF contained 129 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.8 mM MgSO4, 1.6 mM CaCl2, 21 mM NaHCO3, and 10 mM glucose, pH 7.4. After at least 1 h of recovery at room temperature (25 ± 1°C), an individual slice was transferred to a submerged recording chamber and continuously superfused with ACSF at room temperature (25 ± 1°C) at a rate of 3–4 mL/min. All solutions were saturated with 95% O2/5% CO2 (vol/vol).
Electrophysiological Recording
Recordings in brain slice were made with a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) in conjunction with Digidata 1322A digitizer (Molecular Devices) and Clampex 10.0 software. Low‐Mg2+/high‐K+‐induced SLEs were obtained according to a previous study with some modifications 20. SLEs were induced by omitting of Mg2+ from ACSF (low‐Mg2+ condition), and the extracellular K+ concentration ([K+]o) was elevated to 8 mM (high‐K+ condition). 4‐AP‐induced SLEs were induced by adding 50 μM 4‐AP into ACSF. When the current‐clamp mode was used to record the changes in cell membrane potential and firing properties, an SLE was defined as at least five consecutive action potentials 21.
Chemicals, other electrophysiological recordings, lateral ventricle cannulation and injection, animal models, and data analysis were as described in Data S1.
Results
More recent studies have shown that H2S is present in mammalian tissues up to 50–160 μM 22, 23 as measured in rat, human, and bovine brain tissues in physiological conditions. The level of H2S in the brain after stroke is about one time more than control 12, 14. Thus, 200 μM H2S was used to mimic pathophysiological concentration in vivo and in vitro.
H2S Facilitates Pentylenetetrazole (PTZ)‐ and Pilocarpine‐Induced SLEs in Rat In Vivo
We investigated the role of H2S in SLEs in vivo using two epilepsy models as mentioned in Data S1. First, PTZ‐induced epilepsy model was adopted 24. As shown in Figure 1A, the seizure severity was increased in NaHS‐treated groups compared with control. In our experiments, 2 of 8 rats in 75 mg/kg PTZ group were dead; thus, a lower dose of 60 mg/kg PTZ was further employed. In addition, the seizure latency was significantly shortened by NaHS treatment from 152.5 ± 8.5 s to 96.2 ± 5.9 s (Student's t‐test, P < 0.01 vs. control; Figure 1B), and the duration of seizure was obviously increased from 15.3 ± 2.3 s to 61.8 ± 10.8 s by NaHS treatment (Student's t‐test, P < 0.01 vs. control; Figure 1C).
Figure 1.
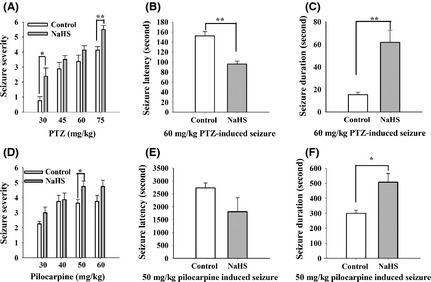
NaHS facilitates pentylenetetrazole (PTZ)‐ and pilocarpine‐induced epilepsy in rat. (A) The seizure severity was increased in NaHS‐treated group than in thecontrol group, especially for the seizure induced by 30 mg/kg and 75 mg/kg PTZ (n = 8, Mann–Whitney rank sum test, *P < 0.05, **P < 0.01). (B) The seizure latency was shortened in NaHS‐treated group (control: n = 4, NaHS: n = 6, unpaired t‐test, P < 0.01). (C) The seizure duration was prolonged in NaHS‐treated group (control: n = 4, NaHS: n = 6, unpaired t‐test, P < 0.01). (D) The seizure severity was increased in NaHS‐treated group than in the control group, especially for the seizure induced by 50 mg/kg pilocarpine (n = 8, Mann–Whitney rank sum test, *P < 0.05). (E) The seizure latency was not changed in NaHS‐treated group (n = 4, unpaired t‐test). (F) The seizure duration was prolonged in NaHS‐treated group (n = 4, unpaired t‐test, P < 0.05).
To further confirm whether H2S could facilitate SLEs in vivo, the type 1 muscarinic receptor agonist pilocarpine‐induced epilepsy model was used. As shown in Figure 1D, the seizure severity was increased in NaHS‐treated groups, especially the seizure induced by 50 mg/kg pilocarpine (P < 0.05 vs. control). Moreover, NaHS prolonged the duration of seizure from 300.00 ± 20.41 to 507.0 ± 58.0 s (Student's t‐test; P < 0.05 vs. control; Figure 1F), but failed to affect the seizure latency (control: 2737.5 ± 191.1 s, NaHS: 1807.5 ± 541.2 s; Student's t‐test; Figure 1E). However, the seizure severity was not induced by NaHS alone (n = 8), indicating that NaHS facilitates, but not induced SLEs in rat in vivo.
H2S Aggravates low‐Mg2+/high‐K+‐ and 4‐AP‐Induced SLEs in Rat Brain Slices In Vitro
Next, we investigated the role of H2S in SLEs in vitro using two SLE models. The EC receives input from the parahippocampus, prefrontal cortex, and frontal cortex and then projects to the hippocampus via the perforant path and the temporoammonic path. It serves as a gateway to the hippocampus and plays a pivotal role in epileptogenesis and seizures. Furthermore, electrical stimulation of EC interferes with the activity of specific brain regions, including the amygdaloid nucleus, piriform cortex, and hippocampus. EC is a potential target for low‐frequency stimulation to interfere with amygdaloid‐kindling seizures in vivo 25. We used low‐Mg2+/high‐K+ models of epilepsy in EC to explore the effect of H2S on seizures 26. As shown in Figure 2A, the onset latency of first SLEs was significantly shorter in 200 μM NaHS‐treated slices when compared with control slices (236 ± 43 s, NaHS: 417 ± 64 s; Student's t‐test, P < 0.05; Figure 2B). The duration of SLEs was significantly longer in NaHS‐treated slices than that of control slices (control: 70 ± 19 s, NaHS: 173 ± 32 s; Student's t‐test, P < 0.01; Figure 2C).
Figure 2.
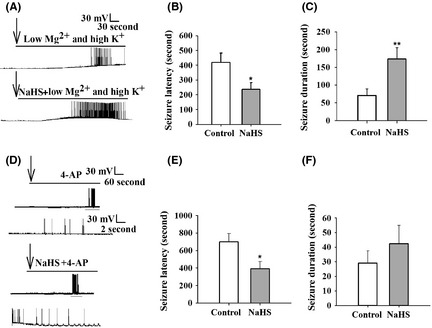
NaHS facilitates low‐Mg2+/high‐K+‐ and 4‐AP‐induced SLEs in rat brain slices. (A) Representative traces of SLEs in control and 200 μM NaHS‐treated slices using the current‐clamp mode. The arrows indicated treatment. (B) The latency of seizure was shortened in NaHS‐treated group. (C) The duration of seizure was increased in NaHS‐treated group. Control: n = 14, NaHS: n = 10, unpaired t‐test. *P < 0.05; **P < 0.01 versus control. (D) Representative traces of SLEs in control and 200 μM NaHS‐treated slices. The arrows indicated 4‐AP and NaHS treatment. The lowers are the expanded traces within gray line of the uppers. (E) The latency of seizure induced by 4‐AP was shortened by NaHS treatment. (F) The duration of seizure induced by 4‐AP was not changed in NaHS‐treated group. Control: n = 10, NaHS: n = 11, unpaired t‐test. *P < 0.05 versus control.
We further investigated the effect of NaHS on 4‐AP‐induced SLE. It is well known that 4‐AP interferes with different types of K+ channels, including D‐ and A‐type K+ currents and a subportion of delayed rectifier currents 27. As shown in Figure 2D, we found that NaHS decreased the onset latency of SLEs significantly (control: 698 ± 95 s, NaHS; 391 ± 83 s; Student's t‐test, P < 0.05; Figure 2E), although the duration of SLEs was not obviously increased by NaHS treatment (control: 29 ± 8 s, NaHS: 42 ± 13 s; Student's t‐test; Figure 2F), indicating that NaHS facilitates SLEs in vitro.
H2S Increases the Excitability of EC Neurons in Rat Brain Slices
Next, we used current clamp to inject depolarizing current and counted action potentials fired (i.e., input–output determination) to explore the excitability of neurons. Injection of constant current into neurons of NaHS‐treated EC slices resulted in more action potentials fired at the same time compared with controls (Figure 3A, B), indicating that NaHS increases the membrane excitability of EC neurons.
Figure 3.
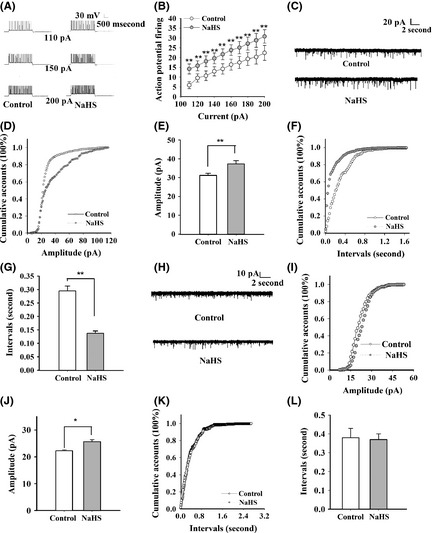
NaHS facilitates the action potential firing and the neuron network activity. (A) Representative traces showing the firing of action potentials by successively greater depolarizing current injections (110, 150, and 200 pA/3.5 s) in control and 200 μM NaHS‐treated groups. (B) The number of action potential firing was increased in NaHS‐treated group (control: 5.9 ± 1.8, NaHS: 14.2 ± 2.5, 110 pA/3.5 s; control: 14.2 ± 2.6, NaHS: 21.7 ± 2.7, 150 pA/3.5 s; control: 22.4 ± 3.7, NaHS: 30.8 ± 3.4, 200 pA/3.5 s). n = 10, paired t‐test, **P < 0.01 versus control. (C) Representative sEPSC traces in control and NaHS‐treated slices. (D, E) The amplitude of sEPSC was increased in NaHS‐treated group (n = 6, P < 0.01). (F, G) The interval of sEPSC was decreased in NaHS‐treated group (n = 6, P < 0.01). (H) Representative mEPSC traces in control and NaHS‐treated slices. (I, J) The amplitude of mEPSC was increased in NaHS‐treated group (n = 5, P < 0.05). (K, L) The interval time of mEPSC was not obviously increased in NaHS‐treated group (n = 5). *P < 0.05; **P < 0.01 versus control; Kolmogorov–Smirnov test.
We then wondered whether neuronal network excitability was changed. sEPSCs were recorded at a holding potential of −70 mV, in the presence of 100 μM picrotoxin. It was shown that the mean amplitude of sEPSCs was significantly increased in NaHS‐treated slices compared with control (control: 31.16 ± 1.09 pA, NaHS: 37.32 ± 1.68 pA; Kolmogorov–Smirnov test; P < 0.01; Figure 3C–E), indicating that NaHS affects the postsynaptic function. Meanwhile, the interevent interval sEPSC was significantly decreased in NaHS‐treated slices compared with control (control: 0.29 ± 0.02 s, NaHS: 0.14 ± 0.01 s; Kolmogorov–Smirnov test; P < 0.01; Figure 3F, G).
The decreased time interval might be resulted from the increased presynaptic transmitter release. To further confirm whether presynaptic transmitter release was changed, mEPSC was recorded in the presence of 1 μM TTX. It was shown that NaHS increased the mEPSC amplitude (control: 22.24 ± 0.38 s, NaHS: 25.69 ± 0.66 pA; Kolmogorov–Smirnov test; P < 0.05; Figure 3H–J), but not the time interval of mEPSC (control: 0.38 ± 0.05 s, NaHS: 0.37 ± 0.03 s; Kolmogorov–Smirnov test; Figure 3K, L). These results suggest that the changes in action potential and postsynaptic mechanisms contribute to NaHS‐facilitated SLEs.
H2S Activates Voltage‐Gated Sodium Channel of EC Neurons in Rat Brain Slices
Voltage‐gated sodium currents contribute to intrinsic membrane hyperexcitability, including action potential of neurons. To test whether sodium current was affected by H2S, sodium currents were recorded from EC neurons of P6–P8 rat pups, which allowed for adequate clamping of sodium currents and excluded synaptic involvement. Voltage‐gated sodium currents were evoked by voltage commands as described in Data S1. A major finding of our studies was the profound increase in the peak sodium current amplitude (2–9 nA) induced by NaHS after 2–5 min of treatment (121.31 ± 3.92% of control; Student's t‐test; P < 0.01; Figure 4A, B). We also found that NaHS did not affect the shape of I–V curve, the steady‐state activation, steady‐state inactivation, and steady‐state reactivation curves of voltage‐gated sodium current (Figure 4C–F). As voltage‐gated sodium current is important for action potential burst and membrane excitability, it is likely that alterations in this current contribute to NaHS‐induced neuronal hyperexcitability.
Figure 4.
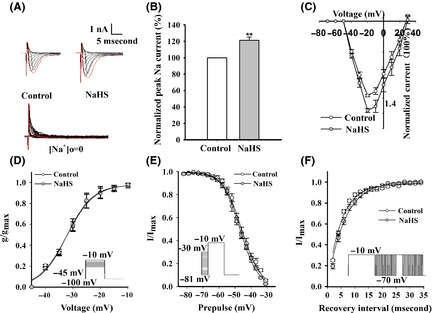
Effects of H2S on voltage‐gated sodium channel of EC neurons in rat brain slices. (A) Representative traces of voltage‐gated sodium currents, which were elicited by stepping from −70 mV to +30 mV. The red trace represented the maximum current. Voltage‐gated sodium currents cannot be elicited, while sodium chloride was removed by choline chloride in ACSF as shown in the bottom. (B) The normalized current–voltage curves before and after 200 μM NaHS treatment (n = 6). (C) NaHS increased the peak sodium currents of EC neurons (n = 6). (D) NaHS had no obvious effects on the steady‐state activation of sodium current (n = 5). Voltage dependence of activation V1/2 (mV) was −32.12 ± 0.85 in control group and −32.32 ± 0.88 in NaHS‐treated group. Schematic diagram of protocol is on the right. (E) NaHS did not shift the steady‐state inactivation of sodium current (n = 5). Voltage dependence of inactivation V1/2 (mV) was −45.95 ± 0.28 in control group and −46.87 ± 0.20 in NaHS‐treated group. Schematic diagram of protocol is on the left. (F) NaHS had no effect on the steady‐state reactivation of sodium current (n = 7). τ was 5.03 ± 0.8 ms in control group and 6.32 ± 0.9 ms in NaHS‐treated group. Schematic diagram of protocol is on the right. *P < 0.05; **P < 0.01 versus control, unpaired t‐test.
H2S Facilitates the Function of AMPAR and NMDAR in EC Neurons
NaHS increased the amplitude of mEPSC, and we next explored the effect of H2S on the function of postsynaptic receptors. eEPSCs were recorded as mentioned in Data S1. We showed that the amplitude of eEPSC was significantly increased in NaHS‐treated groups compared with control after 2–5 min of treatment (NaHS‐treated: 146.4 ± 10.9%, washout: 127.2 + 10.0%; F 2,24 = 7.394, P = 0.003; Figure 5A, B). Then, both AMPAR‐ and NMDAR‐mediated eEPSCs were recorded. We found that the amplitude of AMPAR‐mediated eEPSC was significantly increased in NaHS‐treated group compared with control (NaHS‐treated: 143.5 ±6.6%, washout: 124.2 + 5.0%; F 2,24 = 20.567, P < 0.001; Figure 5C, D). Furthermore, the amplitude of NMDAR‐mediated eEPSC was increased in NaHS‐treated group (NaHS‐treated: 128.1 ± 3.1%, washout: 114.0 + 3.1%; F 2,18 = 30.05, P < 0.001; Figure 5E, F). These results indicate that NaHS facilitates the postsynaptic function via AMPAR and NMDAR.
Figure 5.
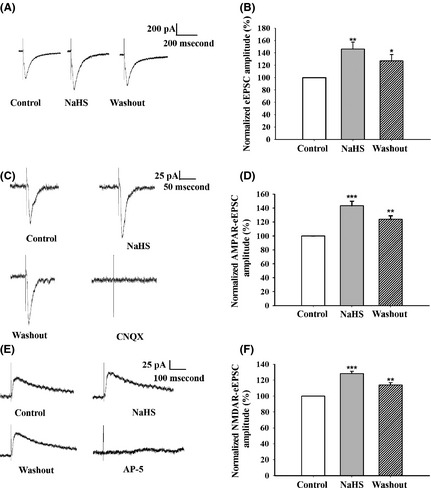
NaHS facilitates the function of postsynaptic glutamate receptors. (A) Representative traces of eEPSC under the condition of control, NaHS, and washout. (B) The amplitude of eEPSC was increased in NaHS‐treated group (n = 9). (C) Representative traces of AMPAR‐mediated eEPSC under the condition of control, NaHS, washout, and AMPAR antagonist 20 μM CNQX. (D) The amplitude of AMPAR‐mediated eEPSC was increased in NaHS‐treated group (n = 9). (E) Representative traces of NMDAR‐mediated eEPSC under the condition of control, NaHS, washout, and NMDAR antagonist 100 μM AP‐5. (F) The amplitude of NMDAR‐eEPSC was increased in NaHS‐treated group (n = 7). *P < 0.05, **P < 0.01, ***P < 0.001 versus control, one‐way ANOVA.
The Effects of H2S are Reversed by Blockade of Voltage‐Gated Sodium Channel, NMDAR, and AMPAR In Vitro and In Vivo
We have found that H2S facilitated SLEs in vitro and in vivo via enhancing the function of voltage‐gated sodium channel, NMDAR, and AMPAR. However, whether blocking these channels will reverse or reduce the effects of H2S is still unknown. To test the hypothesis, we carried out the experiments in low‐Mg2+/high‐K+‐ and PTZ‐induced SLEs.
Interestingly, it was found that TTX prevented low‐Mg2+/high‐K+‐induced change in the cell membrane potential and firing properties and also inhibited the effects of H2S (Figure 6A). Moreover, in the presence of 100 μM AP‐5 or 20 μM CNQX, although the action potentials were still induced, NaHS cannot cause SLEs (Figure 6B, C)
Figure 6.
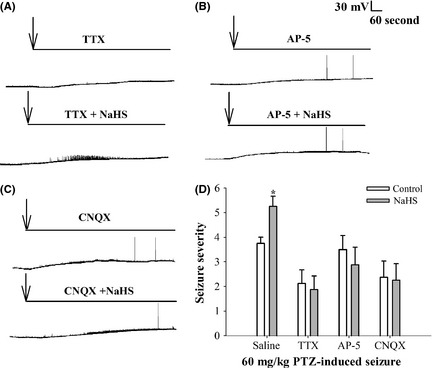
The effects of H2S are reversed by blockade of voltage‐gated sodium channel, NMDAR, and AMPAR in vitro and in vivo. (A) Representative traces of 1 μM TTX and 1 μM TTX + 200 μM NaHS‐treated slices in low‐Mg2+/high‐K+ model using the current‐clamp mode. The arrows indicated the treatment. (B) Representative traces of 100 μM AP‐5 and 100 μM AP‐5 + 200 μM NaHS‐treated slices in low‐Mg2+/high‐K+ model using the current‐clamp mode. The arrows indicated the treatment. (C) Representative traces of 20 μM CNQX and 20 μM CNQX +200 μM NaHS‐treated slices in low‐Mg2+/high‐K+ model using the current‐clamp mode. The arrows indicated the treatment. (D) The seizure severity was increased in NaHS‐treated group than in the control group. TTX, AP‐5, or CNQX inhibited NaHS‐aggregated seizure severity (n = 8, Mann–Whitney rank sum test, *P < 0.05 vs. control).
Next, we tested the effects of blocking agents on PTZ model. Sixty‐four rats were randomly divided into eight groups. NaHS and 60 mg/kg PTZ were intraperitoneally injected 5 min after lateral ventricle injection of blocking agents or equal volume saline as mentioned in supplemental materials and methods. We found that the seizure severity was increased in the presence of NaHS (n = 8, Mann–Whitney rank sum test, *P < 0.05); however, the effect was inhibited by pretreatment with TTX, AP‐5, or CNQX (Figure 6D). These results suggest that blockade of voltage‐gated sodium channel, NMDAR, or AMPAR could prevent the facilitated effect of NaHS on SLEs.
Discussion
H2S is recognized as a toxic gas for centuries. For decades, more and more attention has been paid to the physiological and pathophysiological of H2S, including cerebral ischemia 12, 13, 14. Few reports focused on the effect of H2S on SLEs. In the present study, we provided evidence for the first time that H2S may serve as a pathological risk factor that facilitated SLEs in vivo and in vitro. The major findings in the present study are as follows: (1) NaHS, a donor of H2S, in a pathological concentration (200 μM), facilitated PTZ‐ and pilocarpine‐induced epilepsy of rat, (2) NaHS facilitated low‐Mg2+/high‐K+‐ and 4‐AP‐induced SLEs in rat brain slices, (3) NaHS enhanced the excitability of neuron and neuronal network via facilitating the function of voltage‐gated sodium channel, AMPAR, and NMDAR, and (4) the effect of H2S is reversed by blocking voltage‐gated sodium channel, NMDAR, and AMPAR.
Animal models have played a substantial role in understanding the pathophysiology and treatment of human epilepsies. The PTZ test identifies drugs with efficacy against generalized nonconvulsive seizures 28, 29. The pilocarpine‐treated rat model is also adopted in many studies 30, and the changes along with NMDAR upregulation in parahippocampal areas such as the EC have been reported in this model 31. In the low‐Mg2+/high‐K+‐induced epilepsy model, the generation of SLEs has been attributed to the reduced surface charge of neuronal membrane and the disinhibition of NMDAR followed by calcium influx 32. 4‐AP can induce SLEs by inhibition of potassium channel and facilitation of action potential 27. Thus, both neuronal and network hyperexcitabilities have been induced in these models. 200 μM NaHS facilitates SLE in these models mainly via modulating the function of voltage‐gated ion channels and glutamate receptors.
Neuronal excitability is determined by the properties of the ion channels in the neuronal membrane, so that the aberrant excitability associated with an epileptic discharge is necessarily mediated by voltage‐gated and ligand‐gated ion channels and may even be the result of defects in the function of these channels. Voltage‐gated sodium channels play a significant role in epilepsy, and an enhanced function of these channels results in SLE 33. H2S is known to target various ion channels, such as facilitating KATP channels in vascular smooth muscle cells, stimulating L‐type Ca2+ channels in neurons, inhibiting big‐conductance KCa channels, and activation of Cl− channels 16. We found that NaHS (200 μM) increases amplitude but did not affect the shape of I–V curve, the steady‐state activation, steady‐state inactivation, and steady‐state reactivation curves of voltage‐gated sodium current in neurons. The increase in Na+ peak current may be from increased number of Na+ channels available for opening at threshold, accounting for reaching action potential threshold quickly, increased conduction velocity in neurons. Strege et al. 34 found NaHS (1 and 10 mM) increases native Na+ peak currents in human jejunum smooth muscle cells and interstitial cells of Caj and shifts the half‐point (V 1/2) of steady‐state activation and inactivation, respectively. This difference is thoughtful, because NaHS in lower concentration (200 μM) does not where in higher concentrations (1, 10 mM) does affect steady‐state activation and inactivation curves of voltage‐gated sodium current. Moreover, different doses of NaHS may have distinctive effects on different tissues and cells. Chao et al. 18 found NaHS increases Na+ influx in a concentration‐dependent manner in the cortex. NaHS (150 μM)‐evoked Na+ influx could be entirely blocked by TTX, while NaHS (300 μM) could be partially blocked by TTX or NMDAR blocker MK 801. We found NaHS (200 μM) enhances the function of voltage‐gated sodium channels and NMDAR, which is in accordance with their finding. Moreover, we provided direct evidence for the first time that NaHS enhances the function of TTX‐sensitive voltage‐gated sodium channels of neurons in brain slices via electrophysiological methods. Thus, we conclude that H2S aggravates SLEs via regulation of neuronal excitation.
Apart from voltage‐gated sodium channels, ligand‐gated ion channels, such as AMPAR and NMDAR, are also particularly important in an epileptic discharge 33. The AMPAR is large multisubunit protein complex that spans the membrane and has an ion‐selective central pore that is closed to ion flow in the absence of glutamate. Binding of glutamate causes the AMPAR to gate open, which allows cations to flux across the postsynaptic membrane, resulting in a brief depolarization known as the excitatory postsynaptic potential (EPSP) 35. AMPAR is thought to play a key role in inducing seizures by initiating and synchronizing glutamatergic transmission. AMPA itself, administered systemically or by cerebroventricular infusion, is able to elicit seizures in preclinical models, thereby supporting a role for AMPA receptors in the development of seizures 33. Activation of the NMDAR has been implicated in a number of disease states including stroke and neurodegenerative and seizure disorders. Increased NR2B in both extrasynaptic and presynaptic neuronal compartments and a concomitant decrease in this subunit in postsynaptic density‐95 (PSD‐95) protein contribute to excitotoxicity in epileptogenesis 36. H2S is shown to maintain the excitatory/inhibitory balance in neurotransmission. It selectively stimulates NMDAR‐mediated currents via activation of adenylyl cyclase and the subsequent cyclic adenosine monophosphate (cAMP)–protein kinase A (PKA) cascades 37. Here, we found that NaHS (200 μM) enhanced the function of NMDAR, which is in consistent with the results from Abe and Kimura 15. Moreover, the function of AMPAR was also enhanced. Partly because H2S activates cAMP–PKA cascades and then induces GluR1 serine 845, phosphorylation enhances the function of AMAPR 38. It is interesting that NaHS at a short period (2–5 min) of drug treatment was able to influence the synaptic receptors. Moreover, the effect of NaHS persisted after washout, suggesting that the effect of NaHS on glutamate receptors is largely mediated through a direct not an indirect way. Recent studies show that cysteine residues can be sulfhydrated by H2S and this redox‐dependent posttranslational modification (PTM) controls the activity of ion channels 39, 40. H2S can exert its function by sulfhydrating cysteine residues of key potassium channels in a DTT‐sensitive manner 40. The effect of H2S on NaV1.5 is blocked by DTT pretreatment in human jejunum smooth muscle cells 34. Thus, we hypothesize that H2S modulates glutamate receptor activity via a direct sulfhydration‐dependent mechanism. These results indicate that H2S facilitates SLEs partly via enhancing the postsynaptic function of AMPAR and NMDAR.
The effect of H2S on SLEs was prevented by the blockers of voltage‐gated sodium channel, AMPAR, and NMDAR, indicating that some AEDs, including felbamate, topiramate, or zonisamide, exert their effects by blocking of Na+ channel 33, AMPAR antagonists such as perampanel 41, and NMDAR antagonists such as traxoprodil 42 which could also be used in the treatment with H2S‐aggregated SLEs.
Although our data focus only on voltage‐gated sodium channel, NMDAR, and AMPAR and our previous study showed that NaHS (200 μM) facilitated voltage‐gated calcium channel while suppressed voltage‐gated potassium channel (data not shown), other targets could be investigated including hyperpolarization‐activated cyclic nucleotide‐gated cation (HCN) channel, voltage‐gated chloride channel, acid‐sensing ion channel, nicotinic cholinergic receptor, glycine receptor, G‐protein‐coupled receptors, and electrotonic coupling through gap junctions which present various modalities of brain wiring 33. In other studies, NaHS has been reported to upregulate the expression of GABARB subunits 1 and 2 43. Future studies will also address whether the function of GABAR is changed after H2S treated in SLEs.
Epilepsy is difficult to diagnosis promptly, usually by history or electroencephalogram (EEG) when epilepsy is happening 44. According to our results, H2S and its regulatory enzymes that control H2S biosynthesis are potential biomarkers in diagnosis of epilepsy. Moreover, inhibiting H2S biosynthesis enzymes could be an interesting strategy for poststroke epilepsy treatment.
In conclusion, our results demonstrated that the H2S aggregated SLEs, in vivo and in vitro, involvement of facilitating voltage‐gated sodium, enhancing function of NMDAR and AMPAR. Our studies indicated that H2S or enzymes that control H2S biosynthesis are potential new targets for treating epilepsy.
Disclosure
The authors declare that they have no competing financial interests.
Supporting information
Data S1. Supplemental materials and methods.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC) to Dr. F. Wang (No.81222048), the National Basic Research Program of China (973 Program, No.2013CB531303), and the International Science & Technology Cooperation Program of China (No.2011DFA32670) to Dr. J.G. Chen.
The first two authors contributed equally to this work.
References
- 1. Sander JW. Some aspects of prognosis in the epilepsies: a review. Epilepsia 1993;34:1007–1016. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt D, Rogawski MA. New strategies for the identification of drugs to prevent the development or progression of epilepsy. Epilepsy Res 2002;50:71–78. [DOI] [PubMed] [Google Scholar]
- 3. Jacobs MP, Leblanc GG, Brooks‐Kayal A, et al. Curing epilepsy: progress and future directions. Epilepsy Behav 2009;14:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Reuck J, Van Maele G. Status epilepticus in stroke patients. Eur Neurol 2009;62:171–175. [DOI] [PubMed] [Google Scholar]
- 5. Hu LF, Lu M, Hon Wong PT, Bian JS. Hydrogen sulfide: neurophysiology and neuropathology. Antioxid Redox Signal 2011;15:405–419. [DOI] [PubMed] [Google Scholar]
- 6. Kimura H, Nagai Y, Umemura K, Kimura Y. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal 2005;7:795–803. [DOI] [PubMed] [Google Scholar]
- 7. Eto K, Asada T, Arima K, Makifuchi T, Kimura H. Brain hydrogen sulfide is severely decreased in Alzheimer's disease. Biochem Biophys Res Commun 2002;293:1485–1488. [DOI] [PubMed] [Google Scholar]
- 8. Hu LF, Lu M, Tiong CX, Dawe GS, Hu G, Bian JS. Neuroprotective effects of hydrogen sulfide on Parkinson's disease rat models. Aging Cell 2010;9:135–146. [DOI] [PubMed] [Google Scholar]
- 9. Hu LF, Wong PT, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide‐induced inflammation by inhibition of p38 mitogen‐activated protein kinase in microglia. J Neurochem 2007;100:1121–1128. [DOI] [PubMed] [Google Scholar]
- 10. Kida K, Yamada M, Tokuda K, et al. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson's disease. Antioxid Redox Signal 2011;15:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang XQ, Yang CT, Chen J, et al. Effect of hydrogen sulphide on beta‐amyloid‐induced damage in PC12 cells. Clin Exp Pharmacol Physiol 2008;35:180–186. [DOI] [PubMed] [Google Scholar]
- 12. Qu K, Chen CP, Halliwell B, Moore PK, Wong PT. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006;37:889–893. [DOI] [PubMed] [Google Scholar]
- 13. Wong PT, Qu K, Chimon GN, et al. High plasma cyst(e)ine level may indicate poor clinical outcome in patients with acute stroke: possible involvement of hydrogen sulfide. J Neuropathol Exp Neurol 2006;65:109–115. [DOI] [PubMed] [Google Scholar]
- 14. Ren C, Du A, Li D, Sui J, Mayhan WG, Zhao H. Dynamic change of hydrogen sulfide during global cerebral ischemia‐reperfusion and its effect in rats. Brain Res 2010;1345:197–205. [DOI] [PubMed] [Google Scholar]
- 15. Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 1996;16:1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang G, Wu L, Wang R. Interaction of hydrogen sulfide with ion channels. Clin Exp Pharmacol Physiol 2010;37:753–763. [DOI] [PubMed] [Google Scholar]
- 17. Shivanthan MC, Perera H, Jayasinghe S, et al. Hydrogen sulphide inhalational toxicity at a petroleum refinery in Sri Lanka: a case series of seven survivors following an industrial accident and a brief review of medical literature. J Occup Med Toxicol 2013;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao D, He X, Yang Y, et al. Hydrogen sulfide induced disruption of Na+ homeostasis in the cortex. Toxicol Sci 2012;128:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen YJ, Zhang M, Yin DM, et al. ErbB4 in parvalbumin‐positive interneurons is critical for neuregulin 1 regulation of long‐term potentiation. Proc Natl Acad Sci USA 2010;107:21818–21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovacs R, Rabanus A, Otahal J, et al. Endogenous nitric oxide is a key promoting factor for initiation of seizure‐like events in hippocampal and entorhinal cortex slices. J Neurosci 2009;29:8565–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qi J, Wang Y, Jiang M, Warren P, Chen G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J Physiol 2006;571:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goodwin LR, Francom D, Dieken FP, et al. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J Anal Toxicol 1989;13:105–109. [DOI] [PubMed] [Google Scholar]
- 23. Warenycia MW, Reiffenstein RJ, Goodwin LR, Dieken FP. Brain sulfide levels in anaesthesia: a comparison with hydrogen sulfide intoxication. Toxicol Lett 1989;47:221–224. [DOI] [PubMed] [Google Scholar]
- 24. Bachiega JC, Blanco MM, Perez‐Mendes P, Cinini SM, Covolan L, Mello LE. Behavioral characterization of pentylenetetrazol‐induced seizures in the marmoset. Epilepsy Behav 2008;13:70–76. [DOI] [PubMed] [Google Scholar]
- 25. Xu ZH, Wu DC, Fang Q, et al. Therapeutic time window of low‐frequency stimulation at entorhinal cortex for amygdaloid‐kindling seizures in rats. Epilepsia 2010;51:1861–1864. [DOI] [PubMed] [Google Scholar]
- 26. Holtkamp M, Buchheim K, Elsner M, Matzen J, Weissinger F, Meierkord H. Status epilepticus induces increasing neuronal excitability and hypersynchrony as revealed by optical imaging. Neurobiol Dis 2011;43:220–227. [DOI] [PubMed] [Google Scholar]
- 27. Wahab A, Albus K, Gabriel S, Heinemann U. In search of models of pharmacoresistant epilepsy. Epilepsia 2010;51(Suppl 3):154–159. [DOI] [PubMed] [Google Scholar]
- 28. Higgins GA, Breysse N, Undzys E, et al. Comparative study of five antiepileptic drugs on a translational cognitive measure in the rat: relationship to antiepileptic property. Psychopharmacology 2010;207:513–527. [DOI] [PubMed] [Google Scholar]
- 29. Hu WW, Fang Q, Xu ZH, et al. Chronic h1‐antihistamine treatment increases seizure susceptibility after withdrawal by impairing glutamine synthetase. CNS Neurosci Ther 2012;18:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baraka AM, Hassab El Nabi W, El Ghotni S. Investigating the role of zinc in a rat model of epilepsy. CNS Neurosci Ther 2012;18:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Guzman P, Inaba Y, Baldelli E, de Curtis M, Biagini G, Avoli M. Network hyperexcitability within the deep layers of the pilocarpine‐treated rat entorhinal cortex. J Physiol 2008;586:1867–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones RS, Heinemann U. Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium‐free medium in vitro. J Neurophysiol 1988;59:1476–1496. [DOI] [PubMed] [Google Scholar]
- 33. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics 2007;4:18–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strege PR, Bernard CE, Kraichely RE, et al. Hydrogen sulfide is a partially redox‐independent activator of the human jejunum Na+ channel, Nav1.5. Am J Physiol Gastrointest Liver Physiol 2011;300:G1105–G1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 2011;11:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frasca A, Aalbers M, Frigerio F, et al. Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis 2011;43:507–515. [DOI] [PubMed] [Google Scholar]
- 37. Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun 2000;267:129–133. [DOI] [PubMed] [Google Scholar]
- 38. Hu H, Real E, Takamiya K, et al. Emotion enhances learning via norepinephrine regulation of AMPA‐receptor trafficking. Cell 2007;131:160–173. [DOI] [PubMed] [Google Scholar]
- 39. Peers C, Bauer CC, Boyle JP, Scragg JL, Dallas ML. Modulation of ion channels by hydrogen sulfide. Antioxid Redox Signal 2012;17:95–105. [DOI] [PubMed] [Google Scholar]
- 40. Mustafa AK, Sikka G, Gazi SK, et al. Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 2011;109:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russo E, Gitto R, Citraro R, Chimirri A, De Sarro G. New AMPA antagonists in epilepsy. Expert Opin Investig Drugs 2012;21:1371–1389. [DOI] [PubMed] [Google Scholar]
- 42. Naspolini AP, Cocco AR, Villa Martignoni F, et al. Traxoprodil decreases pentylenetetrazol‐induced seizures. Epilepsy Res 2012;100:12–19. [DOI] [PubMed] [Google Scholar]
- 43. Han Y, Qin J, Chang X, Yang Z, Bu D, Du J. Modulating effect of hydrogen sulfide on gamma‐aminobutyric acid B receptor in recurrent febrile seizures in rats. Neurosci Res 2005;53:216–219. [DOI] [PubMed] [Google Scholar]
- 44. Friedman D, Honig LS, Scarmeas N. Seizures and epilepsy in Alzheimer's disease. CNS Neurosci Ther 2012;18:285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental materials and methods.